
Characterization of P-Glycoprotein Inhibitors for Evaluating the Effect of P-Glycoprotein on the Intestinal Absorption of Drugs

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Animals
2.4. Transport/Inhibition Experiments
2.5. Preparation of Drug Solution for In Vivo Study
2.6. Analytical Methods
- 0–15.0 min, 45% acetonitrile
- 15.0–25.0 min, 45–70% acetonitrile
- 25.0–30.0 min, 70% acetonitrile
- 30.0–40.0 min, 70–45% acetonitrile
- 40.0–50.0 min, 45% acetonitrile
2.7. Data Analysis
2.8. Statistical Analysis
3. Results
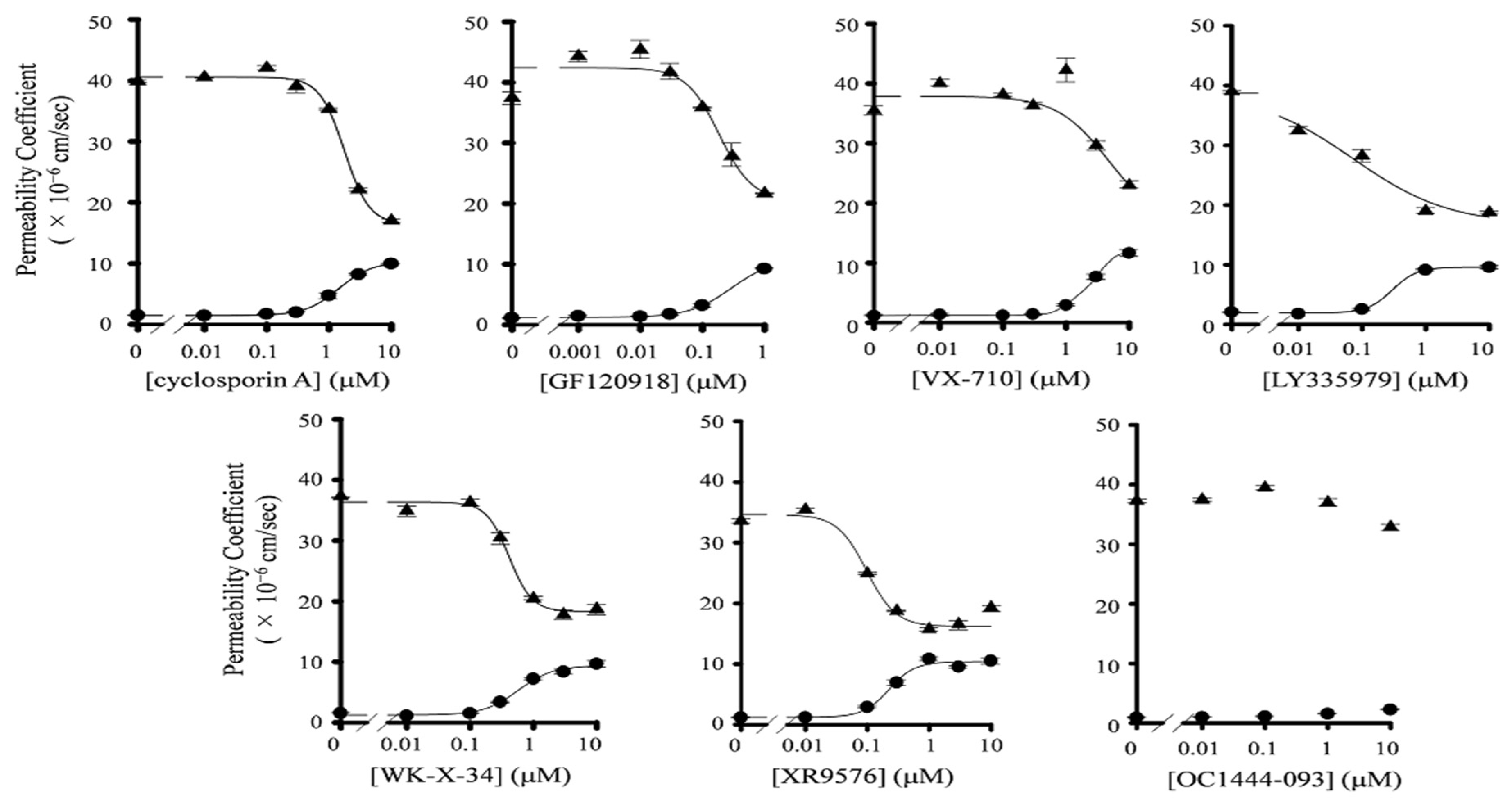
3.1. Inhibitory Effect of P-gp Inhibitors on the Transport of Paclitaxel in Caco-2 Cells
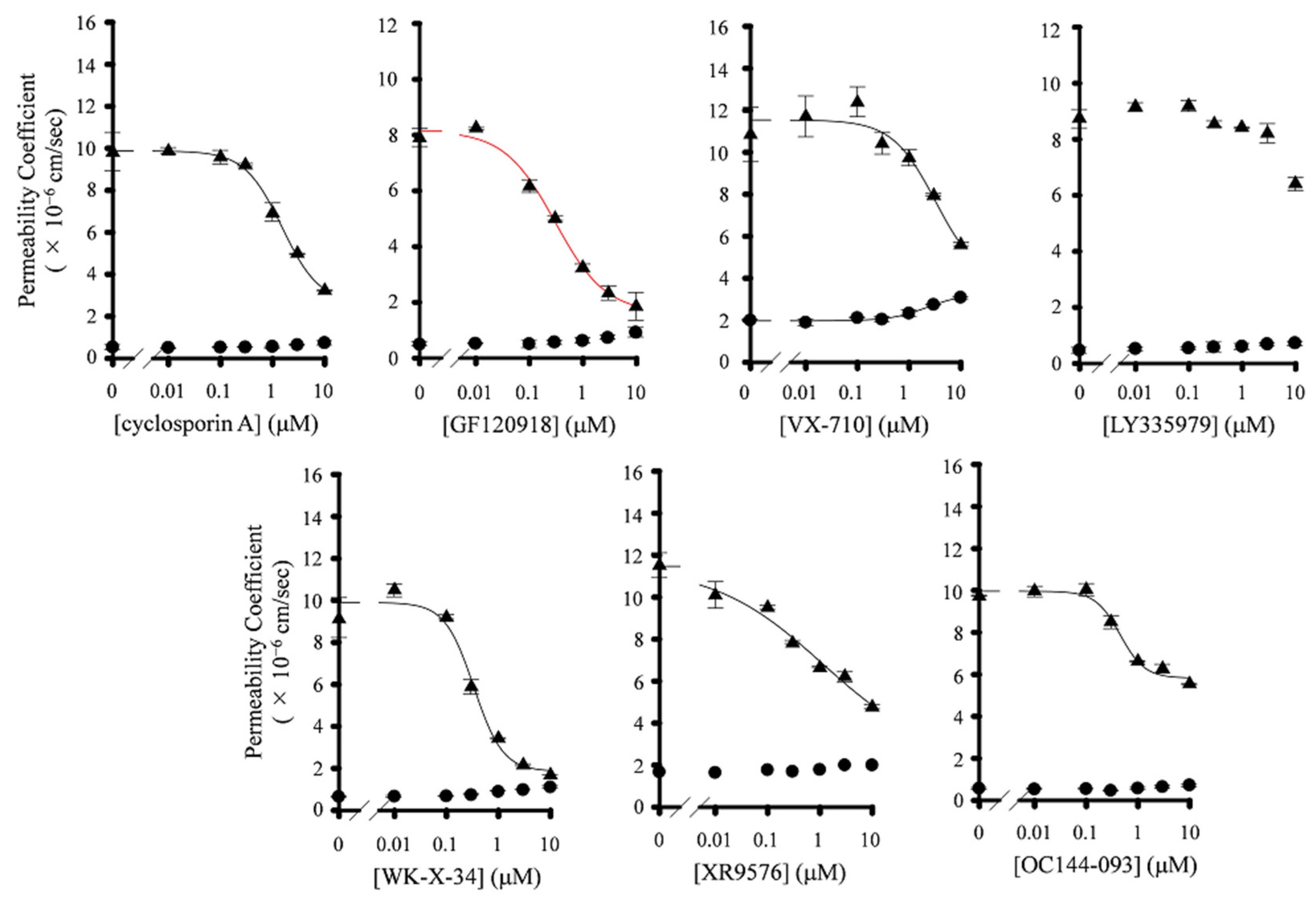
3.2. Effect of P-gp Inhibitors on BCRP-Mediated Drug Efflux in Caco-2 Cells
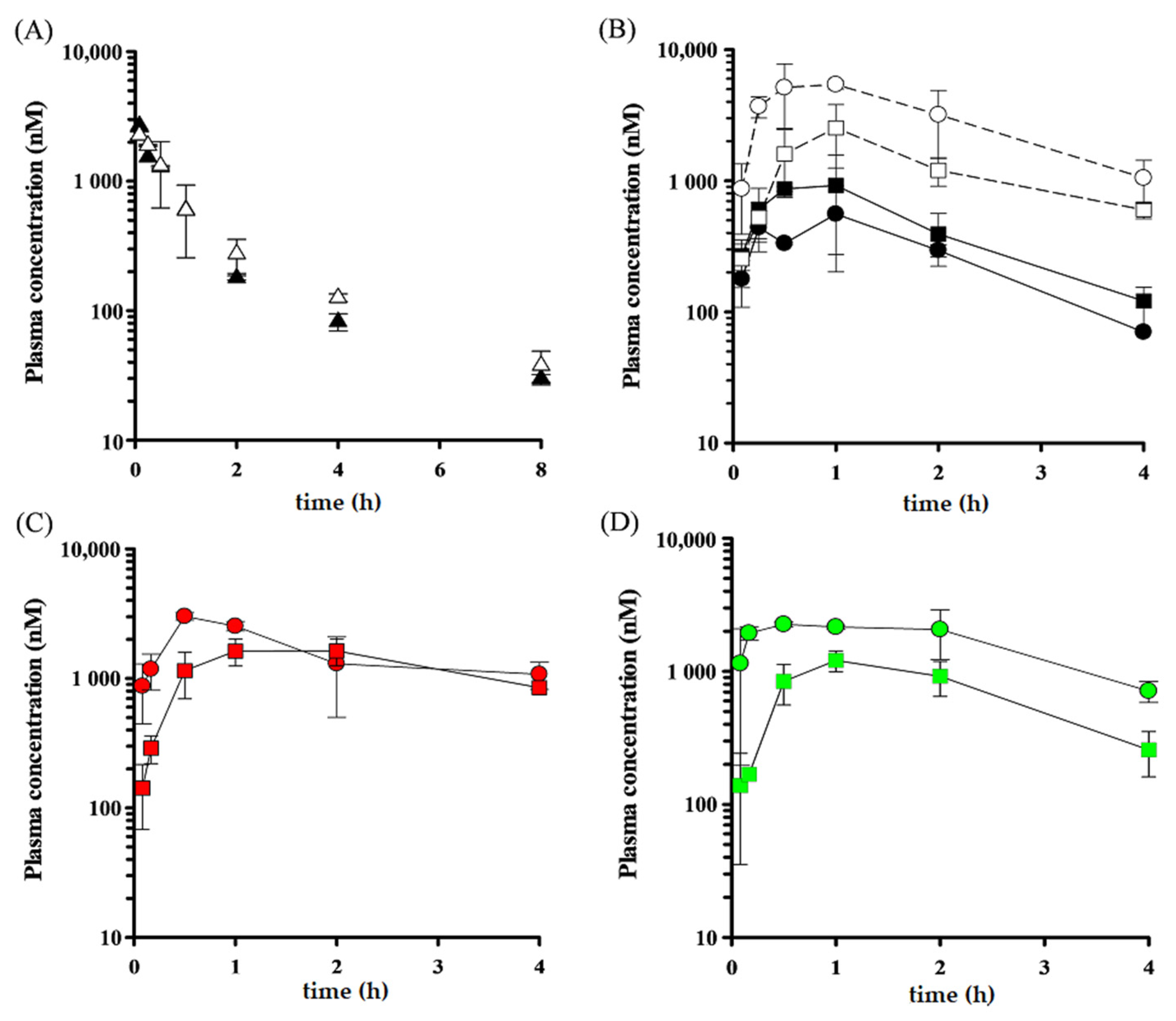
3.3. In Vivo Inhibitory Effect of LY335979 and WK-X-34 on P-gp- and BCRP-Mediated Drug Efflux
3.4. Effect of P-gp Inhibitors on the Absorption Rate of Paclitaxel
3.5. Effect of P-gp Inhibitors on BCRP-Mediated Efflux In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shekhawat, P.B.; Pokharkar, V.B. Understanding Peroral Absorption: Regulatory Aspects and Contemporary Approaches to Tackling Solubility and Permeability Hurdles. Acta Pharm. Sin. B 2017, 7, 260–280. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Konno, Y.; Hashimoto, T.; Nagai, M.; Taguchi, T.; Satsukawa, M.; Yamashita, S. In Vivo Assessment of the Impact of Efflux Transporter on Oral Drug Absorption Using Portal Vein–Cannulated Rats. Drug Metab. Dispos. 2013, 41, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Miyata, K.-I.; Nakagawa, Y.; Kimura, Y.; Ueda, K.; Akamatsu, M. Structure–Activity Relationships of Dibenzoylhydrazines for the Inhibition of P-Glycoprotein-Mediated Quinidine Transport. Bioorganic Med. Chem. 2016, 24, 3184–3191. [Google Scholar] [CrossRef]
- Matheny, C.J.; Lamb, M.W.; Brouwer, K.L.R.; Pollack, G.M. Pharmacokinetic and Pharmacodynamic Implications of P-glycoprotein Modulation. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2001, 21, 778–796. [Google Scholar] [CrossRef]
- Terao, T.; Hisanaga, E.; Sai, Y.; Tamai, I.; Tsuji, A. Active Secretion of Drugs from the Small Intestinal Epithelium in Rats by P-Glycoprotein Functioning as an Absorption Barrier. J. Pharm. Pharmacol. 1996, 48, 1083–1089. [Google Scholar] [CrossRef]
- Jones, C.R.; Hatley, O.J.D.; Ungell, A.-L.; Hilgendorf, C.; Peters, S.A.; Rostami-Hodjegan, A. Gut Wall Metabolism. Application of Pre-Clinical Models for the Prediction of Human Drug Absorption and First-Pass Elimination. AAPS J. 2016, 18, 589–604. [Google Scholar] [CrossRef]
