
Proteomic profiling of the endogenous peptides of MRSA and MSSA
- Published
- Accepted
- Received
- Academic Editor
- Vladimir Uversky
- Subject Areas
- Bioinformatics, Microbiology
- Keywords
- Staphylococcus aureus, Differential endogenous peptidome, Protein, Mass spectrometry, MRSA
- Copyright
- © 2021 Tu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits using, remixing, and building upon the work non-commercially, as long as it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2021. Proteomic profiling of the endogenous peptides of MRSA and MSSA. PeerJ 9:e12508 https://doi.org/10.7717/peerj.12508
Abstract
Staphylococcus aureus is a Gram-positive bacterium that can cause diverse skin and soft tissue infections. Methicillin-resistant Staphylococcus aureus (MRSA) can cause more severe infections than methicillin-susceptible Staphylococcus aureus (MSSA). Nevertheless, the physiological and metabolic regulation of MSSA and MRSA has not been well studied. In light of the increased interest in endogenous peptides and recognition of the important roles that they play, we studied the endogenous peptidome of MSSA and MRSA. We identified 1,065 endogenous peptides, among which 435 were differentially expressed (DE), with 292 MSSA-abundant endogenous peptides and 35 MRSA-abundant endogenous peptides. MSSA-abundant endogenous peptides have significantly enriched “VXXXK” motif of at the C-terminus. MSSA-abundant endogenous peptides are involved in penicillin-binding and immune responses, whereas MRSA-abundant endogenous peptides are associated with antibiotic resistance and increased toxicity. Our characterization of the peptidome of MSSA and MRSA provides a rich resource for future studies to explore the functional regulation of drug resistance in S. aureus and may also help elucidate the mechanisms of its pathogenicity and the development of treatments.
Introduction
Staphylococcus aureus (S. aureus), a Gram-positive bacterium, is a common cause of infections in human worldwide and a significant threat to modern health care systems. As an opportunistic nosocomial and community-associated pathogen, it is a normal part of human flora, covering 30% of the population, and is able to cause diverse skin and soft tissue infections, including pimples, boils, cellulitis, folliculitis, impetigo, scalded skin syndrome, and abscesses, but also cause severe diseases, including bacteremia, acute endocarditis, and meningitis (Monteiro et al., 2012).
The overuse of antibiotics can lead to the development of drug-resistant S. aureus, such as methicillin-resistant Staphylococcus aureus (MRSA), which can evade immune responses and reproduce in human hosts (Singh et al., 2020). Aside from being distinguished from methicillin-susceptible Staphylococcus aureus (MSSA) in their resistance to antibiotics, MRSA also differ in their phenotype and metabolic pathways (Garcia et al., 2017). Previous studies have shown that MRSA have significantly higher biofilm formation abilities than MSSA, as they can easily form small colonies (Akil & Muhlebach, 2018) that colonize for long periods (Jeong, Kang & Cho, 2020) and can develop thickened cell walls (Garcia et al., 2017). MRSA infection can lead to more serious clinical symptoms, inflammatory reactions, and multi-drug resistance (Zhang et al., 2019). It is more common in patients with nosocomial infections, catheter-related infections, or renal insufficiency, all of which require increased medical resources to treat infections. Thus, MRSA infection is a major global health problem (Monteiro et al., 2012).
Given the importance of MRSA and MSSA as human pathogens, numerous proteomics studies have been conducted to characterize differences in the composition of their proteins (Otto et al., 2014). For example, Cordwell et al. (2002) used 2-DE gel-based proteomics methods and identified 377 proteins of S. aureus. The proteins in MRSA included alkali shock protein 23 (Asp23) and the cold-shock protein CspABC, which are not present in MSSA. Ji et al. (2017) used an iTRAQ-based LC-MS/MS quantitative proteomics approach to study the effects of emodin on MRSA and MSSA at the molecular level. However, nearly all previous studies have focused on large proteins produced in S. aureus.
