
Integrated downstream regulation by the quorum-sensing controlled transcription factors LrhA and RcsA impacts phenotypic outputs associated with virulence in the phytopathogen Pantoea stewartii subsp. stewartii
- Published
- Accepted
- Received
- Academic Editor
- Mario Alberto Flores-Valdez
- Subject Areas
- Agricultural Science, Genetics, Microbiology, Molecular Biology, Plant Science
- Keywords
- LrhA, Pantoea stewartii subsp. stewartii, Phytopathogen, Quorum sensing, RcsA, Transcription factor
- Copyright
- © 2017 Duong and Stevens
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2017. Integrated downstream regulation by the quorum-sensing controlled transcription factors LrhA and RcsA impacts phenotypic outputs associated with virulence in the phytopathogen Pantoea stewartii subsp. stewartii. PeerJ 5:e4145 https://doi.org/10.7717/peerj.4145
Abstract
Pantoea stewartii subsp. stewartii is a Gram-negative proteobacterium that causes leaf blight and Stewart’s wilt disease in corn. Quorum sensing (QS) controls bacterial exopolysaccharide production that blocks water transport in the plant xylem at high bacterial densities during the later stage of the infection, resulting in wilt. At low cell density the key master QS regulator in P. stewartii, EsaR, directly represses rcsA, encoding an activator of capsule biosynthesis genes, but activates lrhA, encoding a transcription factor that regulates surface motility. Both RcsA and LrhA have been shown to play a role in plant virulence. In this study, additional information about the downstream targets of LrhA and its interaction with RcsA was determined. A transcriptional fusion assay revealed autorepression of LrhA in P. stewartii and electrophoretic mobility shift assays (EMSA) using purified LrhA confirmed that LrhA binds to its own promoter. In addition, LrhA binds to the promoter for the RcsA gene, as well as those for putative fimbrial subunits and biosurfactant production enzymes in P. stewartii, but not to the flhDC promoter, which is the main direct target of LrhA in Escherichia coli. This work led to a reexamination of the physiological function of RcsA in P. stewartii and the discovery that it also plays a role in surface motility. These findings are broadening our understanding of the coordinated regulatory cascades utilized in the phytopathogen P. stewartii.
Introduction
Pantoea stewartii subsp. stewartii, a Gram-negative rod-shaped, gamma-proteobacterium, is the causal agent of leaf blight and Stewart’s wilt in susceptible varieties of Zea mays. It is primarily transmitted to the plant by the corn flea beetle, Chaetocnema pulicaria (Esker & Nutter, 2002). After being deposited through excrement into wounds generated during insect feeding, the pathogen gains access to the leaf apoplast and causes water-soaked lesions through the Hrp-type III secretion system (Ham et al., 2006; Roper, 2011). In a second phase of the disease, the bacteria then also migrate to the xylem, where they grow to high cell density and form a biofilm that blocks water flow within the plant. This results in wilt disease and even death, if the plants were infected at the seedling phase (Braun, 1982). Quorum sensing (QS), a mechanism of bacterial cell density-dependent communication, controls the virulence, capsule production and surface motility of this pathogen (Roper, 2011; Von Bodman, Bauer & Coplin, 2003).
During QS, P. stewartii produces N-acyl homoserine lactone (AHL) signals due to the activity of EsaI, a LuxI-type protein (Beck von Bodman & Farrand, 1995). The AHL signal then interacts with the master QS regulatory protein EsaR, a LuxR homologue, when the cell density reaches a critical threshold. EsaR is a dual-level transcriptional regulator that binds to DNA at its recognition sites to either activate or repress its downstream targets at low cell density (Beck von Bodman & Farrand, 1995; Von Bodman et al., 2003; Von Bodman, Majerczak & Coplin, 1998). When EsaR and AHL interact at high cell density, the EsaR-AHL complex is unable to bind to the DNA resulting in transcriptional deactivation or derepression of its target genes (Schu et al., 2009; Shong et al., 2013). Multiple approaches have been used to identify several direct targets of EsaR, including classic genetic (Minogue et al., 2005), proteome-level (Ramachandran & Stevens, 2013) and transcriptome-level (Ramachandran et al., 2014) analysis. Two of these direct targets, rcsA and lrhA, are involved in plant virulence and control capsule production and surface motility, respectively (Kernell Burke et al., 2015).
EsaR directly represses the P. stewartii rcsA gene at low cell density, insuring precise control over the timing of capsule synthesis (Carlier & Von Bodman, 2006; Minogue et al., 2005; Von Bodman, Majerczak & Coplin, 1998). At high cell density, gene activation by RcsA leads to production of stewartan, a polymer of galactose, glucose and glucuronic acid in a 3:3:1 ratio, which is the main component of the exopolysaccharide (EPS) (Nimtz et al., 1996). Stewartan is a primary virulence factor of P. stewartii (Carlier, Burbank & Von Bodman, 2009; Minogue et al., 2005; Roper, 2011). Previous work has shown that the lrhA gene is directly activated by EsaR at low cell density and a P. stewartii LrhA deletion mutant exhibits decreased surface motility and intermediate virulence levels in comparison to the wild type (Kernell Burke et al., 2015). However, little is known about the precise role of LrhA and its targets with regard to surface motility and virulence in P. stewartii.
In Escherichia coli, the function of LrhA is better understood. It is the key regulator controlling the expression of flagella, motility and chemotaxis by regulating the synthesis of FlhD2C2, the master regulator of flagella and chemotaxis gene expression (Lehnen et al., 2002). In E. coli, LrhA directly activates its own expression and represses the expression of flhD/flhC, thereby suppressing motility and chemotaxis (Lehnen et al., 2002). LrhA also controls fimA expression (Blumer et al., 2005) and regulates RpoS translation (Gibson & Silhavy, 1999; Peterson et al., 2006), but its binding site is not well defined (Lehnen et al., 2002).
In contrast to E. coli, P. stewartii is only capable of swarming rather than swimming motility. The bacterium’s swarming motility is controlled by QS and contributes to its pathogenicity (Herrera et al., 2008). The surface motility is flagellar-dependent since deletion of fliCI renders the bacterium aflagellar and incapable of swarming (Herrera et al., 2008). There is no evidence demonstrating that EsaR plays a direct role in regulating flagella synthesis. However, EsaR does directly regulate lrhA in P. stewartii and thereby indirectly regulates surface motility and plant virulence (Kernell Burke et al., 2015) through unknown mechanisms. A transcriptome-level analysis of the LrhA regulon in P. stewartii showed that LrhA activates three genes and represses 23 genes four-fold or more (Kernell Burke et al., 2015). In the present study, CKS_0458 and CKS_5211, genes putatively encoding a fimbrial subunit and biosurfactant production enzyme, respectively, have now been confirmed to be direct targets of LrhA. In addition, LrhA has also been demonstrated to repress its own gene and that of RcsA. Follow-up studies led to the finding that RcsA also plays a role in surface motility. This work has helped further reveal how the QS regulatory cascade in P. stewartii coordinately controls genes important for interactions with the plant host.
Materials and Methods
Strains and growth conditions
Table 1 lists strains and plasmids used in this study. E. coli strains were grown in Luria-Bertani (LB) (10 g/l tryptone, 5 g/l yeast extract, and 5 g/l NaCl) broth or plates with 1.5% agar at 37 °C. P. stewartii strains were grown in either LB or Rich Minimal (RM) medium (1 × M9 salts, 2% casamino acids, 1 mM MgCl2, and 0.4% glucose) at 30 °C. Growth medium was supplemented with the following antibiotics: ampicillin (Ap, 100 µg/ml), chloramphenicol (Cm, 35 µg/ml), kanamycin (Kn, 50 µg/ml), nalidixic acid (Nal, 30 µg/ml), or streptomycin (Str, 100 µg/ml) as required (see Table 1).