- Sambuy, Y.; De Angelis, I.; Ranaldi, G.; Scarino, M.L.; Stammati, A.; Zucco, F. The Caco-2 Cell Line as a Model of the Intestinal Barrier: Influence of Cell and Culture-Related Factors on Caco-2 Cell Functional Characteristics. Cell Biol. Toxicol. 2005, 21, 1–26. [Google Scholar] [CrossRef]
- Van Breemen, R.B.; Li, Y. Caco-2 Cell Permeability Assays to Measure Drug Absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef]
- Volpe, D.A. Variability in Caco-2 and MDCK Cell-Based Intestinal Permeability Assays. J. Pharm. Sci. 2008, 97, 712–725. [Google Scholar] [CrossRef]
- Oostendorp, R.L.; Buckle, T.; Beijnen, J.H.; Van Tellingen, O.; Schellens, J.H.M. The Effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP Inhibitors on the in Vivo Absorption, Distribution, Metabolism and Excretion of Imatinib. Investig. New Drugs 2009, 27, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Holmstock, N.; Mols, R.; Annaert, P.; Augustijns, P. In Situ Intestinal Perfusion in Knockout Mice Demonstrates Inhibition of Intestinal P-Glycoprotein by Ritonavir Causing Increased Darunavir Absorption. Drug Metab. Dispos. 2010, 38, 1407–1410. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, I.; Nishikawa, S.; Yamamoto, A.; Kono, Y.; Fujita, T. Assessment of Contribution of BCRP to Intestinal Absorption of Various Drugs Using Portal-Systemic Blood Concentration Difference Model in Mice. Pharmacol. Res. Perspect. 2019, 8, 00544. [Google Scholar] [CrossRef] [PubMed]
- Sababi, M.; Borgå, O.; Hultkvist-Bengtsson, U. The Role of P-Glycoprotein in Limiting Intestinal Regional Absorption of Digoxin in Rats. Eur. J. Pharm. Sci. 2001, 14, 21–27. [Google Scholar] [CrossRef]
- Dey, S.; Gunda, S.; Mitra, A.K. Pharmacokinetics of Erythromycin in Rabbit Corneas after Single-Dose Infusion: Role of P-Glycoprotein as a Barrier to in Vivo Ocular Drug Absorption. J. Pharmacol. Exp. Ther. 2004, 311, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, J.; Müller, F.; Fromm, M.F. Transporters and Drug-Drug Interactions: Important Determinants of Drug Disposition and Effects. Pharmacol. Rev. 2013, 65, 944–966. [Google Scholar] [CrossRef] [Green Version]