With the development of high-throughput technologies, an increasing number of studies have focused on the peptidome, which includes endogenous peptides and can be characterized by peptidomics in a high-throughput way (Romanova & Sweedler, 2015). Although endogenous peptides are small in length, they can also have important functions (Carter, 2012; Cui et al., 2016; Wolf et al., 2011). In eukaryotes, the peptidome has been shown to change during the epithelial-mesenchymal transition of renal epithelial cells (Kanlaya & Thongboonkerd, 2018). The neuropeptides can mediate cell–cell signaling and are potential therapeutic targets (Fricker, 2018). In S.aureus, pheromone peptides can cause activation of virulence regulatory factor agr, leading to the early reproduction and infection spread of S.aureus (Otto, 2001). S.aureus can produce formylated peptides, which play important roles in S.aureus arthritis and may be used for preventing joint destruction in S.aureus arthritis (Gjertsson et al., 2012). Phenolic soluble modules (PSMs) are a family of amphiphilic short peptides produced by S.aureus. PMS can cause the dissolution of human cells, stimulate the inflammatory response, and promote the formation of biofilm in osteomyelitis (Cheung et al., 2014). Thus, systematic analysis of the endogenous peptidome of S. aureus could provide useful insights into its physiology and regulatory networks.
Here, we performed high-throughput liquid chromatography-mass spectrometry (LC-MS) analysis of MSSA and MRSA and identified 1065 endogenous peptides from 282 precursor proteins. After filtering mass spectrum data, and 435 differentially expressed (DE) peptides were obtained. A total of 327 DE peptides of MSSA and MRSA were extracted for further study. The abundance and diversity of DE endogenous peptides in S. aureus indicate that the regulation of its physiological metabolism is complex. DE endogenous peptides of MSSA and MRSA may be closely related to drug resistance and pathogenicity. We analyzed the endogenous peptides of MRSA and MSSA by proteomics, aiming to provide a guidance for further mechanistic studies and treatments of S. aureus. We believe that further research on endogenous peptides will enhance our understanding of their roles in pathogenicity as well as their interaction with the human body.
Materials and Methods
Sample preparation
In this study, MSSA (ATCC 29213) and MRSA (ATCC 43300) samples were purchased from the American Type Culture Collection. Three S. aureus clinical isolates were from the Microbiology Department of Sir Run Run Hospital, Nanjing Medical University, and were identified as MRSA by the BD Phoenix 100 Automated Microbiology System. Three clones from MSSA (ATCC 29213 strains), three clones from MRSA (ATCC 43300 strains), and three above-mentioned S. aureus clinical MRSA isolates were prepared for proteomic analysis. They were placed onto blood agar plates and grown at 37 °C. The colonies were then cultured in 250 mL of M9 minimal medium supplemented with 4 g/L of glucose at 37 °C. Aliquots of growing S. aureus were washed with phosphate-buffered saline three times, pelleted, and stored at −80 °C. Boiling water (500 µL) was added directly to the frozen pellets, and the samples were boiled for 15 min to eliminate proteolytic activity following the methods of a previous study (Slavoff et al., 2013). After cooling to room temperature, the samples were sonicated on ice for 20 s. Acetic acid was then added to the lysate to a final concentration of 0.25% by volume and centrifuged at 20,000 g for 20 min at 4 °C. The supernatant was filtered by a 30-kDa molecular weight cut-off filter (Millipore). The filtered endogenous peptides were quantified by the BCA assay, desalted using StageTip (Thermo Fisher Scientific), and used for mass spectrometry analysis.
LC-MS analysis
Two µg of endogenous peptides of S. aureus were analyzed using an LTQ Orbitrap Velos (Thermo Finnigan, San Jose, CA). First, the peptides were loaded into the precolumn (75 µm × 2 cm, Acclaim Pep Map 100 C18 column, 3 µm, 100 A; Thermo Fisher Scientific) at a flow rate of 10 µL/min and then were transferred to an analytical column (75 µm × 25 cm, Acclaim Pep Map RSLC C18 column, 2 m, 100 A; Thermo Fisher Scientific) at a flow rate of 300 nL/min. The peptides were reverse-phase separated with buffer A (0.51% acetic acid) and buffer B (100% ACN and 0.1% acetic acid) under a 95-min gradient (3–5% buffer B for 5 s, 5–15% buffer B for 40 min, 15–28% buffer B for 34 min and 50 s, 28–38% buffer B for 12 min, 38–100% buffer B for 5 s, and 100% buffer B for 8 min). MS survey scans were performed for mass-to-charge ratios ranging from 350–1,800, and the 20 most intense ions from the survey scans were analyzed by MS/MS spectra in the LTQ, which were determined using Xcalibur mass spectrometer software in real-time. With siloxane (m/z 445.120025) as a lock mass, dynamic mass exclusion windows of 60 s were used similar to previous studies (Wang et al., 2015; Zhou et al., 2015).