Green fluorescent protein fusion (GFP) construction and testing
A transcriptional fusion between the lrhA promoter (903 bp) and the gene for GFP was created through traditional molecular techniques as described previously (Kernell Burke et al., 2015). PCR primers (Table S1) with the restriction sites EcoRI and KpnI added to the 5′ and 3′ ends of the promoter sequence, respectively, were used to facilitate subcloning into the pPROBE′-GFP [tagless] vector (Miller, Leveau & Lindow, 2000). E. coli S17-1 was transformed with this plasmid construct containing PlrhA-gfp, which was then moved into the wild-type P. stewartii DC283, ΔlrhA and ΔlrhA/lrhA+ strains (Table 1) via conjugation. The transconjugates were grown in RM medium supplemented with Kn and Nal overnight, diluted in fresh medium to an OD600 of 0.05 at 30 °C with shaking at 250 rpm to an OD600 of 0.2–0.5, diluted a second time in fresh medium to an OD600 of 0.025 and grown to an OD600 of 0.5. GFP measurements were done as previously described (Kernell Burke et al., 2015) with average relative fluorescence/OD600 from three experiments of triplicate samples, standard errors, and two-tailed homoscedastic Student’s t-test values calculated for each strain.
| Strains | Genotype and notesa | References |
|---|---|---|
| Pantoea stewartiistrains | ||
| DC283 | Wild-type strain; Nalr | Dolph, Majerczak & Coplin (1988) |
| ΔlrhA | Unmarked deletion of lrhA coding sequence from DC283; Nalr | Kernell Burke et al. (2015) |
| ΔlrhA/lrhA+ | ΔlrhA with chromosomal complementation of lrhA and its promoter downstream of glmS; Nalr Cmr | Kernell Burke et al. (2015) |
| ΔrcsA-2015 | Unmarked deletion of rcsA coding sequence from DC283; Nalr, missing 66-kb region | Kernell Burke et al. (2015) |
| ΔrcsA/rcsA+-2015 | ΔrcsA with chromosomal complementation of rcsA and its promoter downstream of glmS; Nalr Cmr, missing 66-kb region | Kernell Burke et al. (2015) |
| ΔrcsA-2017 | Unmarked deletion of rcsA coding sequence from DC283; Nalr | This study |
| ΔrcsA/rcsA+-2017 | ΔrcsA with chromosomal complementation of rcsA and its promoter downstream of glmS; Nalr Cmr | This study |
| ΔCKS_0458-CKS_0459 | Unmarked deletion of both CKS_0458 and CKS_0459 coding sequence from DC283; Nalr | This study |
| ΔCKS_0458-CKS_0459 /CKS_0458+ | ΔCKS_0458-CKS_0459 with chromosomal complementation of CKS_0458 and its promoter downstream of glmS; Nalr Cmr | This study |
| ΔCKS_0458-CKS_0459 /CKS_0458-CKS_0459+ | ΔCKS_0458-CKS_0459 with chromosomal complementation of CKS_0458-CKS_0459 and their promoter downstream of glmS; Nalr Cmr | This study |
| ΔCKS_5208 | Unmarked deletion of CKS_5208 coding sequence from DC283; Nalr | This study |
| ΔCKS_5208/CKS_5208+ | ΔCKS_5208 with chromosomal complementation of CKS_5208 and its promoter downstream of glmS; Nalr Cmr | This study |
| ΔCKS_5211 | Unmarked deletion of CKS_5211 coding sequence from DC283; Nalr | This study |
| ΔCKS_5211/CKS_5211+ | ΔCKS_5211 with chromosomal complementation of CKS_5211 and its promoter downstream of glmS; Nalr Cmr | This study |
| ΔCKS_5211/ΔCKS_5208 | Unmarked deletion of both CKS_5211 and CKS_5208 coding sequences from DC283; Nalr | This study |
| Escherichia colistrains | ||
| Top 10 | F−mcrAΔ(mrr-hsdRMS-mcrBC) Φ80dlacZΔM15ΔlacX 74 deoR recAI araD139Δ(ara-leu)7697 galU galK rpsL ( Strr) endA1 nupG | Grant et al. (1990) |
| DH5 αλpir | F−endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupGΦ80dlacZΔM15Δ(lacZYA-argF)U169 hsdR17(rK- mK+)λpir | Kvitko et al. (2012) |
| S17-1 | recA pro hsdR RP4-2-Tc::Mu-Km::Tn7 | Simon, Priefer & Pühler (1983) |
| S17-1 λpir | recA pro hsdR RP4-2-Tc::Mu-Km::Tn7λpir | Labes, Puhler & Simon (1990) |
| BL21-DE3 | fhuA2 [lon] ompT gal (λ DE3) [dcm]ΔhsdSλ DE3=λ sBamHIoΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21Δnin5 | Studier & Moffatt (1986) |
| Plasmids | ||
| pGEM-T | Cloning vector, Apr | Promega |
| pET28a | Expression vector, Knr | Novagen |
| pDONR201 | Entry vector in the Gateway system, Knr | Life Technologies |
| pAUC40 | Suicide vector pKNG101::attR-ccdB-CmR; Cmr, Strr, sacB | Carlier, Burbank & Von Bodman (2009) |
| pEVS104 | Conjugative helper plasmid, tra trb; Knr | Stabb & Ruby (2002) |
| pUC18R6K-mini-Tn7-cat | Tn7 vector for chromosomal integration into the intergenic region downstream of glmS; Cmr, Apr | Choi et al. (2005) |
| pPROBE′-GFP[tagless] PlrhA | pPROBE′-GFP[tagless] vector with the promoter of lrhA; Knr | This study |
Notes:
- Apr
-
ampicillin resistance
- Nalr
-
nalidixic acid resistance
- Knr
-
kanamacyin resistance
- Cmr
-
chloramphenicol resistance
- Strr
-
streptomycin resistance
Overexpression of LrhA
The lrhA coding sequence was amplified using primers with BamHI and HindIII sites (Table S1), cloned into pGEM-T (Promega, Madison, WI, USA), and sequenced. After double digestion with BamHI and HindIII, the construct was ligated into pET28a (Novagen, Madison, WI, USA) and transformed into E. coli (BL21-DE3) (Studier & Moffatt, 1986) to express LrhA with a His6 tag at the N-terminus (37 kDa). Induction of protein expression with 0.1 M isopropyl β-D-1-thiogalactopyranoside (IPTG) was performed at an OD600 of 0.5–0.8, 19 °C, overnight, shaking at 250 rpm. Cells were pelleted by centrifugation at 5,000 rpm in a JA-10 rotor (Beckman Coulter, Brea, CA, USA) for 20 min at 4 °C, snap-frozen with liquid nitrogen and stored at −75 °C. The cell pellet was then resuspended in Ni-NTA wash buffer (50 mM Tris–HCl, 300 mM NaCl, 50 mM imidazole) and sonicated to release proteins. Ultracentrifugation at 40,000 rpm in a Beckman Ti70 rotor for 1 h at 4 °C was used to subsequently remove the cell debris. The protein was purified using a Ni-NTA column (HisTrap HP, GE Healthcare) with Ni-NTA elution buffer (50 mM Tris–HCl, 300 mM NaCl, 500 mM imidazole). The protein purity was observed through standard SDS-PAGE electrophoresis.
Electrophoretic mobility shift assays (EMSA)
Promoter regions of genes of interest were amplified with FAM-labeled primers (Table S1) and extracted from a 1% agarose gel to examine the specific binding with purified His6-LrhA over a range of concentrations. Twenty µl reactions with purified His6-LrhA, 5 nM FAM-DNA in 1X EMSA buffer (10% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 10 mM Tris–HCl, 50 µg/ml poly (dI-dC) and 150 µg/ml BSA) were incubated at room temperature for 1 h before loading on to 1 × TBE (10.8 g/l Tris–HCl, 5.5 g/l boric acid, 2 mM EDTA, pH 8.0) 4%, 5%, or 6% acrylamide native gels followed by electrophoresis at 80 V for 2–3 h. Images were visualized on a Typhoon Trio Scanner (GE Healthcare). Experiments were done in duplicate.
Construction of unmarked deletion mutant strains
Chromosomal deletions of CKS_0458/CKS_0459, CKS_5208, CKS_5211, and CKS_5211/CKS_5208 were constructed based on the Gateway system (Life Technologies) and suicide vectors as described previously (Kernell Burke et al., 2015), but with primers listed in Table S1. In addition, another chromosomal deletion of rcsA was constructed using the same approach as described in a previous study (Kernell Burke et al., 2015), due to a deletion of ∼66-kilobases (kb) in the chromosome of the original construct.
Construction of chromosomal complementation strains
Complementation strains were constructed by generating a chromosomal insertion of the promoter and coding regions of the target gene into the neutral region downstream of glmS on the P. stewartii chromosome using the pUC18R6K-mini-Tn7-cat vector system (Choi et al., 2005) as previously described (Kernell Burke et al., 2015), but with primers listed in Table S1.
Phenotypic surface motility assay
Swarming motility for the wild-type, deletion and complementation strains was investigated under strict conditions to ensure a reproducible phenotype as previously described (Kernell Burke et al., 2015). Briefly, five µl of cell culture at an OD600 of 0.5 were spotted directly on the agar surface of LB 0.4% agar quadrant plates supplemented with 0.4% glucose (Herrera et al., 2008). Plates were incubated in a closed plastic box at 30 °C for 2 days prior to observation.
Phenotypic capsule production assay
Bacterial strains were grown overnight in LB broth supplemented with the appropriate antibiotics at 30 °C with shaking. The overnight cultures were subcultured in fresh LB medium to an OD600 of 0.05 and grown to an OD600 of 0.5 at 30 °C with shaking. The strains were then cross-streaked with sterilized wooden sticks on 1.5% agar plates containing 0.1% casamino acids, 1% peptone and 1% glucose (CPG) (Kernell Burke et al., 2015; Von Bodman, Majerczak & Coplin, 1998). Plates were incubated at 30 °C, lid-up for 2 days to observe the capsule production and visualized using the Bio-Rad Gel Doc imager system.