- Wacher, V.J.; Wu, C.-Y.; Benet, L.Z. Overlapping Substrate Specificities and Tissue Distribution of Cytochrome P450 3A and P-Glycoprotein: Implications for Drug Delivery and Activity in Cancer Chemotherapy. Mol. Carcinog. 1995, 13, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Kirn, R.B.; Wandel, C.; Leake, B.; Cvetkovic, M.; Fromm, M.F.; Dempsey, P.J.; Roden, M.M.; Belas, F.; Chaudhary, A.K.; Roden, D.M.; et al. Interrelationship Between Substrates and Inhibitors of Human CYP3A and P-Glycoprotein. Pharm. Res. 1999, 16, 408–414. [Google Scholar] [CrossRef]
- Varma, M.V.; Ashokraj, Y.; Dey, C.S.; Panchagnula, R. P-Glycoprotein Inhibitors and Their Screening: A Perspective from Bioavailability Enhancement. Pharmacol. Res. 2003, 48, 347–359. [Google Scholar] [CrossRef]
- Modok, S.; Mellor, H.; Callaghan, R. Modulation of Multidrug Resistance Efflux Pump Activity to Overcome Chemoresistance in Cancer. Curr. Opin. Pharmacol. 2006, 6, 350–354. [Google Scholar] [CrossRef]
- Jekerle, V.; Klinkhammer, W.; Scollard, D.A.; Breitbach, K.; Reilly, R.M.; Piquette-Miller, M.; Wiese, M. In Vitro Andin Vivo Evaluation of WK-X-34, a Novel Inhibitor of P-Glycoprotein and BCRP, Using Radio Imaging Techniques. Int. J. Cancer 2006, 119, 414–422. [Google Scholar] [CrossRef]
- Lin, J.H. Drug–Drug Interaction Mediated by Inhibition and Induction of P-Glycoprotein. Adv. Drug Deliv. Rev. 2003, 55, 53–81. [Google Scholar] [CrossRef]
- Yahanda, A.M.; Alder, K.M.; Fisher, G.A.; Brophy, N.; Halsey, J.; Hardy, R.I.; Gosland, M.P.; Lum, B.L.; Sikic, B.I. Phase I Trial of Etoposide with Cyclosporine as a Modulator of Multidrug Resistance. J. Clin. Oncol. 1992, 10, 1624–1634. [Google Scholar] [CrossRef]
- Guns, E.S.; Denyssevych, T.; Dixon, R.; Bally, M.B.; Mayer, L. Drug Interaction Studies between Paclitaxel (Taxol) and OC144-093—A New Modulator of MDR in Cancer Chemotherapy. Eur. J. Drug Metab. Pharmacokinet. 2002, 27, 119–126. [Google Scholar] [CrossRef]
- Callies, S.; De Alwis, D.P.; Wright, J.G.; Sandler, A.; Burgess, M.; Aarons, L. A Population Pharmacokinetic Model for Doxorubicin and Doxorubicinol in the Presence of a Novel MDR Modulator, Zosuquidar Trihydrochloride (LY335979). Cancer Chemother. Pharmacol. 2003, 51, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Litman, T.; Brangi, M.; Hudson, E.; Fetsch, P.; Abati, A.; Ross, D.D.; Miyake, K.; Resau, J.H.; Bates, S.E. The Multidrug-Resistant Phenotype Associated with Overexpression of the New ABC Half-Transporter, MXR (ABCG2). J. Cell Sci. 2000, 113, 2011–2021. [Google Scholar]
