Synthesis, Characterization and Reaction of New Ruthenium Aromatic Thioamide Nitrosyls with some Nitrogen Donor Ligands
K.K. Singh
Department of Chemistry, T.D.P.G. College, Jaunpur) India.
Corresponding Author E-mail : kksingh5678@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/320426
Article Received on :
Article Accepted on :
Article Published : 08 Aug 2016
Reactions of LH=RCSNHCOOEt (R= 4-tolyl, 1-pyrrolyl, and 2- thiophenyl) with RuCl3. 3H2O and NO leads to the formation of diamagnetic compounds of the formula [L2Ru(NO)(Cl)J. These compounds have been characterised by elemental analysis and IR, UV and Visible and magnetic measurements. The presence of (NO) at 1835-1855-cm-1 indicates a {Ru-NO}6 configuration. The substitution of Cl bound cis to NO by N-hetrocycles bases (a-picoline, y-picoline and or pyridine) and CH3CN has significant effect on the electronic properties of complexes.
KEYWORDS:Ruthenium nitrosyl; Ruthenium; hypsochromic shift
Download this article as:| Copy the following to cite this article: Singh K. K. Synthesis, Characterization and Reaction of New Ruthenium Aromatic Thioamide Nitrosyls with some Nitrogen Donor Ligands. Orient J Chem 2016;32(4). |
| Copy the following to cite this URL: Singh K. K. Synthesis, Characterization and Reaction of New Ruthenium Aromatic Thioamide Nitrosyls with some Nitrogen Donor Ligands. Orient J Chem 2016;32(4). Available from: http://www.orientjchem.org/?p=20352 |
Introduction
The chemistry of metal nitrosyls that release NO has gained an intensive area of research interest during the recent years, since such species could be used in certain biological processes, like neurotransmission, tumor growth inhibition, hepatic metabolism, cell differentiation and blood pressure regulation.1,2 The metal nitrosyls that release NO under photochemical condition has attracted much attention. For example NO donors that are capable of releasing NO upon illumination could find use as antitumor agents in Photo Dynamic Theraphy (PDT).3-5 Non heme iron nitrosyls such as sodium nitropruside Na2[Fe(CN)5NO] and Roussian’s salts (NH4[Fe3S3(NO)7], Na2[Fe2S2 (NO)4] are also known to release NO under photochemical condition Unfortunately release of NO from these NO donors is dependent on enzymatic reaction, heat or pH and hence target specificity is mostly precluded6. Release of auxiliary ligands from metal base NO donors like SNP also leads to deleterious side effect. In contrast, the compounds that release NO strictly upon exposure to light could provide a more selective means of NO delivery. Ruthenium nitrosyls are particularly attractive in this regard owing to their thermal stability and NO release upon exposure to light.7-9 The NO release, in most of the octahedral ruthenium nitrosyls having {RuII NO+} moiety, can be induced by one electron reduction by photolysis or PDT. In general, it has been found that NO release depends markedly on the p– acceptor strength of the ligand coordinated trans to NO and increase in plane ligand field strength.
Despite the large number of ruthenium nitrosyls described in the literature10-13, a few examples of ruthenium nitrosyls in sulphur rich environment14-16 are found to release NO upon irradiation to visible light. This prompts us to explore the chemistry of ruthenium nitrosyls complexes in sulphur rich environment. Further, aromatic thioamides can form stable complexes with transition metal ions.17-19 The impetus for this and similar work arises from the wide ranging applications of sulphur and nitrogen containing ligands in medicine.20-22
The title ligand (LH) is capable of possessing diverse coordinating ability on changing R,
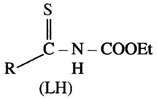
R = 4- tolyl, N-carboethoxy-4-toluenethioamide (Hctt)
R= 1-pyrrolyl, N-carboethoxy-1-pyrrolethioamide (Hcpt)
R= 2- thiophenyl, N- carboethoxy-2-thiophenethioamide ( Hcett)
As a part of our continuing investigations on the transition metal complexes of aromatic thioamides, herein we report the synthesis, characterization and possible structures of ruthenium nitrosyls of the title ligand.