Plant virulence assay
Virulence assays with P. stewartii strains in Zea mays seedlings were adapted from established methods (Von Bodman, Majerczak & Coplin, 1998; Kernell Burke et al., 2015) with some modifications. In this study, Zea mays cv. Jubilee, 2B seeds were planted in Sunshine mix #1 or Promix soil for seven or six days, respectively, in a 28 °C growth chamber with ∼100–200 µE m−2 s−1 light intensity, 16 h light/8 h dark and ∼80% relative humidity (Percival Scientific, Inc.). Fifteen seedlings between 6 and 10 cm of height with two separated leaves were inoculated with five µl (∼3 × 105 CFU) of bacterial culture grown to an OD600 of 0.2 in LB broth (∼6 × 107 CFU/ml). Prior to plant inoculation, the bacterial cells were washed and resuspended in phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4, pH 7.4). Wild-type strain and PBS controls were included in each trial, then, accumulated numbers of control-inoculated plants across all experiments were analyzed. A sterile needle (26G 5/8, 15.9 mm, SUB-Q Becton, Dickinson and Company) was used to make an ∼1 cm incision in the stem ∼1 cm above the soil line and the bacteria were added to the plant by slowly pipetting the inoculum while moving across the wound five times. The plants were observed on day 12 post-infection to assess the virulence by two independent observers. Symptom severity was scored based on a five-point scale with 0 = no symptoms; 1 = few scattered lesions; 2 = scattered water soaking symptoms; 3 = numerous lesions and slight wilting; 4 = moderately severe wilt; 5 = death. The data for each treatment were averaged together and used to calculate the mean and standard error. A Student’s t-test was used to calculate the p-value for experimental treatments compared to the wild-type treatment.
Results
LrhA autorepresses its own gene expression in P. stewartii
A GFP reporter was used to measure levels of transcription from the lrhA promoter in the wild-type, ΔlrhA and ΔlrhA/lrhA+ strains of P. stewartii DC283 (Table 1). Expression levels of GFP in the ΔlrhA strain were significantly higher than the wild-type strain (Fig. 1, p = 0.00001) indicating that LrhA normally represses its own expression in the wild-type strain. The expression level of the lrhA promoter in the complementation ΔlrhA/lrhA+ strain was restored to levels closer to those of the wild-type strain, and was also significantly different than the deletion strain (Fig. 1, p = 0.00002).
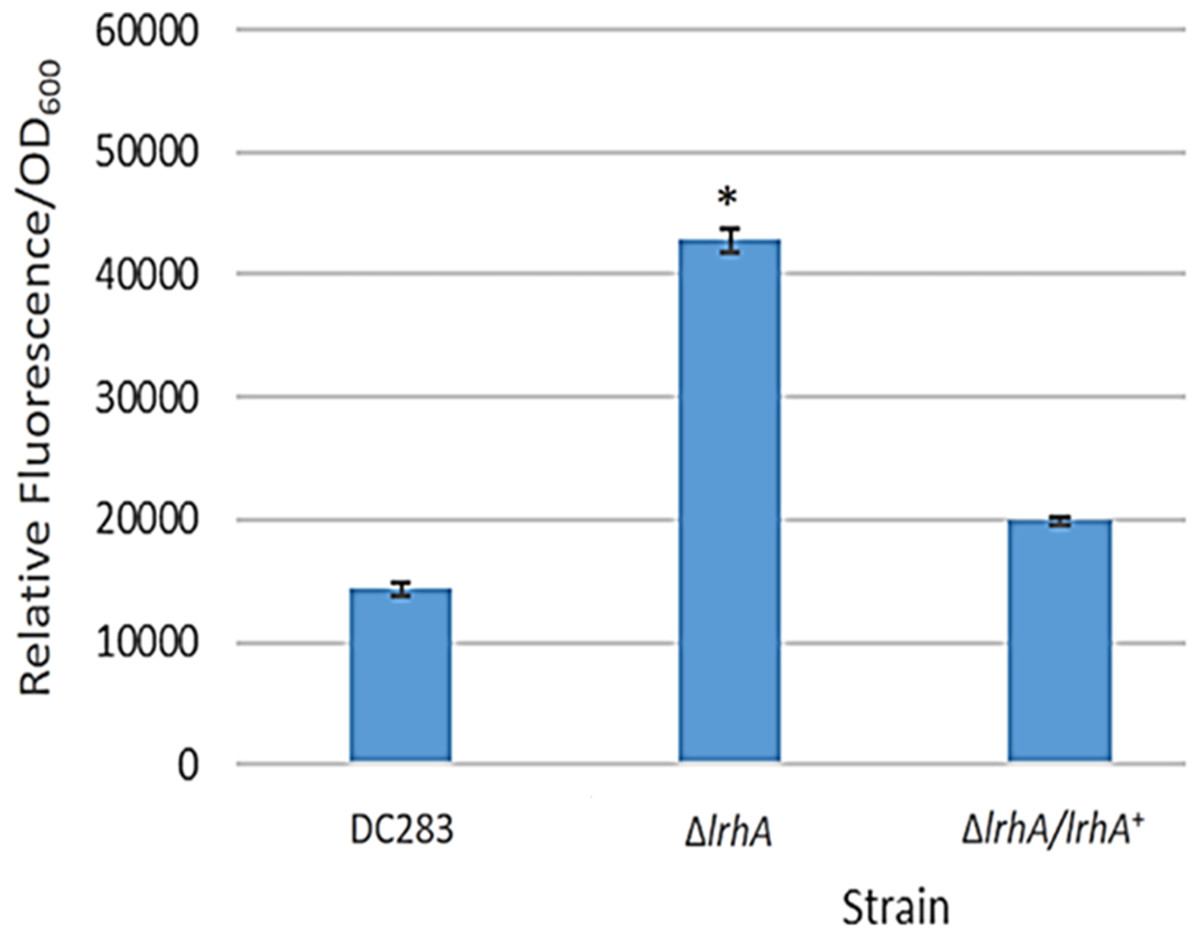
Figure 1: Expression levels of a lrhA promoter-gfp transcription reporter in three P. stewartii strains.
The wild-type DC283 and ΔlrhA and ΔlrhA/lrhA+ strains in the same genetic background (containing pPROBE′-GFP[tagless] PlrhA) were grown to an OD600 of 0.5 and GFP expression levels from the lrhA promoter-gfp transcription reporter were measured as average relative fluorescence/OD600. Data represents three experimental samples analyzed in triplicate. Error bars denote standard error. The asterisk (∗) represents a statistically significant difference (p < 0.05) between the ΔlrhA and both the wild-type and ΔlrhA/lrhA+ strains using a two-tailed homoscedastic Student’s t-test.{kind=link}
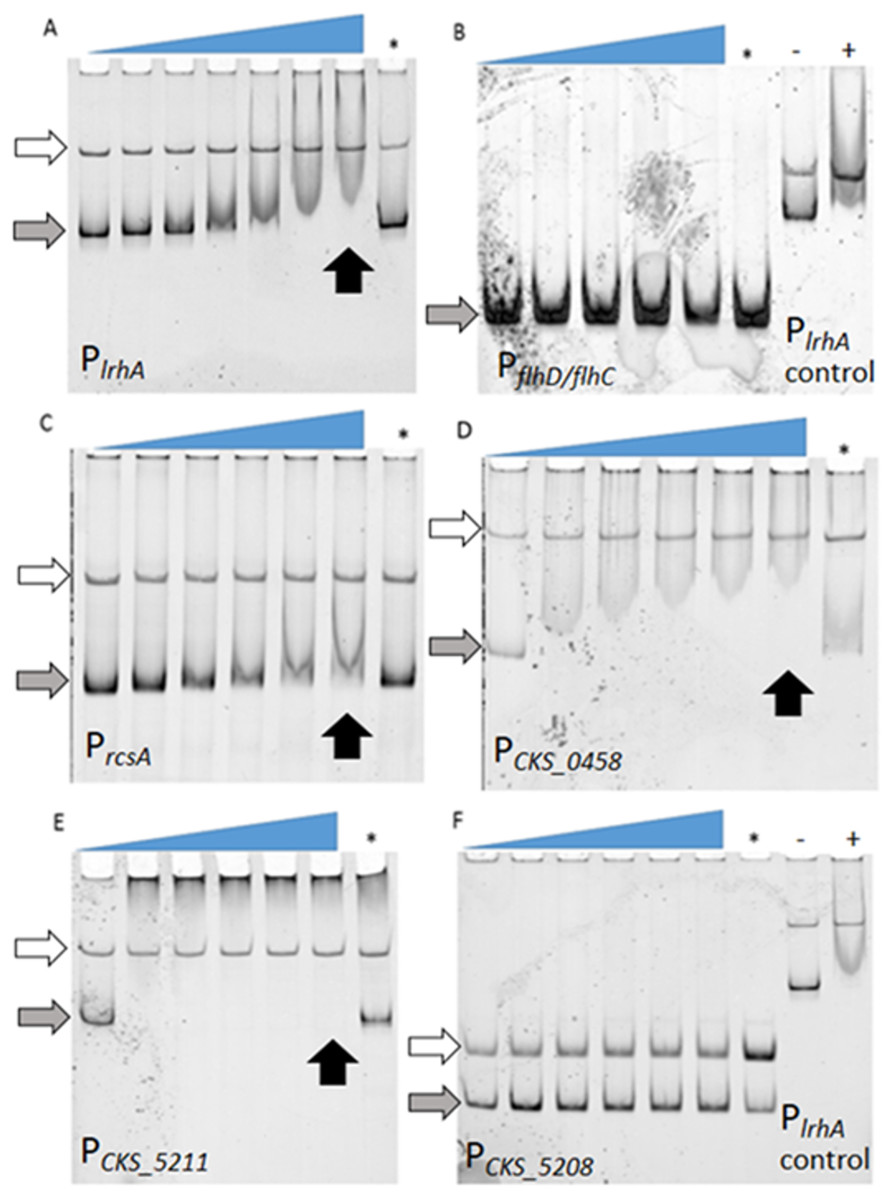
Figure 2: Examination of binding of LrhA to select target promoters via EMSA.