- Kodaira, H.; Kusuhara, H.; Ushiki, J.; Fuse, E.; Sugiyama, Y. Kinetic Analysis of the Cooperation of P-Glycoprotein (P-gp/Abcb1) and Breast Cancer Resistance Protein (Bcrp/Abcg2) in Limiting the Brain and Testis Penetration of Erlotinib, Flavopiridol, and Mitoxantrone. J. Pharmacol. Exp. Ther. 2010, 333, 788–796. [Google Scholar] [CrossRef] [Green Version]
- De Vries, N.A.; Zhao, J.; Kroon, E.; Buckle, T.; Beijnen, J.H.; Van Tellingen, O. P-Glycoprotein and Breast Cancer Resistance Protein: Two Dominant Transporters Working Together in Limiting the Brain Penetration of Topotecan. Clin. Cancer Res. 2007, 13, 6440–6449. [Google Scholar] [CrossRef] [Green Version]
- Newman, M.J.; Rodarte, J.C.; Bendatoul, K.D.; Romano, S.J.; Zhang, C.; Krane, S.; Moran, E.J.; Uyeda, R.T.; Dixon, R.; Guns, E.S.; et al. Discovery and Characterization of OC144-093, a Novel Inhibitor of P-Glycoprotein-Mediated Multidrug Resistance. Cancer Res. 2000, 60, 2964–2972. [Google Scholar]
- Zhang, H.; Xu, H.; Ashby, C.R., Jr.; Assaraf, Y.G.; Chen, Z.-S.; Liu, H.-M. Chemical Molecular-Based Approach to Overcome Multidrug Resistance in Cancer by Targeting P-Glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef]
- Hamid, K.A.; Lin, Y.; Gao, Y.; Katsumi, H.; Sakane, T.; Yamamoto, A. The Effect of Wellsolve, a Novel Solubilizing Agent, on the Intestinal Barrier Function and Intestinal Absorption of Griseofulvin in Rats. Biol. Pharm. Bull. 2009, 32, 1898–1905. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, I.; Nishikawa, S.; Yamamoto, A.; Kono, Y.; Fujita, T. The Impact of Breast Cancer Resistance Protein (BCRP/ABCG2) on Drug Transport Across Caco-2 Cell Monolayers. Drug Metab. Dispos. 2020, 48, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, E.; Lagarce, F.; Garcion, E.; Benoit, J.-P. Reciprocal Competition between Lipid Nanocapsules and P-GP for Paclitaxel Transport across Caco-2 Cells. Eur. J. Pharm. Sci. 2010, 40, 422–429. [Google Scholar] [CrossRef]
- Sugihara, N.; Kuroda, N.; Watanabe, F.; Choshi, T.; Kamishikiryo, J.; Seo, M. Effects of Catechins and Their Related Compounds on Cellular Accumulation and Efflux Transport of Mitoxantrone in Caco-2 Cell Monolayers. J. Food Sci. 2017, 82, 1224–1230. [Google Scholar] [CrossRef]
- Hoffman, D.J.; Seifert, T.; Borre, A.; Nellans, H.N. Method to Estimate the Rate and Extent of Intestinal Absorption in Conscious Rats Using an Absorption Probe and Portal Blood Sampling. Pharm. Res. 1995, 12, 889–894. [Google Scholar] [CrossRef]