Experimental
Materials and Measurements
All the chemicals used were either of AR or chemically pure grade. A literature method was used to prepare LH23, Elemental analyses (C, H and N) were carried out by Microanalytical section of I.I.T. Kanpur (U.P.) India. Sulphur chloride, fluoride and boran, were estimated by standard methods.24 The IR (4000-250 cm) spectra of the ligand and complexes were recorded in KBr pellets with a Perkin-Elmer FT IR spectrometer. UV and Visible spectra of the ligand and complexes were recorded on a cystronic 108 UV and Visible spectrometer from 200 – 900 nm. RuCl3. XH2O was purchased from Aldrich chemicals Co. and was treated several times with concentrated HCl to prepare the starting material RuCl3. 3H2O. All the solvents were purified and or dried by standard techniques and distilled prior to use. Nitric Oxide was generated by driping concentrated sulphuric acid on NaNO2, passing it through a saturated aqueous solution of NaOH and then bubbling through solution/ suspension.
Preparation of Complexes
[(ctt)2Ru(NO)(Cl)]
To a 30 ml DMF solution of LH (0.7Ig, 3.2 mmol) was added slurry of NaH (0.08 g, 3.2 mmol) in 5mL DMF and the resultant solution was stirred for one hour. The colour of solution turned to orange brown when evolution of gas stopped. Solid RuCl3.3H2O (O.42g, 1.6 mmol) was added to this solution. The color of the reaction mixture changed to red brown immediately. It was refluxed for 5h under N2 atmosphere and cooled to room temperature and filtered through a cintered glass crucible to remove NaCl. Purified NO gas was allowed to pass and the resultant solution was stirred for 5h, then concentrated to 10 mL.Et2O (10 mL) was added to this solution where by a reddish purple crystalline precipitate of the complex was formed. It was filtered off and washed with dry Et2O and dried in vacuo. Yield: 0.73g (75%).
[(cpt)2Ru(NO)Cl]
This complex was prepared by the procedure described in 1 except that Hept (0.63 g, 3.2 mmol), NaH (0.08, 3.2 mmol) and RuCl3. 3H2O (0,42, 1.6 mmol) were used in a total volume of 35 mL dry DMF. It was refluxed for 3h in N2 atmosphere and filtered off to remove NaCl. The solution was cooled and purified NO gas was bubbled for Ih; the reaction mixture was concentrated to lOmL. Et2O lOmL was added to the solution whereby a pale reddish brown crystalline precipitate of the complex was formed. It was filtered and washed with Et2O and dried in vacuum. Yield: 0.67g, (75 %).
[(cett)2Ru(NO)Cl]
This complex was prepared by the same method as described in 1 except that Hcett (0.68g, 3.2 mmol), NaH (0.08, 3.2 mmol) and RuCl3. 3H20 (0.42g, 1.6 mmol) were used in a total volume of 35 mL dry DMF. It was refluxed for 8h in N2 atmosphere and filtered off in a centered glass crucible. The solution was cooled to 10°c and purified, NO gas was bubbled for 2h. The reaction mixture was concentrated to 10 mL whereby a red brown crystalline precipitate of the complex separated. It was filtered and washed with a little amount of Et2O and dried in vacuum yield: 0.67g, (70%).
[(ctt)2Ru(NO)(B)] BF4 (B= a-picoline, g- picoline, or pyridine)
To a 25mL CH3CN solution of [(ctt)2Ru(NO)Cl] (Immol) was added 5mL CH3CN solution of AgBF4 (Immol). This reaction mixture was heated to reflux for 20h. The precipitated AgCl during this time was removed by filtration through a cintered glass crucible. 0.15g of ligands (B) was added to the stirred solution (filtrate). The resultant solution was further stirred for 6h whereby a red brown solution is formed. It was reduced to 10mL and on adding dry Et2O a crystalline precipitate of the complex was formed. It was filtered and washed with Et2O and dried in Vacuo. Yield: 0.59g, (80%).
[(ctt)2Ru(NO)(CH3CN)]BF4
This complex was prepared by the same procedure as described in pyridine complex. The resultant solution was stirred for 6h and concentrated to 10 mL. On adding Et2O (10 mL), a yellow brown crystalline complex was formed which was filtered and washed with Et2O and dried in vacuum. Yield: 0.45g, (65%).
