
Comparative analysis of prophage-like elements in Helicobacter sp. genomes
- Published
- Accepted
- Received
- Academic Editor
- M. Pilar Francino
- Subject Areas
- Genomics, Microbiology, Virology
- Keywords
- Helicobacter, Prophage, Phylogeny, Comparative genomics
- Copyright
- © 2016 Fan et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2016. Comparative analysis of prophage-like elements in Helicobacter sp. genomes. PeerJ 4:e2012 https://doi.org/10.7717/peerj.2012
Abstract
Prophages are regarded as one of the factors underlying bacterial virulence, genomic diversification, and fitness, and are ubiquitous in bacterial genomes. Information on Helicobacter sp. prophages remains scarce. In this study, sixteen prophages were identified and analyzed in detail. Eight of them are described for the first time. Based on a comparative genomic analysis, these sixteen prophages can be classified into four different clusters. Phylogenetic relationships of Cluster A Helicobacter prophages were investigated. Furthermore, genomes of Helicobacter prophages from Clusters B, C, and D were analyzed. Interestingly, some putative antibiotic resistance proteins and virulence factors were associated with Helicobacter prophages.
Introduction
Prophages, a type of phage that integrates into and remains in a bacterial genome, play an important role in the genomic diversification and fitness cost of bacteria to the infected host. As a class of genetic elements, some prophages can mediate horizontal gene transfer in the evolution of bacterial genomes (Lang, Zhaxybayeva & Beatty, 2012). Because they carry virulence genes, some prophages make outstanding contributions to bacterial pathogenesis (Penadés et al., 2015) and some have also contributed to the fitness cost of bacteria to the infected host (Fan et al., 2013). Therefore, it is essential to search for the presence of prophages in the bacterial genomes and to analyze them. To date, studies have identified prophages in a diverse range of hosts, such as Moraxella catarrhalis (Ariff et al., 2015), Lawsonia intracellularis (Vannucci, Kelley & Gebhart, 2013), Bifidobacterium spp. (Lugli et al., 2016; Ventura et al., 2009), Lactococcus spp. (Ventura et al., 2007), Mycobacterium spp. (Fan, Abd Alla & Xie, 2015; Fan et al., 2014), Streptococcus spp. (Tang et al., 2013), and some plant-pathogenic bacteria (Varani et al., 2013). However, a systemic investigation of genomic information and function of Helicobacter prophages is largely lacking.
Helicobacter is a genus of Gram-negative bacteria, most frequently found in the upper gastrointestinal tract of mammals. One well-known species of the genus is Helicobacter pylori, a carcinogen identified by the World Health Organization (Uemura et al., 2001). H. pylori infection may be associated with gastritis, peptic ulcer, and gastric cancer (Peek & Blaser, 2002). Other non-pylori Helicobacter species such as H. suis, H. felis, H. bizzozeronii and H. salomonis have been reported and also exhibit carcinogenic potential in animals (O’rourke, Grehan & Lee, 2001). Previous research suggests that Helicobacter phages and prophages are unusual (Canchaya, Fournous & Brüssow, 2004). Information on Helicobacter prophages is becoming increasingly available. Two prophage-like elements were detected in Helicobacter acinonychis str. Sheeba (Eppinger et al., 2006). One prophage-like element was found within Helicobacter felis ATCC 49179 (Arnold et al., 2011). One prophage, phiHP33, which can be induced by UV irradiation, was found in H. pylori B45 (Lehours et al., 2011). Luo and colleagues (2012) found that the H. pylori str. HP1961 chromosome contains a full-length prophage 1961P. Luo also found that H. pylori Cuz20, H. pylori India7, H. pylori B38, H. pylori F16, and H. pylori Gambia94/24 chromosomes all contain a prophage-like element (Luo et al., 2012). In addition, two potential prophages were described in H. pylori str. Egypt (Abdel-Haliem & Askora, 2013). These findings suggest that prophages are common within the Helicobacter genomes. Vale et al. (2015) have demonstrated that prophages play a role in the diversity of H. pylori. The function of Helicobacter prophages is nonetheless ill-defined. Some researchers suggest that it is possible to use Helicobacter phages to control some diseases caused by H. pylori (Abdel-Haliem & Askora, 2013). However, if virulence factors and antibiotic resistance genes are found associated with Helicobacter phages or prophages, it is worth reconsidering phage therapy as treatment of H. pylori infections. As of 1 Oct 2015, eighty-one Helicobacter species genomes have been sequenced and assembled. These comprise an essential dataset for researching the presence of Helicobacter prophages.