- Tabata, K.; Yamaoka, K.; Fukuyama, T.; Nakagawa, T. Evaluation of Intestinal Absorption into the Portal System in Enterohepatic Circulation by Measuring the Difference in Portal–Venous Blood Concentrations of Diclofenac. Pharm. Res. 1995, 12, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.W.; Lee, J.; Park, J.H.; Kwon, Y.; Na, Y.; Lee, H.J. Intestinal P-Glycoprotein Inhibitors, Benzoxanthone Analogues. J. Pharm. Pharmacol. 2018, 70, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Sun, X.; Lustburg, M.B.; Sparreboom, A.; Hu, S. Predicting Paclitaxel Disposition in Humans With Whole-Body Physiologically-Based Pharmacokinetic Modeling. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 931–939. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, X.; Sagawa, K.; Morris, M.E. Flavonoids Chrysin and Benzoflavone, Potent Breast Cancer Resistance Protein Inhibitors, Have No Significant Effect on Topotecan Pharmacokinetics in Rats or MDR1A/1B (−/−) Mice. Drug Metab. Dispos. 2004, 33, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.; Morris, T. Physiological Parameters in Laboratory Animals and Humans. Pharm. Res. 1993, 10, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Bardelmeijer, H.A.; Ouwehand, M.; Beijnen, J.H.; Schellens, J.H.M.; van Tellingen, O. Efficacy of Novel P-Glycoprotein Inhib-Itors to Increase the Oral Uptake of Paclitaxel in Mice. Investig. New. Drugs. 2004, 22, 219–229. [Google Scholar] [CrossRef]
- Troutman, M.D.; Thakker, D.R. Efflux Ratio Cannot Assess P-Glycoprotein-Mediated Attenuation of Absorptive Transport: Asymmetric Effect of P-Glycoprotein on Absorptive and Secretory Transport across Caco-2 Cell Monolayers. Pharm. Res. 2003, 20, 1200–1209. [Google Scholar] [CrossRef]
- Jonker, J.W.; Smit, J.W.; Brinkhuis, R.F.; Maliepaard, M.; Beijnen, J.H.; Schellens, J.H.M.; Schinkel, A.H. Role of Breast Cancer Resistance Protein in the Bioavailability and Fetal Penetration of Topotecan. J. Natl. Cancer Inst. 2000, 92, 1651–1656. [Google Scholar] [CrossRef]
- Lagas, J.S.; Van Waterschoot, R.A.; Van Tilburg, V.A.; Hillebrand, M.J.; Lankheet, N.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. Brain Accumulation of Dasatinib Is Restricted by P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) and Can Be Enhanced by Elacridar Treatment. Clin. Cancer Res. 2009, 15, 2344–2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.C.; Nguyen, L.N.; Sparidans, R.W.