FAM-DNA probes were incubated with increasing concentrations of His6-LrhA (LrhA) from left to right, corresponding to the slope of the triangles, to investigate the mobility shift upon specific binding to the protein. The competition reaction (indicated by the asterisk, ∗) was conducted with 25 nM unlabeled DNA of PlrhA to prove the specificity of the interaction. Autoregulation of LrhA was confirmed with the direct binding between purified LrhA to its promoter (A). Shifted bands were also observed with PrcsA (C), PCKS_0458 (D), and PCKS_5211 (E). There were no shifted bands observed for PflhDC (B) and PCKS_5208 (F), while the positive controls for LrhA activity showed a shift (−: reaction with PlrhA probe in the absence of LrhA, +: reaction with PlrhA probe in the presence of 200 nM LrhA). Concentrations of LrhA tested for PlrhA (A) are 0, 25, 50, 100, 200, 400, and 800 nM. Concentrations of LrhA tested for PflhDC (B) are 0, 400, 600, 800, and 1,000 nM. Concentrations of LrhA tested for PrcsA (C), PCKS_0458 (D), PCKS_5211 (E) and PCKS_5208 (F) are 0, 200, 400, 600, 800, and 1,000 nM. Grey arrows highlight unbound DNA probes. White arrows indicate unbound DNA generated during PCR reactions that do not interact specifically with LrhA. Black arrows point to the lane with specific binding at the highest concentration of LrhA.{kind=link}
Identification of LrhA direct targets through EMSAs
To determine if the observed lrhA autorepression occurred directly or indirectly, electrophoretic mobility shift assays (EMSAs) were performed. First, direct binding of LrhA to the promoter of its own gene was demonstrated by EMSA analysis (Fig. 2A). Next, the ability of LrhA to directly regulate additional gene targets was explored, using the lrhA promoter as a positive control for the His6-LrhA activity and unlabeled PlrhA DNA to prove the specificity of the binding. In E. coli, LrhA is known as a repressor of motility by direct interaction with the promoter region of flhD/flhC, whose products promote the expression of flagellar gene synthesis (Lehnen et al., 2002). However, RNA-Seq data of expression levels of flhD/flhC in P. stewartii showed less than a two-fold difference between wild-type and ΔlrhA strains (Kernell Burke et al., 2015) suggesting a lack of transcriptional regulation. Here, EMSA analysis showed that His6-LrhA does not bind to the promoter of flhD/flhC (Fig. 2B), explaining the observed lack of transcriptional regulation.
Additional analysis of the LrhA-regulated transcriptome in P. stewartii revealed that LrhA repressed the expression level of several more downstream targets, including rcsA, CKS_0458, CKS_5208 and CKS_5211 (Kernell Burke et al., 2015). RcsA activates capsule production, a known virulence factor in P. stewartii (Kernell Burke et al., 2015; Minogue et al., 2005; Poetter & Coplin, 1991; Wehland et al., 1999). The putative roles of genes for fimbria encoded by CKS_0458, annotated as a putative fimbrial subunit, (and CKS_0459 located downstream in an operon) and for surfactant expression encoded by CKS_5208 and CKS_5211, annotated as a rhamnosyltransferase I subunit B and a putative alpha/beta superfamily hydrolase/acyltransferase, respectively, in plant colonization and/or virulence had not been established. However, it seemed plausible that they might also play roles in host association as they were some of the most highly LrhA-repressed genes, four-fold or greater (Kernell Burke et al., 2015). Therefore, the binding of LrhA to the promoters of these genes was also examined via EMSA. The direct binding of His6-LrhA to PrcsA, PCKS_0458 and PCKS_5211 (Figs. 2C–2E), was demonstrated via EMSAs while PCKS_5208 did not interact with His6-LrhA in vitro (Fig. 2F). Collectively, these findings identified four directly controlled gene targets in the LrhA regulon. The lack of LrhA regulation of FlhD2C2, the master regulator of flagellar-based motility and chemotaxis in E. coli, indicates a different role for LrhA in controlling P. stewartii motility. The direct binding of LrhA to the promoter of rcsA further links LrhA to P. stewartii pathogenesis. The role of the two other LrhA direct targets CKS_0458 and CKS_5211 remained to be established.
Examining the role of putative fimbrial and surfactant production genes in the surface motility and virulence of P. stewartii
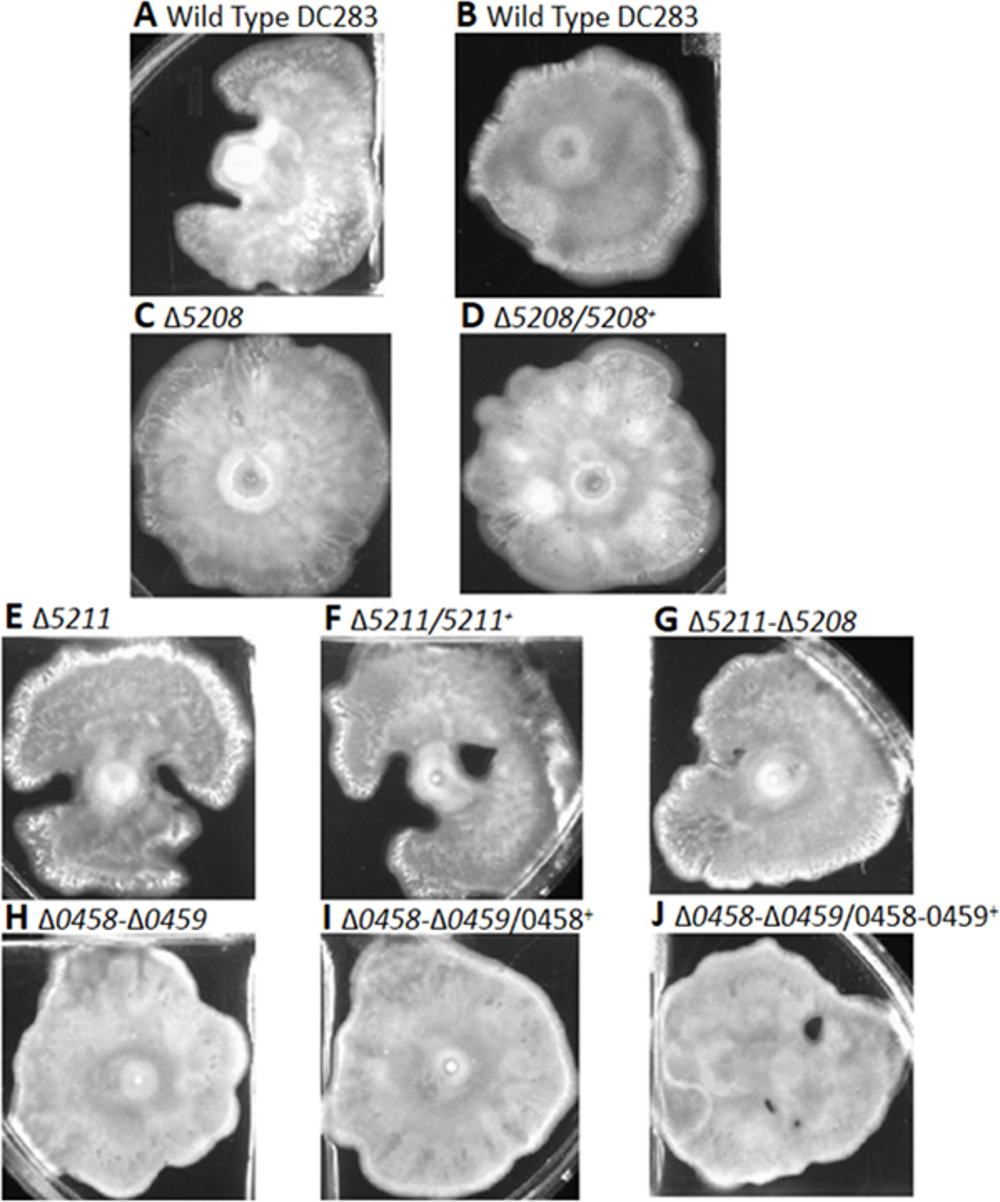
To further investigate the role of the downstream targets of LrhA putatively involved in production of fimbriae and surfactant, a reverse genetic approach was utilized. Markerless deletions of CKS_0458/CKS_0459, CKS_5208, CKS_5211 and CKS_5211/CKS_5208 were successfully generated. Corresponding chromosomal complementation strains were also generated with the exception of a double deletion mutant of CKS_5211/CKS_5208 complementation strain, due to the length constraint of the DNA fragment containing the adjacent genes. In surface motility assays, the P. stewartii wild-type strain showed either uni-directional (Fig. 3A and Fig. S1A) or omni-directional (Fig. 3B and Fig. S1B) expansion from the inoculum sites as had been previously observed (Herrera et al., 2008; Kernell Burke et al., 2015). In comparison to the wild type, there is no obvious difference between the various deletion and complementation strains; they all possessed similar level of expansion on the agar surface (Figs. S1C –S1J). Therefore, these genes do not appear to play any detectable role in surface motility via this assay.