As mentioned above, it is important that “hidden” Helicobacter prophages are identified. In this study, we screened all the available complete Helicobacter sp. genome sequences deposited in GenBank for the presence of prophages. We here report the results of our comparative genomic analysis, genome content analysis, and prophage-encoded virulence and antibiotic resistance gene analysis of Helicobacter prophages.
Materials and Methods
Data collection and identification of Helicobacter prophages
Eighty-one complete Helicobacter genomes were downloaded from NCBI (the National Center for Biotechnology Information). Helicobacter prophages were identified using a previously reported method (Fan et al., 2014). In the first place, we used PHAST (http://phast.wishartlab.com/index.html) to analyze bacterial genomes to find candidate prophages. Next, we screened integrase gene from prophage genomes to drop false positives results. Finally, based on the presence of significant homology between ORFs (open reading frames) and known phage genes, we obtain Helicobacter prophages.
Genomic and comparative genomic analyses of Helicobacter prophages
Prophage flanking sites attL and attR were identified using DNAMAN. Prophage genes were annotated using Glimmer (Delcher et al., 2007). Dot plot comparisons of Helicobacter prophage genomes were carried out with Geneious software (Kearse et al., 2012). Global genome comparison was performed using BLASTn, at NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi), and results were shown by ACT software. For all software, default settings were used.
Results and Discussion
Prophages in Helicobacter sp. genomes
Eighty-one complete Helicobacter sp. genomes (Table S1) were retrieved. Thirteen prohages (Table 1) were detected using a previously reported method (Fan et al., 2014), eight of them were novel, and five of them have been described in the literature (Luo et al., 2012). Moreover, seven reported prophages (Table 1) from Helicobacter genomes were not detected in the screen (Arnold et al., 2011; Eppinger et al., 2006; Lehours et al., 2011; Luo et al., 2012). Two of them, contained in the genomes of H. acinonychis str. Sheeba and H. felis ATCC 49179, have not been designated. We named them phiHac_1 and phiHFELIS_1, respectively. It is worth noting that phiHac_1, phiHFELIS_1 and two other prophages from H. pylori str. Egypt, ΦHPE1 and ΦHPE2, all lack sequence information. The original papers where these prophages were identified did not provide the sequence information and we cannot retrieve it from the corresponding genomes using our screening method. We therefore discarded them during follow-up analyses. In general, sixteen prophages are analysed.
| Prophages | Cluster | Host | Accession numbers of bacteria | Coordinates | Size | Putative attB regions of prophage-like elements | References |
|---|---|---|---|---|---|---|---|
| phiK747_1a | Cluster A2 | Helicobacter pylori UM032 | CP005490.3 | 1500592–1515028 | 14.4 kb | AAACAAATTTTTAAAA | this study |
| phiK749_1a | Cluster A2 | Helicobacter pylori UM299 | CP005491.3 | 487627–502064 | 14.4 kb | AAACAAATTTTTAAAA | this study |
| phiK750_1a | Cluster A1 | Helicobacter pylori UM037 | CP005492.3 | 1184664–1213258 | 28.6 kbd | ATTGATAGAAATAAT | this study |
| phiK748_1a | Cluster A2 | Helicobacter pylori UM298 | CP006610.2 | 167091–181528 | 14.4 kb | AAACAAATTTTTAAAA | this study |