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Increased Oral Availability and Brain Accumulation of the ALK Inhibitor Crizotinib by Coadministration of the P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) Inhibitor Elacridar. Int. J. Cancer 2013, 134, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pan, M.; Dai, Y.; Liu, B.; Cui, J.; Shi, W.; Qiu, Q.; Huang, W.; Qian, H. Design, Synthesis and Biological Evaluation of LBM-A5 Derivatives as Potent P-Glycoprotein-Mediated Multidrug Resistance Inhibitors. Bioorganic Med. Chem. 2016, 24, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Minderman, H.; O’Loughlin, K.L.; Pendyala, L.; Baer, M.R. VX-710 (Biricodar) Increases Drug Retention and Enhances Chemosensitivity in Resistant Cells Overexpressing P-Glycoprotein, Multidrug Resistance Protein, and Breast Cancer Resistance Protein. Clin. Cancer Res. 2004, 10, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Shepard, R.L.; Cao, J.; Starling, J.J.; Dantzig, A.H. Modulation of P-Glycoprotein but not MRP1-or BCRP-Mediated Drug Resistance by LY335979. Int. J. Cancer 2002, 103, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Pick, A.; Müller, H.; Wiese, M. Structure–Activity Relationships of New Inhibitors of Breast Cancer Resistance Protein (ABCG2). Bioorg. Med. Chem. 2008, 16, 8224–8236. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, J.J.; Lagas, J.S.; Rosing, H.; Schellens, J.H.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein and Cytochrome P450 3A Act Together in Restricting the Oral Bioavailability of Paclitaxel. Int. J. Cancer 2012, 132, 2439–2447. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Chae, S.W.; Xia, Y.; Kim, N.H.; Kim, H.J.; Rhie, S.; Lee, H.J. Effect of Coumarin Derivative-Mediated Inhibition of P-Glycoprotein on Oral Bioavailability and Therapeutic Efficacy of Paclitaxel. Eur. J. Pharmacol. 2014, 723, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Korzekwa, K.R.; Grogan, J.; Gonzalez, F.J.; Harris, J.W. Selective Biotransformation of Taxol to 6 α-Hydroxytaxol by Human Cytochrome P450 2C8. Cancer Res. 1994, 54, 5543–5546. [Google Scholar] [PubMed]
- Sonnichsen, D.S.; Liu, Q.; Schuetz, E.G.; Schuetz, J.D.; Pappo, A.; Relling, M.V. Variability in Human Cytochrome P450 Paclitaxel Metabolism. J. Pharmacol. Exp. Ther. 1995, 275, 566–575. [Google Scholar] [PubMed]
- Dantzig, A.H.; Shepard, R.L.; Law, K.L.; Tabas, L.; Pratt, S.; Gillespie, J.S.; Binkley, S.N.; Kuhfeld, M.T.; Starling, J.J.; Wrighton, S.A. Selectivity of the Multidrug Resistance Modulator, LY335979, for P-Glycoprotein and Effect on Cytochrome P-450 Activities. J. Pharmacol. Exp. Ther. 1999, 290, 854–862. [Google Scholar] [PubMed]