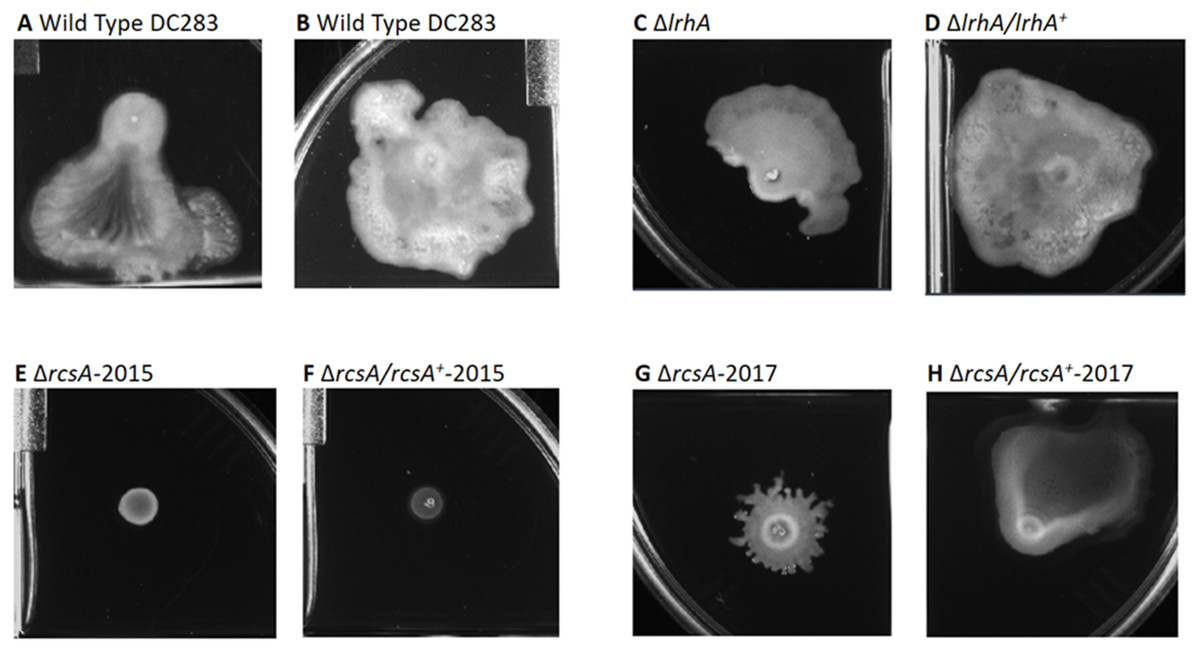
Figure 3: Impact of RcsA and LrhA on surface motility of P. stewartii.
The pictures show the analysis of surface motility in P. stewartii DC283 strains. Examples of wild type unidirectional (A) or omnidirectional surface motility (B) are shown as controls. The ΔlrhA/lrhA+ complementation strain (D) is similar to the control in (B), while the ΔlrhA strain has reduced surface motility expanding over a smaller surface area (C), as has been previously observed (Kernell Burke et al., 2015). Both ΔrcsA strains had dramatically reduced surface motility (E and G) as well as the ΔrcsA/rcsA+-2015 strain (F). The ΔrcsA/rcsA+-2017 strain was complemented for the defect in surface motility (H). All pictures were taken at the same magnification after 2 days of incubation at 30 °C in a closed plastic box.{kind=link}
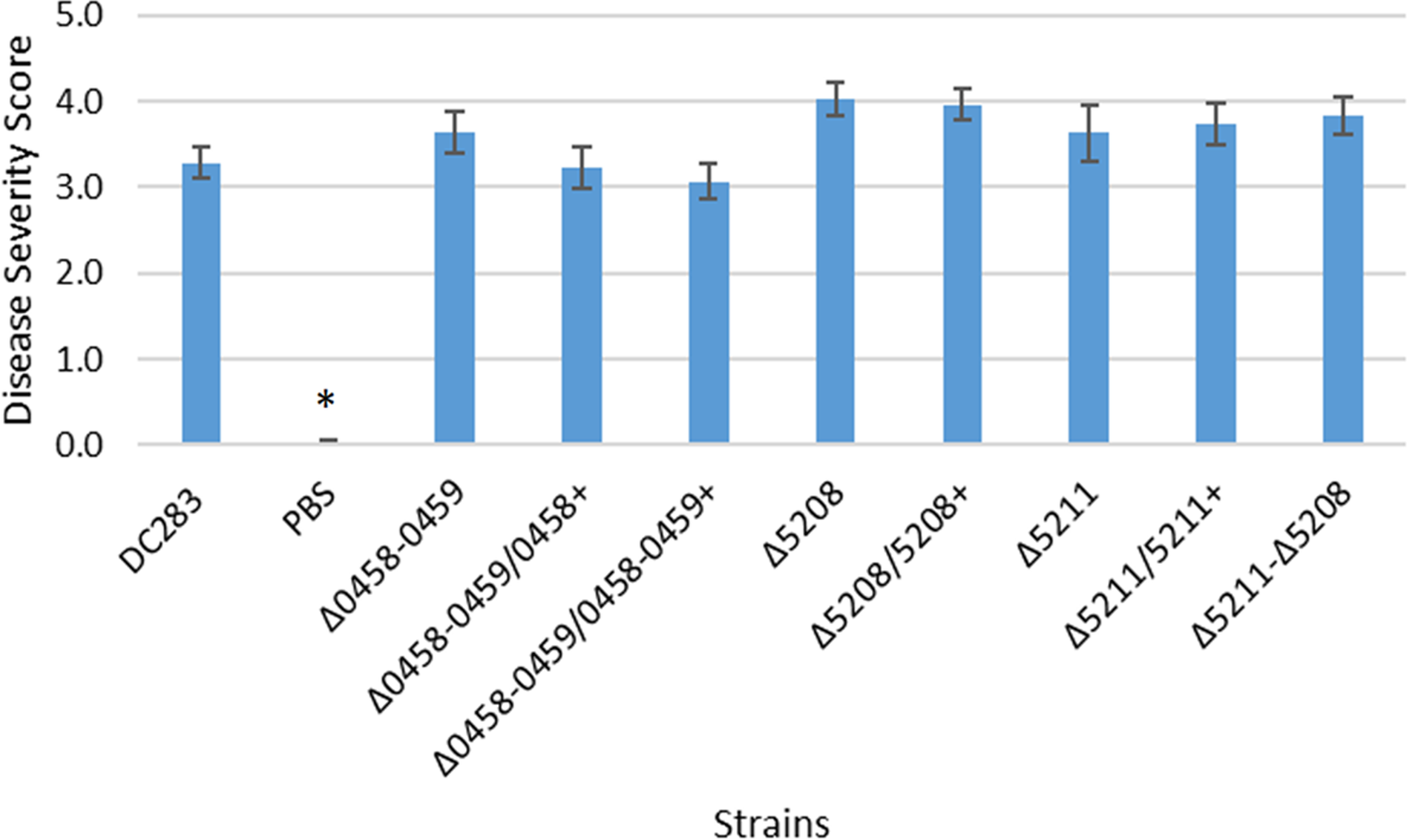
The same deletion and complementation strains of the genes putatively involved in fimbriae and surfactant production were also tested for virulence via in planta xylem infection assays. A lrhA deletion strain caused an intermediate level of disease severity in corn seedlings during xylem-infection assays (Kernell Burke et al., 2015). However, similar to the surface motility assays, no significant impacts on the virulence of P. stewartii were observed in the strains with deletions in either the fimbriae or surfactant synthesis genes (Fig. S2). Hence, the contribution of these genes individually to the virulence of the phytopathogen could not be measured.
Re-examining the role of RcsA in the capsule production, surface motility and virulence of P. stewartii
The important finding that LrhA directly binds to the promoter of rcsA, led to a reexamination of the previous findings about the physiological role of RcsA in P. stewartii. In prior work, rcsA deletion and complementation strains of DC283 had been constructed (ΔrcsA-2015 and ΔrcsA/rcsA+-2015) (Kernell Burke et al., 2015). However, complete assembly of the genome of P. stewartii DC283 (Duong, Stevens & Jensen, 2017) revealed that there is a large deletion, ∼66 kb containing 68 genes (Table S2), in the ΔrcsA-2015 and ΔrcsA/rcsA+-2015 strains. This deletion was not obvious using the incomplete genome sequence (NCBI GenBank accession no. AHIE00000000.1), but was found during a re-analysis of previously generated RNA-Seq data (Kernell Burke et al., 2015) using the new genome sequence (NCBI GenBank accession no. CP017581).