| phiNY40_1a | Cluster A1 | Helicobacter pylori NY40 | AP014523.1 | 523881–555620 | 31.7 kbd | TTTTTGTGATTGAT | this study |
| phiHH_1a | Cluster B | Helicobacter hepaticus ATCC 51449 | AE017125.1 | 732167–748393 | 16.2 kb | AATCAAAGTGAGAGA | this study |
| phiHCD_1a | Cluster C | Helicobacter cetorum MIT 99-5656 | CP003481.1 | 178240–203078 | 24.8 kbd | AAACACTTTTAAA | this study |
| phiHBZC1_1a | Cluster D | Helicobacter bizzozeronii CIII-1 | FR871757.1 | 1613405–1669733 | 39.3 kbd | CTTTATCAAAATGC | this study |
| Cuz20ab | Cluster A1 | Helicobacter pylori Cuz20 | CP002076.1 | 186400–215514 | 29.1 kbd | TTATAGCTTATTTCA | (Luo et al., 2012) |
| India7ab | Cluster A1 | Helicobacter pylori India7 | CP002331.1 | 1217797–1246918 | 29.1 kbd | TTATAGCTTATTTCA | (Luo et al., 2012) |
| B38ab | Cluster A3 | Helicobacter pylori B38 | FM991728.1 | 1513448–1518986 | 5.5 kb | TTATAG (attL)e | (Luo et al., 2012) |
| Gambia94/24ab | Cluster A2 | Helicobacter pylori Gambia94/24 | CP002332.1 | 202163–218412 | 16.3 kb | TTATAGCTAATT (attL) TTATAGCTTATTTCA (attR) | (Luo et al., 2012) |
| phiHac_1bc | c | Helicobacter acinonychis str. Sheeba | AM260522.1 | NM | 11.6 kb | NM | (Eppinger et al., 2006) |
| Sheebaab | Cluster A1 | Helicobacter acinonychis str. Sheeba | AM260522.1 | 1396699–1425613 | 28.9 kbd | AAGATATCTCTTATT | (Eppinger et al., 2006) |
| F16b | Cluster A4 | Helicobacter pylori F16 | AP011940.1 | 470905–485827 | 14.9 kb | TTATAGCTTATTTCA (attL)e | (Luo et al., 2012) |
| phiHP33 (B45)b | Cluster A1 | Helicobacter pylori B45 | JF734911.1 | NM | 24.6 kbd | TTATAGCTTATTTCA (attL) TTATAGCTTATTT (attR) | (Lehours et al., 2011) |
| 1961Pb | Cluster A1 | Helicobacter pylori strain HP1961 | Not found | NM | 26.8 kbd | TTATCTTT | (Luo et al., 2012) |
| phiHFELIS_1bc | c | Helicobacter felis ATCC 49179 | FQ670179.2 | NM | NM | NM | (Arnold et al., 2011) |
| ΦHPE1bc | c | Helicobacter pylori str. Egypt | Not found | NM | NM | NM | (Abdel-Haliem & Askora, 2013) |
| ΦHPE2bc | c | Helicobacter pylori str. Egypt | Not found | NM | NM | NM | (Abdel-Haliem & Askora, 2013) |
The size of all Helicobacter prophage genomes varies between 5.5 kb and 39.3 kb. Based on the presence of predicted prophage proteins and the length of the prophage genomes, nine sequences were designated as full-length prophages, and seven sequences were labeled prophage-like elements.
Comparative genomics of Helicobacter (pro)phages
We carried out a comparative genomics analysis of sixteen Helicobacter prophages with known sequence information using dot plot matrix (Fig. 1). Two Helicobacter phages, KHP30 (Uchiyama et al., 2013) and KHP40 (Uchiyama et al., 2012), were selected as the reference for DotPlots. This revealed that most Helicobacter (pro)phages can be sorted into a common group called a ‘cluster’ (designated ‘Cluster A’) based on the similarities of their genomes. Helicobacter phages of Cluster A can be further divided into subclusters, according to their genomic sequences. These were designated subcluster A1 (containing phiNY40_1, phiK750_1, Sheeba, KHP30, KHP40, 1961P, phiHP33, Cuz20 and India7), subcluster A2 (containing Gambia94/24, phiK747_1, phiK749_1 and phiK748_1), subcluster A3 (B38), and subcluster A4 (F16), respectively. Other Helicobacter phages were grouped into Cluster B (phiHH_1), Cluster C (phiHCD_1), and Cluster D (phiHBZC1_1), as appropriate.