Results and Discussion
Synthesis of the Complexes
In the present work, the reaction of NaH with stirred solution of title ligand (LH) in dry DMF resulted the deprotonation of ligand. The reaction is given as
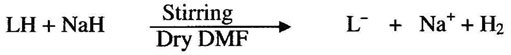
RuCl3H2O was allowed to react with deprotonated ligand (L”) whereby after prolonged heating the color of the reaction mixture changed to with the formation of 2 equivalents of NaCl
![]()
This suggest that [L2Ru(DMF)Cl] type species is present in the solution. After passing NO, through the cold solution of this intermediate, desired metal nitrosyls (1-3) were obtained in 70-75% yield
![]()
Reaction of chloro complex [(ctt)2Ru(NO)Cl] (L= ctt) with AgBF4 results in the formation of CH3CN bound yellow brown intermediate [ctt)2Ru(NO)(CH3CN)]BF4 type species by halide displacement reaction.
![]()
on adding N hetrocycles bases B to this yellow brown solution displacement of CH3CN by B occurs with the formation of [(ctt)2Ru(NO)(B)] .
[(ctt)2Ru(NO)(CH3CN)]BF4 + B →[(ctt)2Ru(NO)(B)] BF4 + CH3CN
IR Spectra
The ligand LH contains a thioamide25-27 group ( H-N-C=S and give rise to four characteristic thioamide bands, namely I, II, III and IV having contributions from d (C-H) + d (N-H), u(C=S) + u(C=N) + u(C-H), u(C-N) + u(C-S) and u
(C—S) modes of vibrations respectively (Table 2) The disappearance of u(NH) bands of the ligand (3200-3240) cm-1 from the spectra of all the complexes indicate the displacement of N-H hydrogen by Ru+3 and the formation of metal nitrogen bond. All the complexes (1-6) display strong carbonyl stretching frequencies (CO) in the range 1750-1770 cm-1, indicating a blue shift of (CO) in complexes compared to the free ligand (Table 2). This confirms the coordination of the ligands in their deprotonated forms in these complexes and non involvement of carbonyl group of COOEt in bonding. It is highly unlikely for the oxygen atom of -OEt group to participate in the complex formation. The thioamide band I of the ligands at 1540-1550 cm-1 shift to lower wave number by 20-25 cm-1 in the spectra of complexes. This indicates M-N bond formation as the thioamide band I has principal contribution from d (C-H) + d (N-H).
Thioamide band II of the ligands (1340-1400) shilfts to lower wave number as a result of coordination through thiocarbonyl sulphur. The thioamide band III is relatively weak in the spectra of the complexes. There is a red shift of 15-20 cm-1 in thioamide band IV (major contribution from u(C=S) in the spectra of all the complexes. This further indicates the bonding through thiocartbonyl sulphur. A red slift of 25-55 cm-1 in thioamide band IV usually indicates the bonding through sulphur and a blue shift of 40-90 cm-1 in this band indicates bonding through nitrogen. However, if there is simultaneous coordination through nitrogen and sulphur, there may not be much of the shift in band IV. Hence the small shift in band IV is attributed to simultaneous coordination through nitrogen and sulphur.
Two bands around (685-695 cm-1 and (630-660 cm-1 assigned to t(NH) of the ligand disappear in the spectra of complexes, further supports the deprotonation of NH group of the ligand.19-28
The appearance of new medium intensity bands in the region 300- 450 cm-1 is strong evidence of coordination by N, S and Cl. These bands have been assigned to coupled vibrations of u(Ru-N), u(Ru-Cl) and u(Ru-S).
A single strong NO stretching band u (NO) is observed for each of the 7 complexes15,29 (Table 2). These values fall in the expected range for {RuNO}6 type complexes. In RCS NH COOEt, the electron donating ability of R is expected to be 4-tolyl > 1-pyrolyl > 2-thiophenyl. Thus, the ligand field strength decreases in the order Hctt>Hcpt>Hcett. The stretching frequencies of NO at 1842, 1848 and 1855 in compound 1,2,and 3 respectively, supports the expections in the change of ligand field strength as R is varied. The stretching frequencies of NO in compound 4, 5, 6 and 7 are 1835, 1835, 1840 and 1845 respectively. This is smaller than u(NO) in compound 1-3, indicating a better a donor ability of N hetrocycles and CH3CN than chloride19, 30.