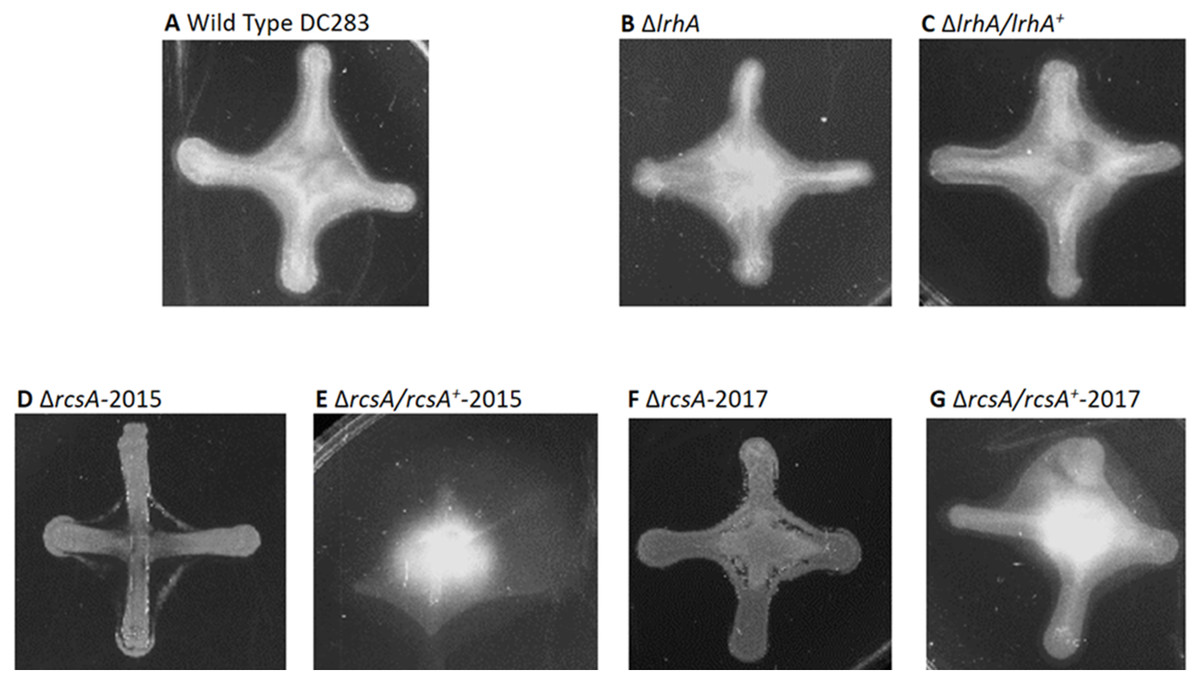
Figure 4: Impact of RcsA and LrhA on capsule production of P. stewartii.
All pictures were taken at the same magnification after two days of incubation at 30 °C after cross-streaking on casamino acid, peptone, glucose (CPG) agar plates. Differences in capsule production are apparent in the regions between the arms of the X-cross streak.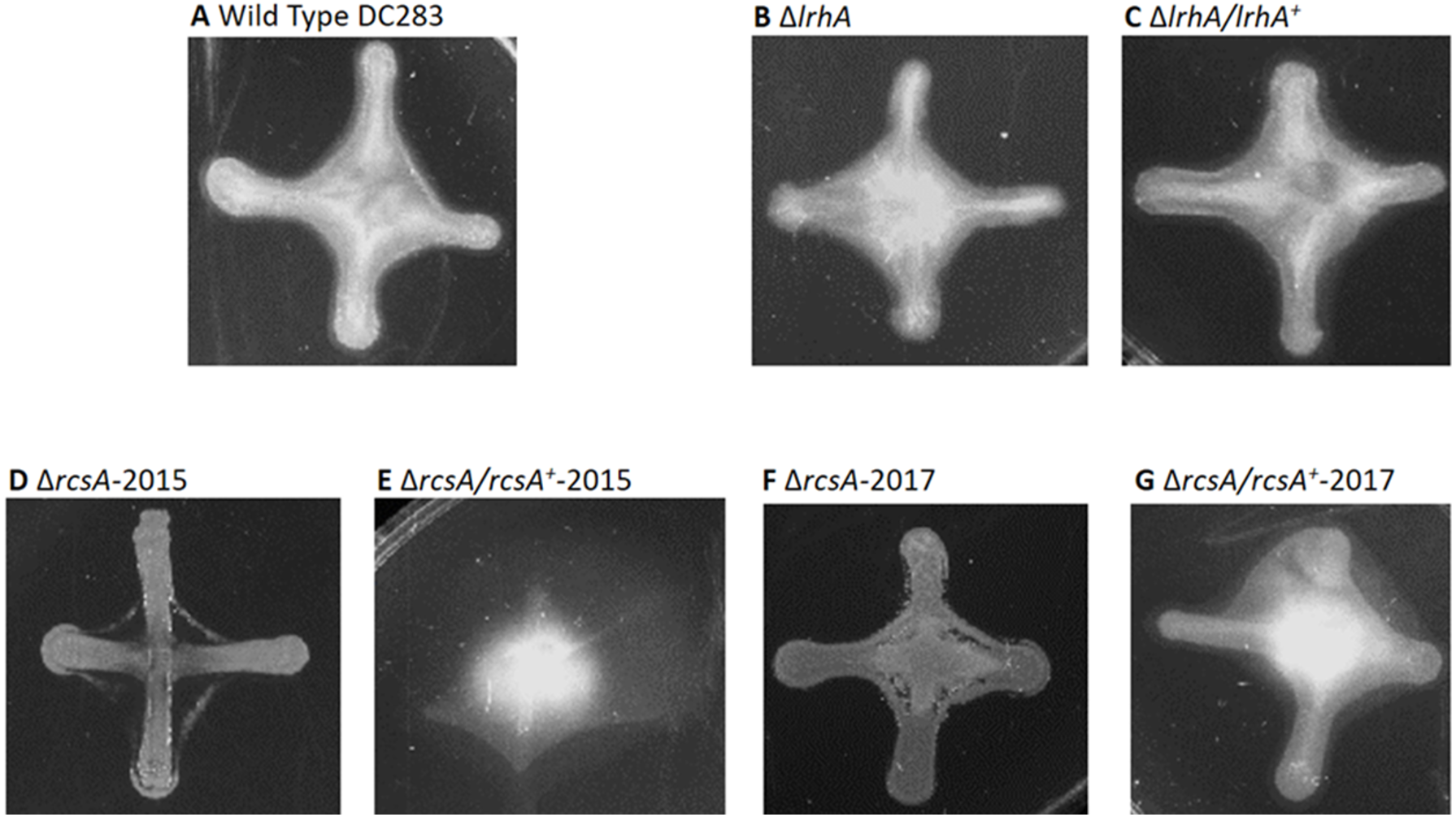{kind=link}
Therefore, a new set of rcsA deletion and complementation strains was re-constructed (ΔrcsA-2017 and ΔrcsA/rcsA+-2017) and shown to include the 66-kb region using PCR (data not shown). These new strains then were subjected to three assays to establish the true phenotypes of the rcsA deletion strain. First, capsule production assays have re-confirmed that RcsA regulates EPS production, as was shown for the 2015 strains (Kernell Burke et al., 2015). Both the ΔrcsA-2015 (Fig. 4D) and ΔrcsA-2017 strains (Fig. 4F) are not as mucoid as the parental wild-type strain (Fig. 4A) or the pair of lrhA deletion and complementation strains (Figs. 4B and 4C) as assessed by visual observation. The chromosomal complementation strains ΔrcsA/rcsA+-2015 (Fig. 4E) and ΔrcsA/rcsA+-2017 (Fig. 4G) had mucoid levels as high or higher than those seen in the wild type (Fig. 4A).
Second, surface motility assays were performed. These original strains ΔrcsA-2015 and ΔrcsA/rcsA+-2015 strains had not previously been examined for surface motility (Kernell Burke et al., 2015), but both were surprisingly defective for this phenotype (Figs. 3E and 3F). The fact that surface motility was not complemented by addition of rcsA back into the chromosome provided further evidence for the importance of the 66-kb region deletion that had been discovered initially through bioinformatics analysis. The new ΔrcsA-2017 also has severely reduced surface movement (Fig. 3G) while its complementation strain (Fig. 3H) restored motility levels similar to the wild-type strain (Figs. 3A and 3B). Thus, it has been demonstrated that RcsA plays a previously unappreciated role in the surface motility of P. stewartii. The defect in surface motility associated with the deletion of rcsA (Fig. 3G) appears to be greater than the defect in ΔlrhA (Fig. 3C). The ΔlrhA/lrhA+ strain has restored levels of surface motility (Fig. 3D), similar to the wild type (Figs. 3A and 3B), as previously reported (Kernell Burke et al., 2015).