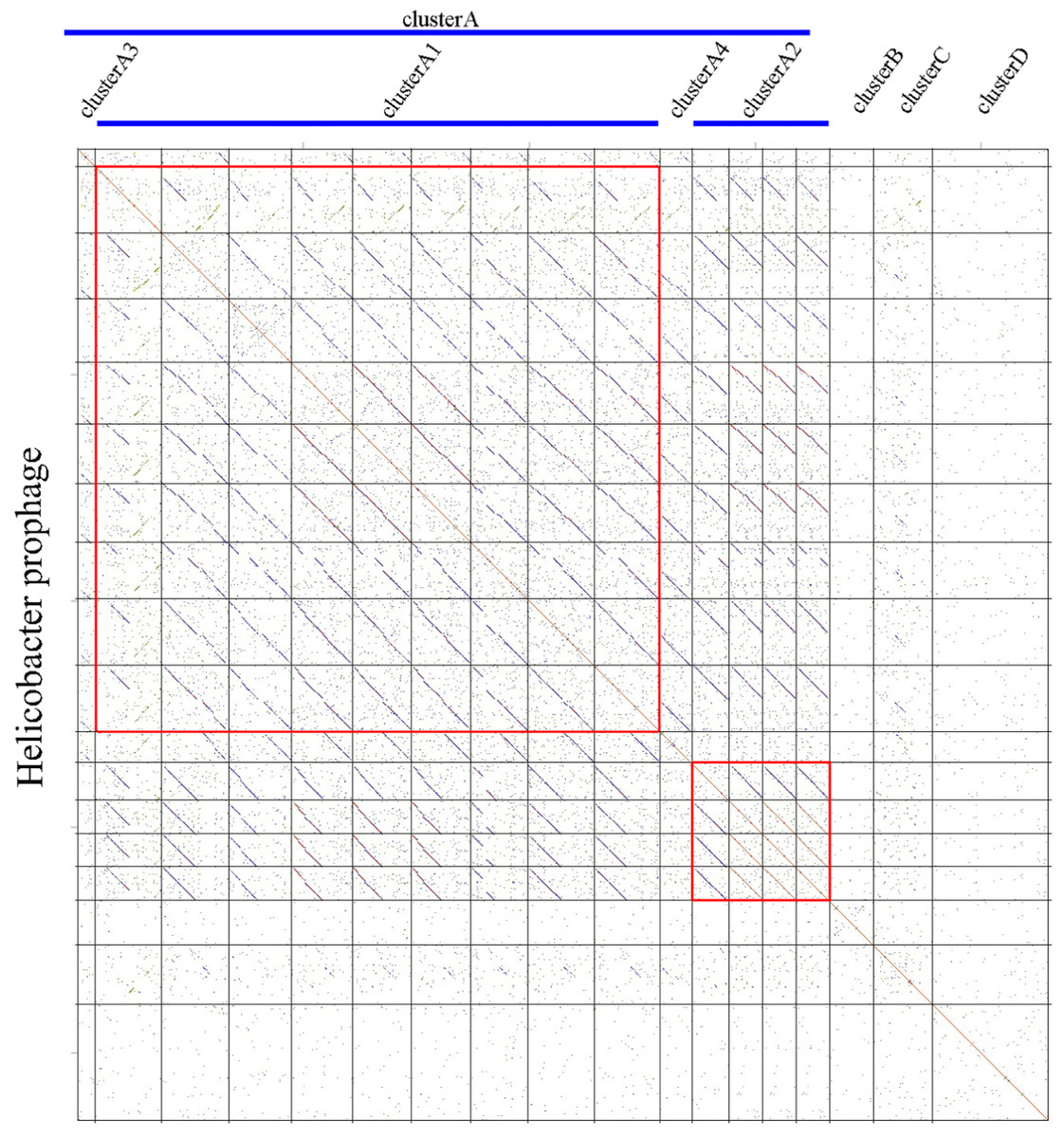
Figure 1: Comparative genomic analyses of Helicobacter prophages.
There are 16 Helicobacter prophages and 2 Helicobacter phages. The order of phages was B38, phiNY40_1, phiK750_1, Sheeba, KHP30, KHP40, 1961P, phiHP33, Cuz20, India7, F16, Gambia94/24, phiK747_1, phiK749_1, phiK748_1, phiHH_1, phiHCD_1 and phiHBZC1_1. The clusters of related phages (Clusters A, B, C and D) are shown in the figure. Geneious software was used to carry out dot plot analysis. The word length used is 13 bp.{kind=link}
Helicobacter phage Cluster A
Based on the similarities of their genomes, Helicobacter Cluster A phages were divided into four subclusters. Phages belonging to one subcluster are more closely related to each other than to phages in the remaining subclusters (Figs. S1 and S2). Some subcluster A1 phages (phiK750_1, Sheeba, KHP30, KHP40, 1961P, phiHP33, Cuz20 and India7) possess 70.57% identity with each other, as determined by multiple genomic sequence alignments in DNAMAN. In addition, a BLASTn comparison of phiNY40_1 and phiK750_1 revealed one major sequence segment (8,953 bp) with 81% identity and three segments (3,550 bp, 3,039 bp, and 1,997 bp) with identity greater than 76%. Based on the multiple genomic sequence alignments, all subcluster A2 phages displayed 82.79% identity between each other.