- Dastvan, R.; Mishra, S.; Peskova, Y.B.; Nakamoto, R.K.; Mchaourab, H.S. Mechanism of Allosteric Modulation of P-Glycoprotein by Transport Substrates and Inhibitors. Science 2019, 364, 689–692. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | IC50 (nM) | Hill Coefficient | ||||
|---|---|---|---|---|---|---|
| Papp,AB | Papp,BA | ER | Papp,AB | Papp,BA | ER | |
| Cyclosporin A | 1973 ± 21 | 1820 ± 126 | 502 ± 126 | −1.63 ± 0.09 | 2.18 ± 0.39 | 1.63 ± 0.20 |
| GF120918 | 319 ± 30 | 239 ± 97 a | 60 ± 21 | −1.28 ± 0.09 | 1.63 ± 1.28 | 1.66 ± 0.51 |
| XR9576 | 234 ± 61 | 64 ± 40 | 46 ± 19 | −2.10 ± 0.64 | 18.4 ± 4.76 | 3.41 ± 0.28 |
| LY335979 | 427 ± 52 | 107 ± 59 a | 115 ± 22 | −2.23 ± 0.31 | 0.57 ± 0.35 | 1.71 ± 0.41 |
| WK-X-34 | 935 ± 33 | 501 ± 132 a | 214 ± 113 | −10.7 ± 6.00 | 1.29 ± 0.42 | 2.07 ± 1.22 |
| VX-710 | 2680 ± 53 | 4496 ± 84 | 871 ± 277 | −1.84 ± 0.07 | 1.08 ± 0.60 | 1.68 ± 0.38 |
| OC144-093 | n.c. | n.c. | n.c. | n.c. | n.c. | n.c. |
| Inhibitor | IC50 (nM) | Hill Coefficient | ||||
|---|---|---|---|---|---|---|
| Papp,AB | Papp,BA | ER | Papp,AB | Papp,BA | ER | |
| Cyclosporin A | n.c. | 2038 ± 13 | 1708 ± 248 | n.c. | 1.25 ± 0.17 | 1.33 ± 0.25 |
| GF120918 | n.c. | 298 ± 13 a | 307 ± 23 | n.c. | 0.93 ± 0.41 | 0.92 ± 0.15 |
| XR9576 | n.c. | 1000 ± 45 | 531 ± 162 | n.c. | 0.41 ± 0.08 | 0.44 ± 0.05 |
| LY335979 | n.c. | >10 μM a | >10 μM | n.c. | n.c. | n.c. |
| WK-X-34 | n.c. | 370 ± 38 a | 328 ± 96 | n.c. | 1.56 ± 0.53 | 1.51 ± 0.41 |
| VX-710 | n.c. | 2675 ± 31 | 1638 ± 520 | n.c. | 1.04 ± 0.40 | 0.46 ± 0.12 |
| OC144-093 | n.c. | n.c. | n.c. | n.c. | 1.83 ± 0.59 | 3.61 ± 0.41 |
| WT | mdr1a/1b KO | WT + WK-X-34 | WT + LY335979 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IV | po | IV | po | po | po | ||||||
| pv | sys | pv | sys | pv | sys | pv | sys | ||||
| Dose | (mg/kg) | 5 | 20 | 5 | 20 | 20 | 20 | ||||
| Cmax | (nM) | --- | 730 | 442 | --- | 2523 | 1179 | 3018 | 1631 | 2258 | 1208 |
| Tmax | (h) | --- | 1 | 1 | --- | 1 | 1 | 0.5 | 2 | 0.5 | 1 |
| AUC | (nM·hr) | 2320 | 2002 | 1089 | 2661 | 6575 | 3086 | 10,615 | 8636 | 7970 | 3201 |
| ke | (h−1) | 0.43 | 0.50 | 0.55 | 0.35 | 0.26 | 0.23 | 0.53 | 0.64 | ||
| t1/2 | (h) | 2.38 | 1.62 | 1.37 | 2.12 | 1.25 | 2.00 | 2.69 | 2.98 | 1.30 | 1.09 |
| MRT | (h) | 1.71 | 2.51 | 2.29 | 1.47 | 2.16 | 3.09 | 4.15 | 4.67 | 2.42 | 2.25 |
| CLtot | (L/h/kg) | 2.52 | 2.20 | ||||||||
| Vdss | (L/kg) | 4.32 | 4.15 | ||||||||
| BA | 11.7 | 29.0 | |||||||||
| FaFg | 16.6 | 66.3 | 36.0 | 86.8 | |||||||
| Fh | 70.6 | 45.7 | |||||||||
| n | +Inhibitor (mg/kg) | Cpv (nM) | Csys (nM) | Cpv − Csys (nM) | Absorption Rate (V) (nmol/min/kg) | |