Processing of MS data
Raw files were processed using MaxQuant (version 1.2.2.5) (Cox & Mann, 2008) and searched against the UniProt Staphylococcus NCTC8325 protein database (2015/12/13; UniProtKB: UP000008816; 2,889 sequences). Enzyme specificity was set to unspecific. The minimum peptide length required was six amino acids. Variable modifications included oxidation (Met) and acetylation (protein N-terminus). Mass tolerances were set at 20 ppm for precursor ions and 0.5 Da for fragment ions. An estimate of the false discovery rate (FDR) of the identification was obtained using the reversed sequences in the databases. All FDRs were set to 0.01 for site, peptide, and protein.
Bioinformatics analysis of the endogenous peptidome
To annotate the features of the peptidome, EMBOSS (Rice, Longden & Bleasby, 2000) was used to calculate the isoelectric point (pI) and molecular weight (Mw) of the peptide sequences. One-way ANOVA was performed to analyze the differential expression of peptides among groups, and a peptide was considered differentially expressed if the FDR-q value was less than 0.01. To analyze the sequence patterns of proteases to generate endogenous peptides, the amino acid sequences surrounding the termini of peptides were extracted, and overrepresented amino acids were analyzed using iceLogo (Colaert et al., 2009). Overrepresented sequence motifs were analyzed using motif-x (Chou & Schwartz, 2011; Schwartz & Gygi, 2005), and a P value of less than 0.0001 was considered significant. Interactions among precursor proteins were annotated by the STRING database (Szklarczyk et al., 2017), and the protein–protein interaction network was constructed using Cytoscape software (Kohl, Wiese & Warscheid, 2011). The database for annotation, visualization, and integrated discovery (DAVID) (version 6.8) (Huang da, Sherman & Lempicki, 2009) was used to identify significantly enriched gene ontology terms in biological processes (BP), molecular function (MF), and cellular component (CC) categories for the precursor proteins of the DE endogenous peptidome.
Results
Identification of endogenous peptides from S. aureus
To characterize the endogenous peptides of MSSA and MRSA, three clones from MSSA (ATCC 29213 strains), three clones from MRSA (ATCC 43300 strains), and three S. aureus clinical MRSA isolates were boiled before protein extraction to ensure that proteases were inactivated and that all peptides identified were S. aureus endogenous peptides (i.e., not artificial proteolytic peptides produced during or after protein extraction). To identify small endogenous peptides, the proteins were filtered using a membrane with a molecular weight cut-off of 30 kD so that only small endogenous peptides remained. The extracted endogenous peptides were separated by liquid chromatography and identified by an LTQ Orbitrap mass spectrometer. We selected high-score reliable sequences with an FDR of 1% for peptides to ensure the reliability of the proteomic identification. Filtering the reverse sequences (2 peptides) and contaminant proteins (23 peptides) resulted in 1040 endogenous peptides, corresponding to 282 precursor proteins from S. aureus. The detailed list of identified endogenous peptides is shown in Table S1. The S. aureus has complex endogenous peptides.
Properties of the endogenous peptidome of S. aureus
With the identified endogenous peptidome, we then analyzed the features of these endogenous peptide sequences. We found that some peptides are shorter forms of longer peptides. For example, there were 6 peptides for precursor protein P48940, including “ANEILDAANNTGGAVK”, “NEILDAANNTGG”, “EILDAANNTGGAVK”, “ILDAANNTGGAVK”, “LDAANNTGGAVK” and “ILDAANNTGGAVK”. The latter five peptides occurred after the first and longest one. The longest peptide “ANEILDAANNTGGAVK” was preserved for the bioinformatics analysis to facilitate subsequent analyses. After filtering, the 274 representative (longest) peptides were obtained. With the filtered peptides, we analyzed the length distribution and found that the peptides ranged from 8 AA to 25 AA (Fig. 1A), with 160 peptides between 10 AA and 13 AA. The length of the endogenous peptides peaked at 11 AA. The endogenous peptides in S. aureus are relatively short in length.