Different subclusters in Helicobacter phage Cluster A possess segments of DNA similarity. Phages of subclusters A2, A3, and A4 all shared sequence similarity with subcluster A1 phages (Fig. 2). These are remnant prophage-like elements that have lost sequence segments during evolution. Subcluster A2 prophages retained an upstream region with many virion-associated genes of the subcluster A1 prophages. Subcluster A3 prophage (prophage B38) retained only an incomplete upstream region (5.5 kb) of subclusters A1 and A2 prophages. Subcluster A4 prophage (prophage F16) retained a downstream region containing many DNA metabolism genes of the subcluster A1 prophages. Genome organization of most Cluster A phages has been reported (Luo et al., 2012).
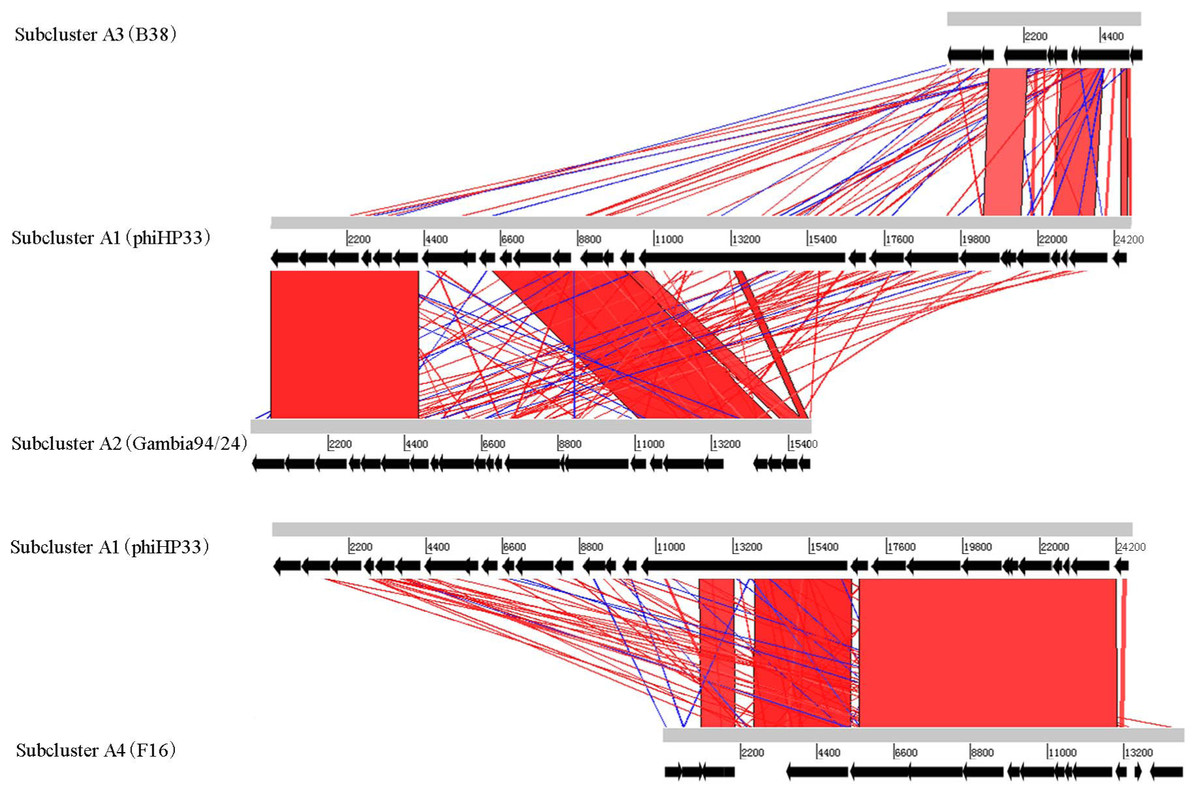
Figure 2: Global comparison of representative phages of Cluster A.
The red shading means that the fragments are homologous to other fragments. The results were obtained by Blast-N and depicted by ACT software. Numbers indicate the length of genomes (bp).{kind=link}
Helicobacter phage Cluster B
Cluster B contains only one Helicobacter prophage, phiHH_1. The genome size of phiHH_1, which lacks the lysin gene, is 16.2 kb. Therefore, phiHH_1 is considered to be a prophage-like element. This prophage is integrated into the H. hepaticus ATCC 51,449 genome, extends from HH_0750 (the integrase gene) to HH_0772 (encoding a carbohydrate-binding protein), and contains twenty-three ORFs (Fig. 3; Table S2). PhiHH_1 prophage is flanked by 15 bp attL and attR sites (Table 1). Twelve ORFs were assigned phage gene status after homologous analysis of protein sequences (Table S2). Based on database searches, nine of these encode specific functions, namely, integrase (HH_0750), DNA transposition protein (HH_0752), host-nuclease inhibitor protein Gam (HH_0754), Rha family transcriptional regulator (HH_0755), DNA-binding protein RdgB (HH_0756), phage Tail Collar Domain family (HH_0761), DNA methyltransferase (HH_0763), tape measure protein (HH_0771), and carbohydrate-binding protein (HH_0772).