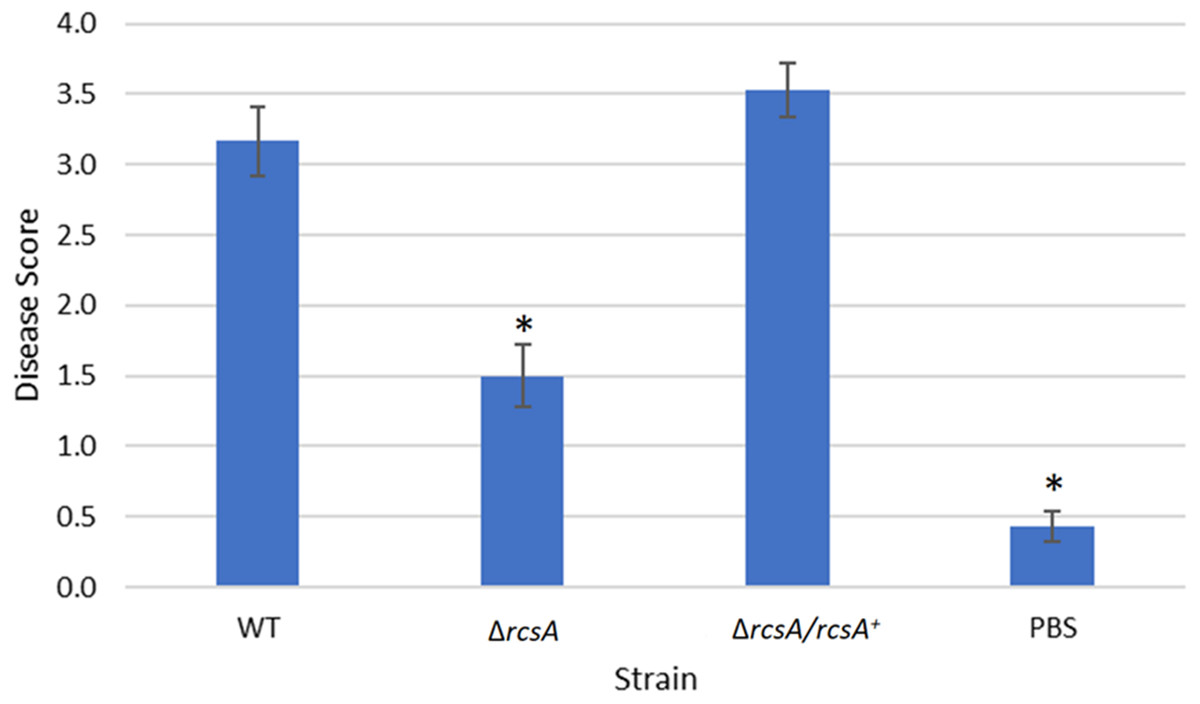
Figure 5: Plant assay testing the role of RcsA in virulence.
Data shown is the average score of disease for Day 12 of an infection assay performed with 15 plants inoculated with P. stewartii DC283 strains: wild type (WT), ΔrcsA-2017, ΔrcsA/rcsA+-2017, or PBS as a negative control. Higher value in the disease score indicates more severe symptoms from the infection. The asterisks (∗) represent strains that are statistically significantly different (p < 0.05) from the wild-type strain using a two-tailed homoscedastic Student’s t-test. Error bars denote standard error.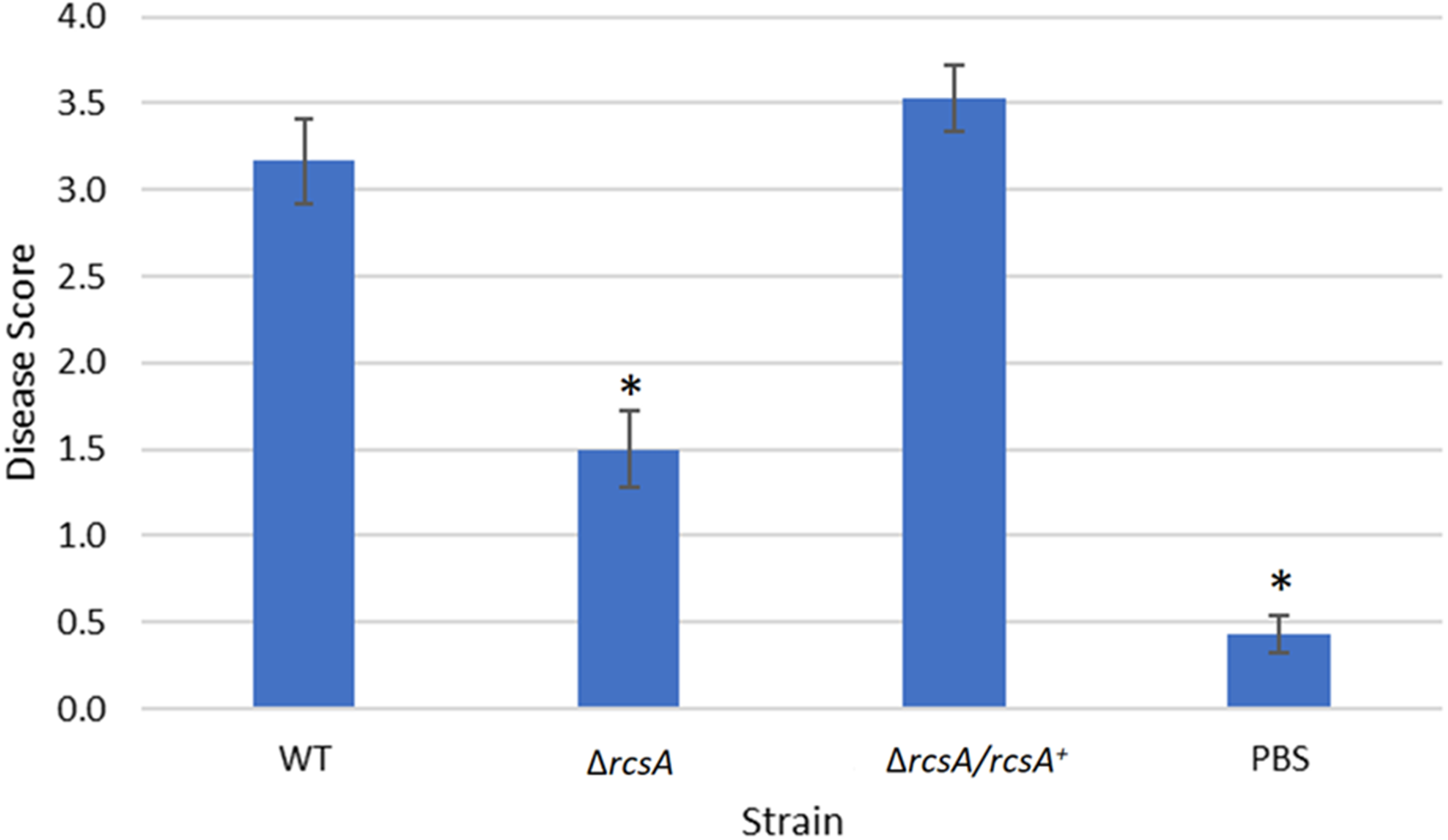{kind=link}
Finally, the xylem infection assays for the newly constructed ΔrcsA-2017 strain and its complement, with the inclusion of the wild-type strain and PBS as controls, indicated that the absence of rcsA significantly (p < 0.05) reduces the severity of the disease compared to the wild-type and complementation strains (Fig. 5) (p < 0.05). These results have similar trends with those reported for the 2015 strains (Kernell Burke et al., 2015) which confirms the role of rcsA in virulence of this phytopathogen. However, strains from the 2015 study that were missing the 66-kb region were reduced in their average disease severity (score ∼0 and ∼1.5 for the deletion and complementation strains, respectively) in comparison to the new 2017 strains (score ∼1.5 and ∼3.5 for the deletion and complementation strains, respectively) while the wild-type control had similar levels in both studies, implicating a role for the 66-kb region in virulence as well as surface motility.
Discussion
The role of the LrhA regulon in P. stewartii was further investigated in this study to understand how it is involved in the surface motility and virulence of the pathogen. Previous studies showed that surface motility in P. stewartii contributes to disease pathogenesis and this process involves both QS-controlled biofilm formation and flagella (Herrera et al., 2008). However, to date, there is no clear evidence to directly connect the synthesis of flagella to QS control in P. stewartii. Unlike E. coli, the QS-controlled transcription factor LrhA in P. stewartii does not regulate FlhD2C2, the master activator of flagellar synthesis. This was suggested by earlier RNA-Seq data (Kernell Burke et al., 2015), but directly tested here through EMSA that confirmed the inability of LrhA to bind to the flhD/flhC promoter. Additionally, LrhA activates its own expression in E. coli whereas autorepression was observed in P. stewartii. Even though P. stewartii LrhA has 77% amino acid identity to E. coli LrhA, the two have clearly evolved distinctive physiological roles in their host organisms.