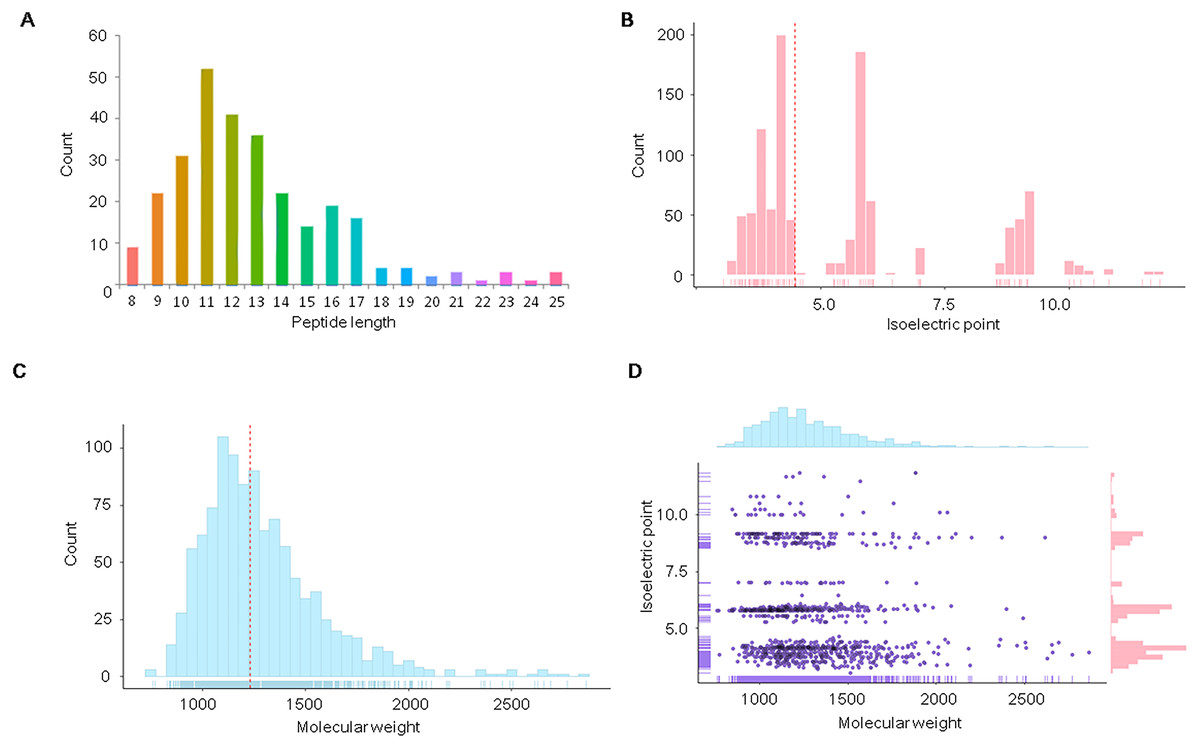
Figure 1: Distribution of physicochemical features of the endogenous peptidome of S. aureus.
The count distribution of peptide length (A), isoelectric point (B), and molecular weight (C) are given separately for all identified endogenous peptides of S. aureus. The two-dimensional distribution of the isoelectric point and molecular weight of the identified peptides is shown in (D).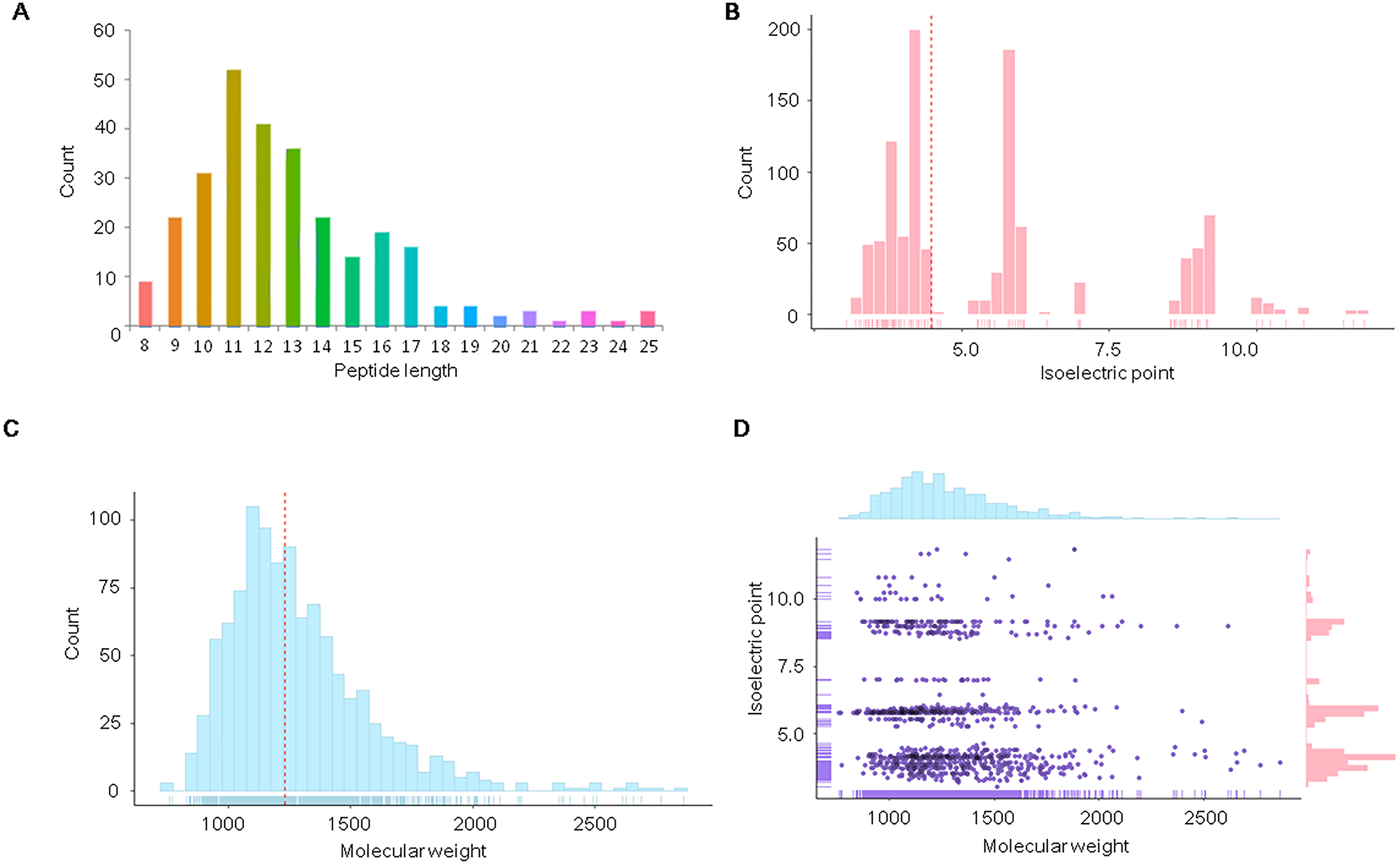{kind=link}
We further analyzed the isoelectric point and molecular weights of the endogenous peptides. The isoelectric point distribution analysis of the endogenous peptidome of S. aureus revealed a wide range of isoelectric point values from 3.05 to 11.82 (Fig. 1B). The median of the isoelectric point was 4.50, indicating that many of the peptides were acidic. The molecular weights of the identified peptides ranged from 759.41 to 2,862.28 Da with a median of 1,229.13 Da (Fig. 1C). A two-dimensional plot of isoelectric point and molecular weight showed that most of the endogenous peptides had molecular weights less than 2 kDa (Fig. 1D). These results suggest that the endogenous peptides are mostly short, and are mainly acidic.
Differential expression of endogenous peptides among S. aureus strains
We further analyzed differences of endogenous peptides among the different strains of S. aureus. Statistical analysis showed that there were 435 DE endogenous peptides between MSSA and MRSA (Table S2). Unsupervised hierarchical cluster analysis of the DE endogenous peptides revealed 6 categories (Fig. 2). Cluster C1 (140 peptides) and cluster C2 (152 peptides) were highly expressed in MSSA. Peptides in cluster C3 (27 peptides), cluster C4 (58 peptides), and cluster C5 (23 peptides) were highly expressed in both MSSA and MRSA with differential expression patterns between standard MRSA and clinical isolates. Peptides in cluster C6 (35 peptides) were highly expressed in MRSA. Endogenous peptides from C1 and C2 (292 peptides) highly expressed in MSSA were designated as MSSA-abundant endogenous peptides (MSSA-AEP), and 35 peptides from C6 were designated as MRSA-abundant endogenous peptides (MRSA-AEP). Differences in the expression of endogenous peptides between MRSA and MSSA may be related to their physiological and functional differences.