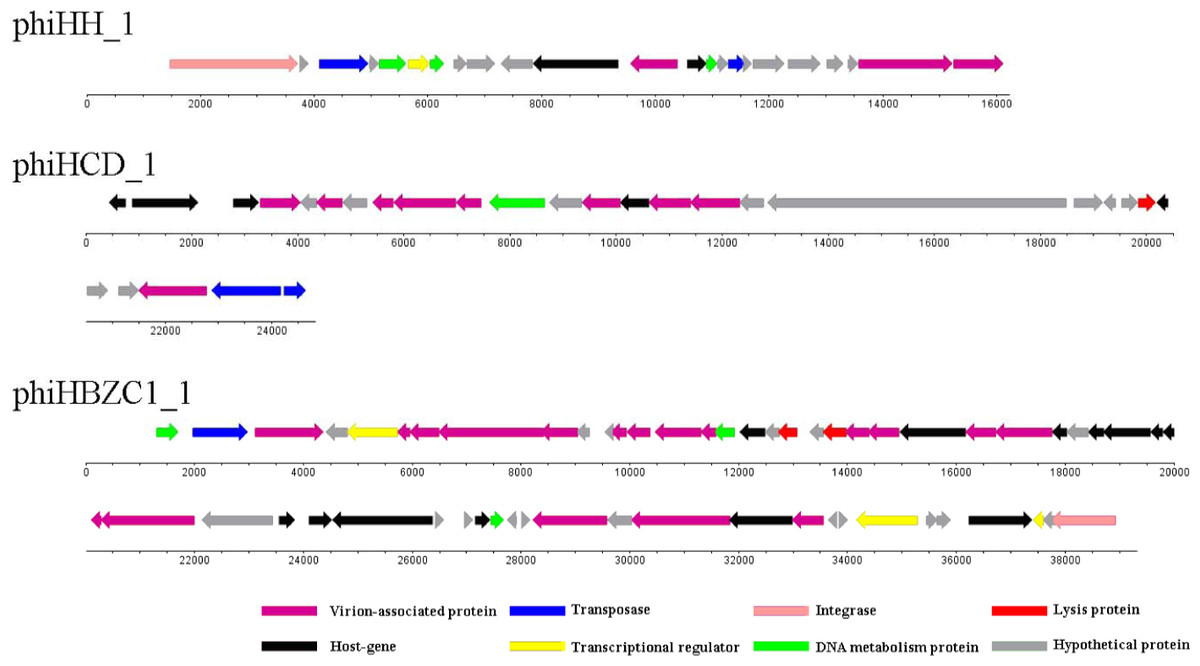
Figure 3: The genomic organization of Helicobacter prophage phiHH_1, phiHCD_1 and phiHBZC1_1.
Helicobacter prophage genes are grouped into eight functional modules: lysis module, DNA packaging and virion-associated modules, DNA metabolism module, transcriptional regulatory module, lysogeny module, host protein module and hypothetical protein module. The functions of the proteins are displayed by color coding. Dnaplotter software was used to draw the figure. Numbers indicate the length of genomes (bp).{kind=link}
Helicobacter phage Cluster C
Although Helicobacter prophage phiHCD_1 displays some similarity to the subcluster A1 and A4 phages, it is not sufficiently closely related to be included in a common cluster. Therefore, phiHCD_1 is categorized into Cluster C. The genome size of phiHCD_1 is 24.8 kb, which renders it a full-length prophage. Prophage phiHCD_1, inserted between HCD_00885 (thioredoxin-encoding) and HCD_01020 (transposase-encoding) in the genome of Helicobacter cetorum MIT 99-5656, contains twenty-eight ORFs (Fig. 3). The prophage has identical 13 bp attL and attR sites (Table 1). Based on amino acid sequence homology, we identified eighteen ORFs that have sequence similarity to genes of other phages. It was possible to assign function to thirteen of them (Table S3). These are, accordingly: terminase (HCD_00900); phage tail tape measure protein (HCD_00910); phage structure protein (HCD_00920, HCD_00930, and HCD_00960); phage major capsid protein (HCD_00925); UV radiation resistance protein (HCD_00935); phage prohead protease (HCD_00945); phage tail protein (HCD_00955); holin (HCD_00990); portal protein (HCD_01010); transposase (HCD_01015, and HCD_01020).