In an attempt to define the function of the genes controlled by LrhA in P. stewartii, a reverse genetics approach was used to examine the role of select LrhA-regulated genes in surface motility and virulence of the phytopathogen. Multiple deletion and complementation strains of genes annotated as being involved in surfactant production (CKS_5208 and CKS_5211, initially annotated as a rhamnosyltransferase I subunit B and putative alpha/beta superfamily hydrolase/acyltransferase, respectively) and fimbriae assembly (CKS_0458 and CKS_0459, annotated as putative fimbrial subunits) were constructed and tested. Interestingly, none of these genes appear to play a fundamental role in surface motility and virulence individually. A LrhA deletion mutant impacting expression of multiple genes in the regulon produced noticeably decreased surface motility, but only intermediate virulence levels in comparison to the wild-type strain (Kernell Burke et al., 2015).

Figure 6: Updated model of the quorum-sensing regulatory network in P. stewartii.
Solid lines indicate known direct regulatory control. Red lines indicate direct control found in this study. Arrows represent activation and T lines represent repression. At low cell density when AHL levels are low, EsaR represses expression of rcsA, wceG2, and CKS_0458, and activates expression of lrhA. LrhA represses its own expression as well as that of rcsA and CKS_0458. At high cell density when EsaR-AHL complexes form, EsaR no longer activates or represses its direct targets. Thus, rcsA expression increases leading to activation of wceG2 and other genes necessary for capsule production. See the text for additional details.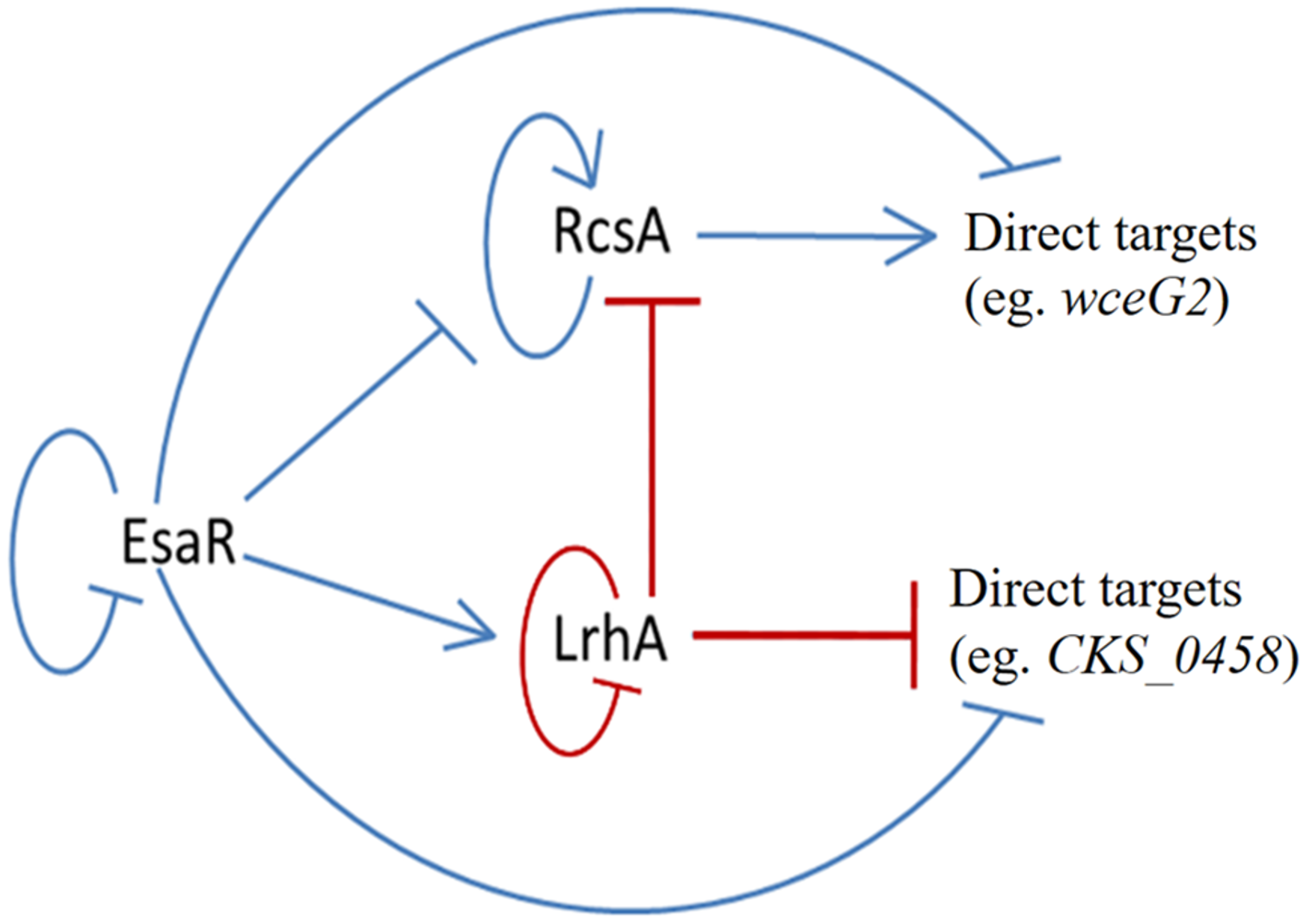{kind=link}
With regard to biosurfactant and fimbriae genes potentially associated with surface motility and adhesion, respectively, P. stewartii appears to utilize multiple levels of repression to ensure that the level of those genes’ expression is minimal. This low level of expression was again confirmed by an in planta RNA-Seq analysis (Packard et al., 2017). In the LrhA deletion strain expression of these genes was elevated. Thus, deletion mutants might actually mimic wild-type levels of the expression of these genes, producing a wild-type phenotype. Alternatively, these genes are not functional in the wild-type strain (indeed the new genome sequence (Duong, Stevens & Jensen, 2017) suggests that CKS_5211 is a pseudogene) or they may serve another function for the bacterium that was not examined in this study. Biofilm/adhesion assays were inconclusive (data not shown). Interestingly, some Pantoea species have been demonstrated to produce biosurfactants when grown on hydrocarbons (Vasileva-Tonkova & Gesheva, 2007). How this might impact bacterial surface motility or survival in planta is unclear.
It has been demonstrated that both RcsA and LrhA play an essential role to the surface motility of the wild-type strain of P. stewartii. The observed intermediate impact of a LrhA deletion on virulence may be due primarily to its direct control of RcsA and thereby its indirect control on the levels of stewartan extracellular polysaccharide produced during growth within the plant. However, it could be that some of the other genes regulated by LrhA that were not examined in this work were actually contributing to the observed phenotypes in the LrhA deletion strain. RNA-Seq analysis of the transcriptome controlled by LrhA revealed 23 additional genes, in addition to the ones examined in this study, that were differentially expressed four-fold or more in comparison to the wild-type strain (Kernell Burke et al., 2015). Overall, the majority of the genes in the LrhA regulon code for hypothetical proteins and phage-related proteins, 57.7% (15/26) and 15.4% (4/26) respectively. The possible role of these genes with regard to surface motility and virulence remains to be established, but LrhA clearly regulates these processes.
The newly discovered connection between RcsA and surface motility suggests coordination of the RcsA and LrhA regulons with regard to bacterial virulence in the corn host beyond promotion of capsule production. Capsule production is thought to be a factor impacting the ability of surface motility to occur in this phytopathogen, which may explain the need for integrated downstream regulation. The fact that the strain with the 66-kb deletion region could not be complemented by rcsA suggests that there are additional genes in this region that are essential to surface motility and virulence. Further work will be needed to identify these genes and to overall correlate to the ability of the phytopathogen to move inside the plant via surface motility in relation to virulence.
Conclusions
The findings of this study have further defined the tightly coordinated gene regulation that occurs in the QS regulon of the corn pathogen P. stewartii. The EsaR-activated transcription factor LrhA was found to directly auto-repress expression of its own gene as demonstrated through GFP-transcription fusions and EMSA experiments. In addition, the direct binding of LrhA to downstream targets, such as the promoters of genes coding for RcsA, and for putative biosurfactant synthesis (CKS_5211) and fimbrial production (CKS_0458), was also shown. This established a hierarchy of gene regulation in the QS network from the master regulator, EsaR, to the downstream transcription factors, RcsA and LrhA, which in turn control the expression of their own targets. Intriguingly, EsaR also directly controls some of these same targets (Ramachandran et al., 2014; Ramachandran & Stevens, 2013) integrating with coherent type two (RcsA) and type three (LrhA) feed forward loops (Mangan & Alon, 2003) to regulate genes in the QS regulon in a manner that ensures precisely synchronized gene expression (Fig. 6).
Supplemental Information
List of 68 genes present in the 66-kb deletion region in ΔrcsA-2015
Impact of putative fimbrial and surfactant genes on surface motility
Surface motility assays for the indicated strains. All pictures were taken at the same magnification after 48 hours of incubation.
{kind=link}
Xylem-infection assays testing the role of putative fimbrial and surfactant genes in virulence
Data shown is the average score of disease for Day 12 of an infection assay performed with 15 plants inoculated with P. stewartii DC283 strains or PBS as a negative control as indicated on the X-axis. Error bars denote standard errors. The asterisk (∗) indicates a statistically significant difference (p < 0.05) between the wild type and the negative control while the remaining strains have p > 0.05 using a two-tailed homoscedastic Student’s t-test.
{kind=link}