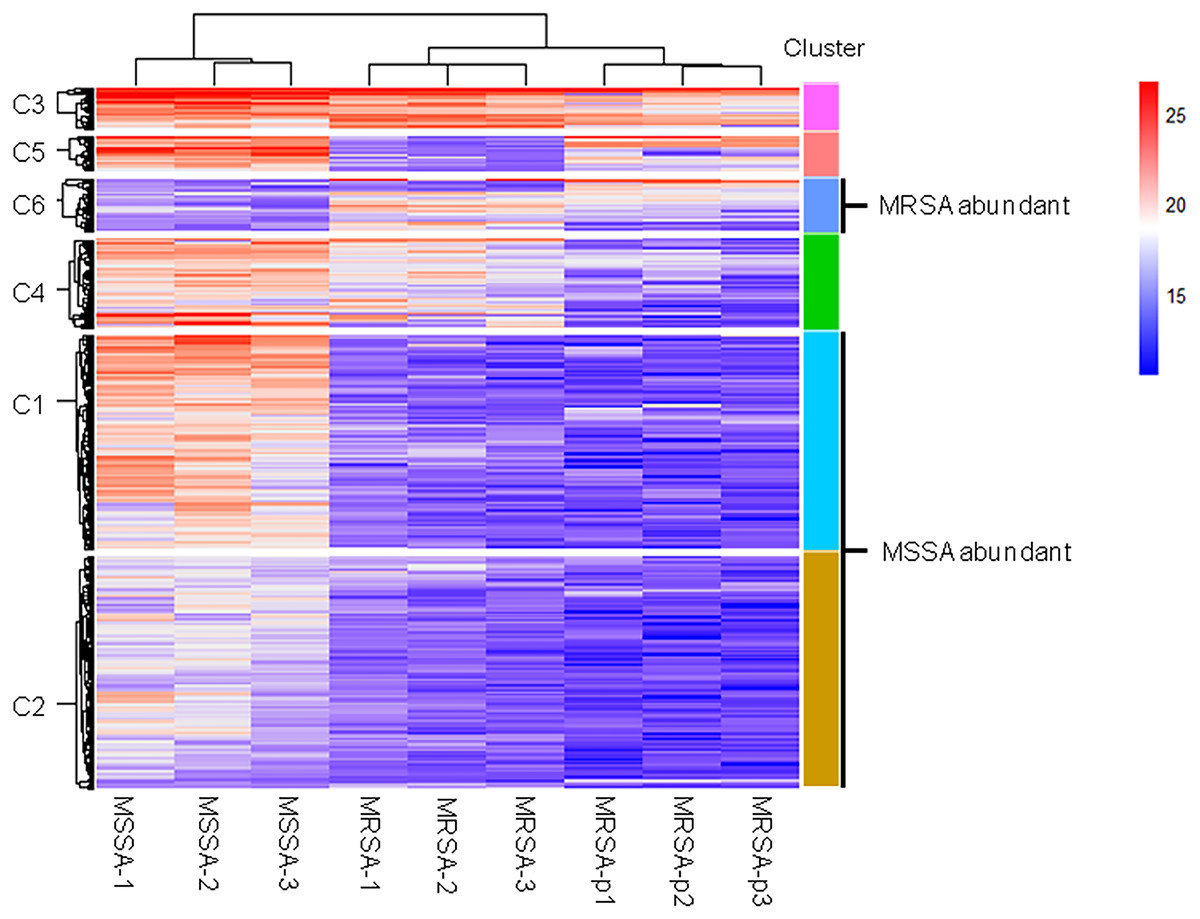
Figure 2: Cluster analysis of the DE endogenous peptidome between MSSA and MRSA.