|---|---|---|---|---|---|---|
| WT mice | 3 | 439 ± 98 | 223 ± 76 | 216 ± 80 | 15.7 | |
| +Cyclosporin A | 5 | 30 | 708 ± 77 | 263 ± 168 | 534 ± 102 | 38.7 |
| +LY335979 | 6 | 30 | 1009 ± 149 | 202 ± 52 | 830 ± 140 | 60.2 |
| +WK-X-34 | 3 | 30 | 746 ± 57 | 196 ± 64 | 549 ± 7.5 | 39.8 |
| mdr1a/1b KO mice | 6 | 967 ± 191 | 218 ± 100 | 749 ± 165 | 54.3 |
| n | +Inhibitor (mg/kg) | Cpv (nM) | Csys (nM) | Cpv − Csys (nM) | Absorption Rate V (nmol/min/kg) | |
|---|---|---|---|---|---|---|
| WT mice | 3 | 328 ± 41 | 135 ± 41 | 165 ± 61 | 12.4 | |
| +Cyclosporin A | 2 | 15 | 540 | 315 | 225 | 16.9 |
| +LY335979 | 3 | 15 | 626 ± 82 | 368 ± 30 | 259 ± 45 | 19.5 |
| +WK-X-34 | 3 | 15 | 674 ± 139 | 238 ± 63 | 437 ± 127 | 32.9 |
| +Cyclosoprin A | 3 | 30 | 1488 ± 146 | 829 ± 166 | 657 ± 38 | 49.4 |
| +LY335979 | 3 | 30 | 1407 ± 70 | 524 ± 58 | 883 ± 59 | 66.4 |
| +WK-X-34 | 3 | 30 | 2069 ± 204 | 757 ± 100 | 1311 ± 87 | 98.6 |
| mdr1a/1b KO mice | 3 | 1092 ± 118 | 245 ± 69 | 847 ± 144 | 63.7 |
| n | +Inhibitor (mg/kg) | Cpv (nM) | Csys (nM) | Cpv − Csys (nM) | Absorption Rate V (nmol/min/kg) | |
|---|---|---|---|---|---|---|
| WT mice | 3 | 196 ± 29 | 179 ± 25 | 17.1 ± 5.7 | 1.29 | |
| +Cyclosporin A | 2 | 30 | 298 | 240 | 58.5 | 4.40 |
| +LY335979 | 3 | 30 | 249 ± 26 | 228 ± 22.3 | 20.5 ± 9.7 | 1.54 |
| +WK-X-34 | 3 | 30 | 464 ± 53 | 388 ± 19 | 76.6 ± 27 | 5.76 |
| mdr1a/1b KO mice | 3 | 236 ± 30 | 207 ± 28 | 28.9 ± 8.4 | 2.17 | |
| bcrp KO mice | 3 | 469 ± 64 | 398 ± 88 | 70.5 ± 20 | 5.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kono, Y.; Kawahara, I.; Shinozaki, K.; Nomura, I.; Marutani, H.; Yamamoto, A.; Fujita, T. Characterization of P-Glycoprotein Inhibitors for Evaluating the Effect of P-Glycoprotein on the Intestinal Absorption of Drugs. Pharmaceutics 2021, 13, 388. https://doi.org/10.3390/pharmaceutics13030388
Kono Y, Kawahara I, Shinozaki K, Nomura I, Marutani H, Yamamoto A, Fujita T. Characterization of P-Glycoprotein Inhibitors for Evaluating the Effect of P-Glycoprotein on the Intestinal Absorption of Drugs. Pharmaceutics. 2021; 13(3):388. https://doi.org/10.3390/pharmaceutics13030388
Chicago/Turabian StyleKono, Yusuke, Iichiro Kawahara, Kohei Shinozaki, Ikuo Nomura, Honoka Marutani, Akira Yamamoto, and Takuya Fujita. 2021. "Characterization of P-Glycoprotein Inhibitors for Evaluating the Effect of P-Glycoprotein on the Intestinal Absorption of Drugs" Pharmaceutics 13, no. 3: 388. https://doi.org/10.3390/pharmaceutics13030388