The weak broad band at 3500 cm-1 in the spectra of [(ctt)2Ru(NO)B]BF4 (B= pyridine, y-picoline and a-picoline are characteristic of pyridine and substituted pyridine, shift towards higher frequency
(+80cm-1) . In these complexes, the bands in free pyridine or substituted pyridine at 605-615 cm-1 (in plane ring deformation) and 405-410 cm-1 (out of plane ring deformation) shift to higher frequencies. In the spectra of complexes, there seems to be extensive mixing between the bands of ligands and those of bases, hence the other characteristic frequencies of N-hetrocycles did not occur at their standard positions.19, 30
UV and Visible spectra
The UV and Visible spectra of the ligands and complexes were recorded in CH3CN from 200-900 nm. The intensity of the bands (e=102-103) indicates that the bands are charge transfer and not d-d transition bands. The systematic assignments of all the bands of ligands and complexes are given in Table 3. The absorption bands at (395-368) are very much dependent on the ligand field strength and basicity of ligands bound cis to NO. Stronger ligand field strength (Hctt)>Hcpt> Hcett) increases the N-O bond length (decreases uNO, lesser NO+ character). This is nicely supported by u(NO) values (Table 2). These bands are assigned to dp (Ru) → p* (NO) (MLCT)9 There is a red shift (max. 5 nm) of the band at 370 nm by ligands B (a- pic, 395; y- pic, 395; py, 390; and CH,CN, 368.) bound cis to NO. This red shift of the CT bands arises from the coordination of more basic N-hetrocycles that increase electron density at the metal center and facilitate dp (Ru) → p* (NO) transition at lower energy.
The other band at 345-360 nm in assigned to dp (Ru) → dp (S) on the basis that this transition requires larger amount of energy than dp (Ru) → p* (NO). This is observed empirically, that all the sulphur containing ligands lie at the lower field end of the spectrochemical series31. From the general point of view -C=S → M bond of prevailing s character should induce a fractional positive charge on the sulphur atom and thereby decrease the bond order of – C=S group, with a consequent decrease of u(C=S). On the other hand, subsequent contribution of metal to sulphur p back donation should diminish the sulphur positive charge and increase the bond order of -C=S group. In presence of only a donating N atom of the ligand trans to sulphur, the dp (Ru) → dp (S) increases28. A small downward shift (10-20 cm-1) of u(C=S) compared to free ligand indicates the presence of N atom of one ctt trams to S atom of other ctt. This supports the extensive back donation of electrons from metal filled dp orbital to sulphur empty dp. orbital.
The bands at 300, 350 and 450 nm in free ligands (Hcpt, Hcett, and Hctt respectively), disappear in the spectra of complexes due to stabilization of the lone pair of electrons on complex formation, and similarly the hypsochromic shift in the bands in the spectra of complexes may be explained as that metal ion is bonded through thiocarbonyl sulphur and nitrogen. The bond formation lowers the energy of non bonding orbital as well as p levels, thus now more energy will be required to promote the electron from then n or p level to p* orbital, hence the observed hypsochromic shift19.
The entire compounds are diamagnetic in solid state and solution.
Thus, on the basis of IR, UV-visible and magnetic studies, we proposed the following tentative structures of complexes.
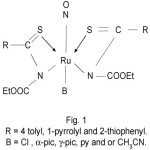 |
Figure 1 Click here to View Figure |
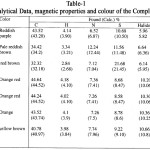 |
Table 1 Click here to View table |
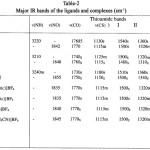 |
Table 2 Click here to View table |
 |
Table 3 Click here to View table |
References
- Bonavida, B.; Khineche, S.; Huerta-yepez, S.; and Garban, h; Therapeutic Potential of Nitric oxide in Cancer. Drug. Resist Updat. 2006, 9, 157-173.
CrossRef - Fricker, S.P.; Platinum Metal Rev. 1995, 59, 150-159.
- Tfouni, E.; Krieger, M.; McGarvey, B.; and Franco, D.W.; Coord. Chem. Rev. 2003, 236, 57-59.
CrossRef - Serli, B.; Zangrando, E.; Lengo, E.;, Mestroni, G.; Yellowleas, L; and Alessio, E.; Inorg. Chem. 2002, 41, 4033-4043.
CrossRef - Rose, M.J.; Fry, N.L.; Marlow, R.; Hinck, L.; and Mascharak, P.K.; J. Am. Chem. Soc. 2008, 130, 8834-8846.
CrossRef - Butler, A.R.; Chem. Rev. 2002, 102, 1155-1165.