Unsupervised hierarchical clustering analysis of all DE endogenous peptides from MSSA (ATCC 29213), MRSA (ATCC 43300), and S. aureus clinical isolates (MRSA-p1 to -p3).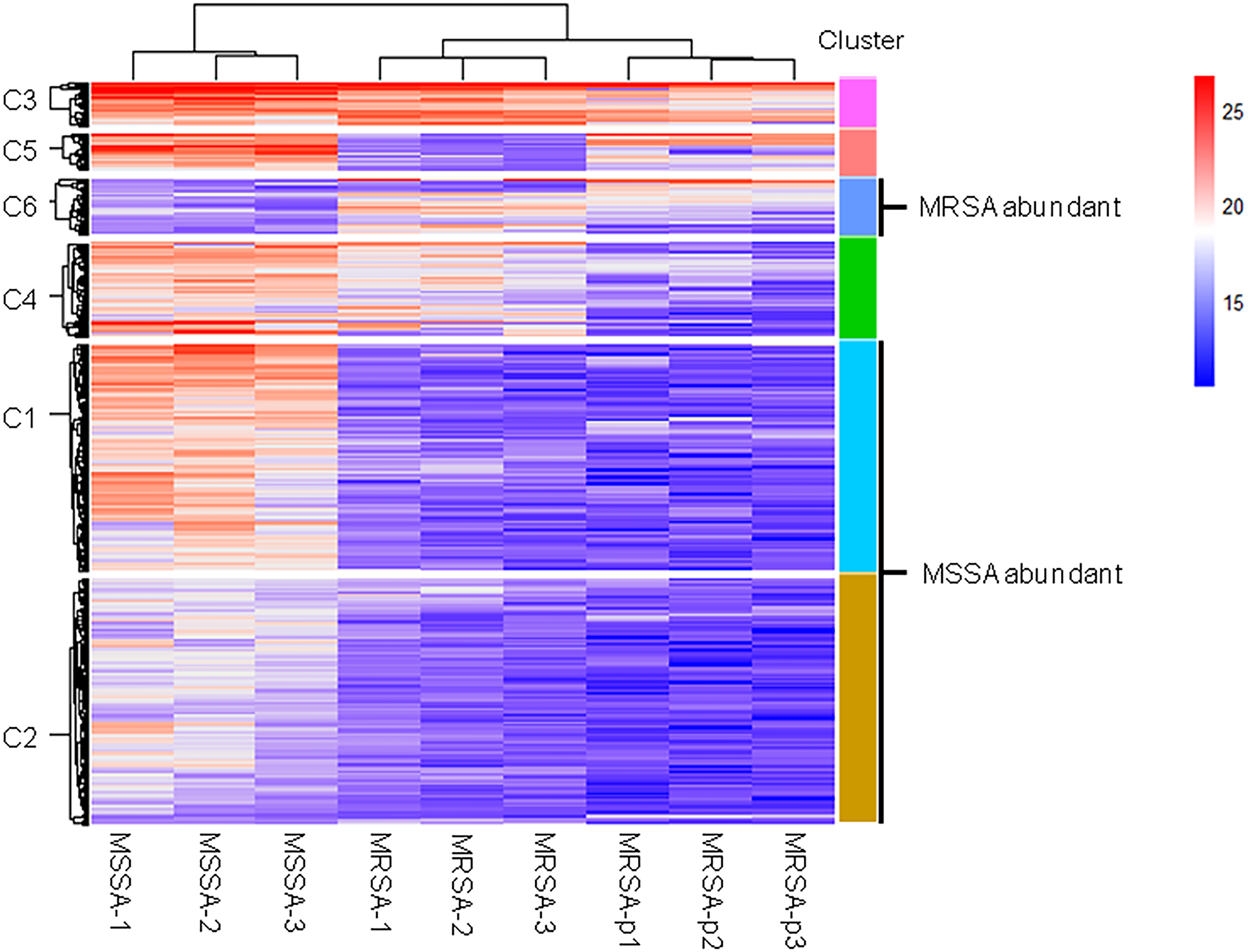{kind=link}
Proteolytic cleavage features of MSSA-AEP and MRSA-AEP
DE peptides may be generated by differential protease activities between MSSA and MRSA. To investigate the differential protease activities, precursor protein cleavage sites of peptides were examined for the frequency of signature amino acids. We extracted sequences of six amino acids surrounding the precursor proteins of MSSA-AEP and MRSA-AEP and analyzed the amino acid distribution at the N-terminus and C-terminus of the DE peptides. Figure 3A shows an enrichment diagram of amino acids around the N-terminus of MSSA-AEP. We found that “A”, “S” and “D” were significantly enriched at the first amino acid of the peptides at the N-terminus; “A” and “S” were significantly enriched at the P1 position; and “A”, “T”, “G” and “Q” were significantly enriched at the P1′ position (p < 0.05). “Q”, “A” and “N” were the most frequently observed amino acids at the C- terminal end of the peptides. “Q” was significantly enriched at the P1′ and P1 positions; “A”, “S” and “Q” were significantly enriched at the P1 position; and “Q”, “L”, “V” and “Y” were significantly enriched at the P1′ position (p < 0.05) (Fig. 3B). Figures 3C and 3D show the enrichment diagram of amino acids around the N-terminus and C-terminus of MRSA-AEP. We found that “I” was enriched at the first amino acid of the peptides at the N-terminus; there was no enrichment of amino acids at the P1 position; and “C” was significantly enriched at the P1′position (p < 0.05). “A”, “Q” and “V” were the most frequently observed amino acids at the C-terminal end of the peptides; “A” and “Q” were significantly enriched at the start of the peptides, “A” and “M” were significantly enriched at the P1 position, and “Q” was significantly enriched at the P1′position (p < 0.05). The uneven distribution of amino acids surrounding the DE endogenous peptides indicated possible protease recognition motifs.