Helicobacter phage Cluster D
PhiHBZC1_1 is found in Helicobacter bizzozeronii CIII-1. It belongs to Cluster D and does not share any similarities with other Helicobacter phages. As a full-length prophage, the genome size of phiHBZC1_1 is 39.3 kb. There are fifty-eight ORFs in this genome (Fig. 3), spanning a region from HBZC1_17420 (DNA invertase-encoding) to HBZC1_17990 (site-specific recombinase integrase-encoding). The prophage is flanked by two 14 bp attL and attR sites (Table 1). Sequence alignment analysis indicated some level of similarity between thirty ORFs of prophage phiHBZC1_1 and other known phage genes. Of these, twenty-eight ORFs could be assigned biological functionalities (Table S4).
The genome of phiHBZC1_1 can be divided into several different functional modules. The lysis module includes HBZC1_17600 and HBZC1_17620, which encode a holin and a lysozyme protein, respectively. The DNA packaging and virion-associated modules consist of HBZC1_17440, coding for a phage terminase large subunit; HBZC1_17470, encoding a phage tail protein; phage tail tape measure proteins-encoding HBZC1_17480, HBZC1_17490, and HBZC1_17500; phage tail proteins-encoding HBZC1_17530, HBZC1_17540, HBZC1_17550, HBZC1_17630, HBZC1_17640, and HBZC1_17660; HBZC1_17560, encoding a phage tail sheath-like protein; HBZC1_17670, encoding a phage baseplate protein; capsid proteins-encoding HBZC1_17740 and HBZC1_17750; HBZC1_17860, encoding a portal protein; HBZC1_17880, encoding a phage terminase large subunit; and HBZC1_17900, encoding a phage baseplate assembly protein V. The DNA metabolism module comprises of three genes (HBZC1_17420, HBZC1_17570, and HBZC1_17830), whose predicted protein products are phage DNA invertase, DNA methyltransferase, and DNA polymerase, respectively. The transcriptional regulatory module is composed of HBZC1_17460 (encoding a phage late control D family protein), HBZC1_17930 (coding for the repressor LexA), and HBZC1_17970 (encoding a YcfA family protein). The lysogeny module appears to be limited to HBZC1_17990, whose predicted protein product is a phage integrase.
Putative antibiotic resistance genes and virulence factors associated with Helicobacter prophages
Except for phiHBZC1_1, none of the other characterized Helicobacter prophages contain known antibiotic resistance genes. The protein encoded by HBZC1_17700 shows high similarity to multidrug resistance protein D (emrD) of Salmonella enterica subsp. enterica serovar Infantis (Table 2). Multidrug resistance protein D belonging to the major facilitator superfamily facilitates the transport of a variety of antibiotics (Shaheen et al., 2015).