CrossRef - Rose, M.J.; Patra, A.K.; Alcid, E.A.; Olmstead, M.M.; and Mascharak, P.K.; Inorg. Chem. 2007, 46, 2328-2338.
CrossRef - Patra, A.K.; Rose, M.J.; Murphy, A.; Olmstead, M.M.; and Mascharak, P.K.; Inorg. Chem. 2004, 43, 4487-4495.
CrossRef - Rose, M.J.; Olmstead, M.M.; and Mascharak, P.K.; J. Am. Chem. Soc. 2007, 129, 5342-5343.
CrossRef - Hayton, T.W.; Legzdins, P.; and Sharp, W.B.; Chem. Rev. 2002, 102(4), 935-992.
CrossRef - Wang, H.; Xie, Y.; King, R.B.; and Schaefer, H.F., Inorg. Chem. 2008, 47, 3045-3055.
CrossRef - Wang, H.; Xie, Y.; Jhang J.D.; King, R.B.; and Schaefer, H.F.; Inorg. Chem. 2007, 46, 1336-1346.
- Golfeto, C.C.; Poethsitz, G.V.; Selistre-de-Araujo, S.H.; de Arays, M.P.; Ellena, J.; Castellano, E.E.; Lopes, L.G.L.; Moreira, I.S.; and Batista, A.A.; Inorg. Biochem., 2010, 104, 489-495.
CrossRef - Moffett, S.M.; Gunter, K.L.; Kreisel, K.A.; Yop. G.P.A.; and Rabinovich, D.; Polyhedron. 2007, 26, 4758-4764.
- Cheung, Wai-Man.; Zhang, Qian-Feng,; Lai, Chui-Ying.; Williams, I.D.; and Leung, Wa-Hung.; Polyhedron. 2007, 26, 4631-4637.
CrossRef - Prakash, R.; Czaja, A.U., Heinemann, F.W.; and Shelmann, D.; J. Am. Chem. Soc. 2005, 127, 13758-13767.
CrossRef - Singh, T.; and Singh, R.N.; React. Inrog. Met-Org. Chem. 1989, 19(3), 251-262.
- Chauhan, Veena.; and Dikshit, S.K.; Synth. React.Inorg. Met-org. Chem. 1986, 16(10), 1435-1452.
- Chauhan, Veena; and Dikshit, S.K.; Bull. Chem. Soc. Jpn. 1987, 60, 3005-3009.
CrossRef - Ferrari, M.B.; Capacchi, S.; Pelosi, G.; Reffo, G.; Tarasconi, P.; Albertini, R.; Pinelli, S.; and Lunghi, P.; Inorg. Chem. Acta. 1999, 286, 134-141.
CrossRef - Canpolat, E.; and Kaya, M.; J. Coord. Chem. 2004, 57, 1217-1223.
CrossRef - Saghatforoush, L.A.; Aminkhani, A.; Ersad, S.; Karimnezhad, G.; Ghammamy, S.; and Kabiri, R.; Molecules. 2008, 13, 804-811.
CrossRef - Papadopouls, E.P.; J. Org. Chem. 1973, 38, 667-674. J. Org. Chem. 1976, 41, 962-972.
- Vogel, A.I. A Text Book of Quantitative Inorganic Analysis. IV ed., ELBS and Longmans. London. 1985, 433-434, 504-507, 494-495, 154-155.
- Singh, T.; and Agarwala, U.; Indian J. Chem. 1980, 19A, 991-993.
- Pandey, R.N.; and Nag, A.K.; Rasayan. J. Chem. 2009, 2, 990-993.
- Chauhan, Veena.; Gupta, H.K.; and Dikshit, S.K.; Polyhedron. 1988, 7, 623-628.
CrossRef - Alessio, E.; Balducci, G.; Calligaris, M.; Costa, G.; Attia, W.M.; and Mestroni, G.; Inorg. Chem. 1991, 30, 609-618.
CrossRef - Andrews, L.; and Citra, A.; Chem. Rev. 2002, 102, 885-991.
CrossRef - Wikinson, G.; and Gilbert, J.D.; J. Chem. Soc.(A), 1969, 1749-1753.
- Huheey, J.E.; Keiter, E.A.; and Keter, R.; Inoranic Chemistry, Principal of structure and reactivity, 4th ed., Addison Longman, Singapore, 2001, 421-424.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