| Prophage | Gene (Accession number) | Putative virulence element | Query coverage | E-value | Identity |
|---|---|---|---|---|---|
| KHP40 | ORF24 ( BAM34796.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 8e–41 | 91% |
| KHP30 | ORF23 ( BAM34765.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 1e–40 | 92% |
| 1961P | gp26 ( AFC61925.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 6e–44 | 96% |
| Cuz20 | HPCU_00990 ( ADO03382.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–42 | 100% |
| India7 | HPIN_06120 ( ADU80418.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–47 | 100% |
| Gambia94/24 | HPGAM_01040 ( ADU81058.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 1e–45 | 100% |
| phiK747_1 | K747_07685 ( AGL67312.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–41 | 92% |
| phiK749_1 | K749_02305 ( AGL67850.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–41 | 92% |
| phiK750_1 | K750_05880 ( AGL70120.1) | DNA methyltransferase (Helicobacter pylori) | 95% | 4e–37 | 87% |
| phiK748_1 | K748_00765 ( AGR63209.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–41 | 92% |
| phiNY40_1 | NY40_0558 ( BAO97577.1) | Type II methylase (Helicobacter pylori) | 100% | 0.0 | 100% |
| phiNY40_1 | NY40_0553 ( BAO97572.1) | Serine/threonine protein kinase (Thiorhodococcus drewsii) | 99% | 6e–56 | 23% |
| phiNY40_1 | NY40_0545 ( BAO97564.1) | DNA methyltransferase (Helicobacter pylori) | 100% | 2e–43 | 100% |
| phiHH_1 | HH_0763 ( AAP77360.1) | DNA methyltransferase (Helicobacter sp. MIT 03-1614) | 84% | 3e–31 | 97% |
| Sheeba | Hac_1629 ( CAK00337.1) | DNA methyltransferase (Helicobacter pylori) | 97% | 1e–29 | 69% |
| phiHCD_1 | HCD_00890 ( AFI05210.1) | Glycosyltransferase (Neisseria meningitidis) | 71% | 3e–71 | 17% |
| phiHBZC1_1 | HBZC1_17570 ( CCB80743.1) | DNA methyltransferase (Helicobacter sp. MIT 03-1614) | 42% | 4e–08 | 55% |
| phiHBZC1_1 | HBZC1_17680 ( CCB80754.1) | Type VI secretion protein (Herbaspirillum sp. B39) | 64% | 2.4 | 28% |
| phiHBZC1_1 | HBZC1_17710 ( CCB80757.1) | DNA methyltransferase (Oceanospirillum beijerinckii) | 89% | 3e–66 | 39% |
| phiHBZC1_1 | HBZC1_17820 ( CCB80754) | Addiction module antitoxin RelB (Burkholderia cenocepacia) | 91% | 2e–24 | 53% |
| phiHBZC1_1 | HBZC1_17770 ( CCB80763.1) | DNA adenine methylase (Campylobacter jejuni subsp. jejuni 2008-979) | 89% | 6e–19 | 39% |
| phiHBZC1_1 | HBZC1_17780 ( CCB80764.1) | DNA adenine methylase (Desulfosporosinus acidiphilus) | 87% | 2e–25 | 44% |
| phiHBZC1_1 | HBZC1_17700 ( CCB80756.1) | Multidrug resistance protein D (Salmonella enterica subsp. enterica serovar Infantis) | 40% | 2e–06 | 31% |
A range of phage-encoded virulence genes was identified within the Helicobacter prophage sequences (Table 2). A DNA methyltransferase-encoding gene was identified in most of the analyzed Helicobacter prophages. DNA methyltransferase is thought to contribute to the specificity of bacterium-host interactions or H. pylori virulence (Vitkute et al., 2001). Furuta and colleagues (2015) found that DNA methyltransferase genes are rapidly evolving in H. pylori genomes, which facilitates H. pylori adaptation to a new host. A protein encoded by phiNY40_1 (NY40_0553) displayed 23% identity with a serine/threonine kinase of Thiorhodococcus drewsii. Phosphorylation of proteins usually occurs during interactions between bacterial cells and host cells and plays a role in bacterial pathogenesis (Cozzone, 2005). Serine/threonine kinases are considered to affect cell survival pathways and contribute to H. pylori pathogenesis (King & Obonyo, 2015). A putative glycosyltransferase is encoded by phiHCD_1. Glycosyltransferases are involved in biosynthesis of LPS (Luke et al., 2010) that can promote proliferation of gastric cancer cells (Tomoda, Kamiya & Suzuki, 2015). An antitoxin component RelB of the addiction toxin-antitoxin (TA) module system RelBE was identified in phiHBZC1_1. The protein plays a role in cell survival (Park, Son & Lee, 2013).
Conclusions
In brief, we present here sixteen Helicobacter prophages. Eight of them were identified for the first time after mining the sequenced Helicobacter sp. genomes, and the other eight had been reported in published literature. Based on comparative genomic analyses, the sixteen phages were sorted into four clusters, Clusters A–D, respectively. Cluster A was further divided into four subclusters, subclusters A1–A4. Different subclusters displayed similarity to each other. Subcluster A1 phages are full-length prophages. Subcluster A2, A3 and A4 phages are remnant prophage-like elements. The genomes and genetic information of the Cluster B, C and D phages were analyzed. Interestingly, several genes encoding antibiotic resistance proteins and virulence factors were found within various prophage genomes. These results highlight an important issue, which needs to be resolved before proceeding with phage therapy for treatment of H. pylori infections. To our knowledge, this is the first systematic analysis of Helicobacter prophages. With more forthcoming Helicobacter genome sequences, more Helicobacter prophages will be identified, and the role of prophages in evolution, adaptations and physiology of Helicobacter sp. will be clarified.