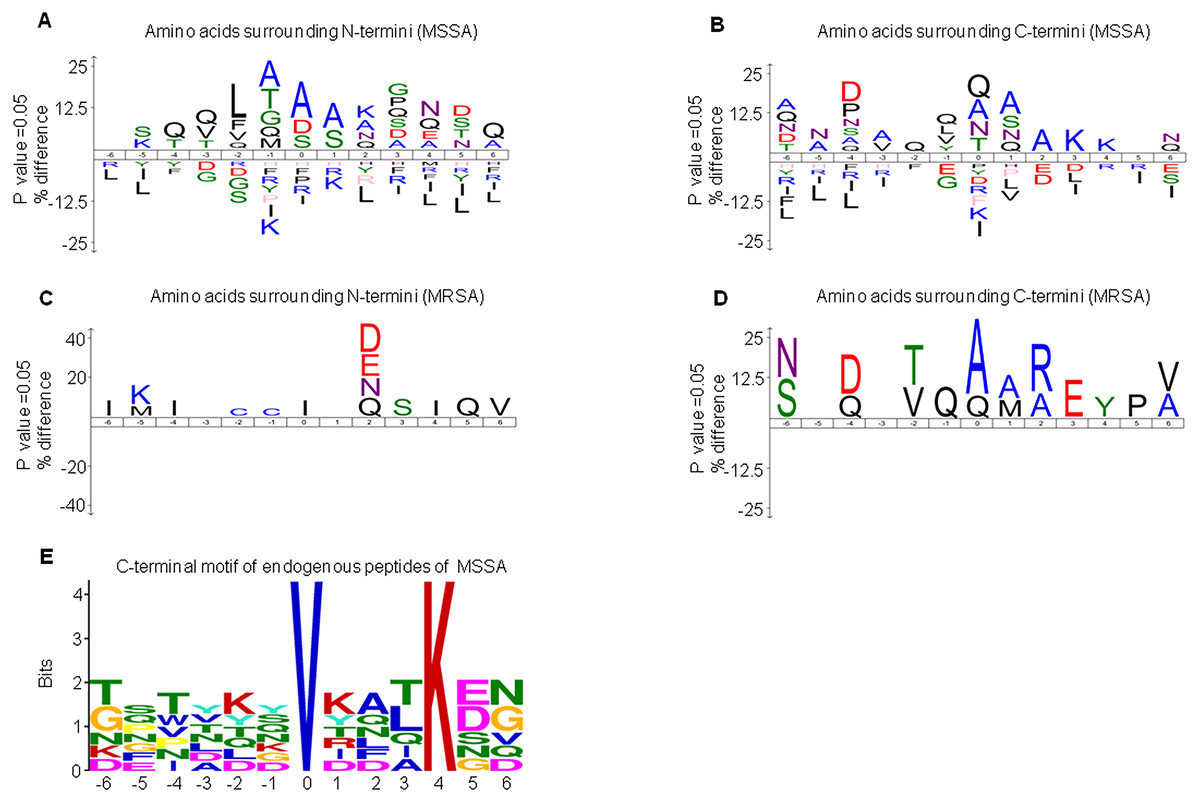
Figure 3: The sequence logos and enrichment of amino acids and motifs surrounding the termini of MSSA-AEP and MRSA-AEP.
The N-terminal consensus sequences (A) and C-terminal consensus sequences (B) of MSSA-AEP, and the N-terminal consensus sequence (C) and C-terminal consensus sequence (D) of MRSA-AEP. According to the motif-x analysis, there was one motif (E) that was enriched in the C-terminal sequences of MSSA-AEP (P < 0.0001). Different types of amino acids highlighted in different colors correspond to their annotations.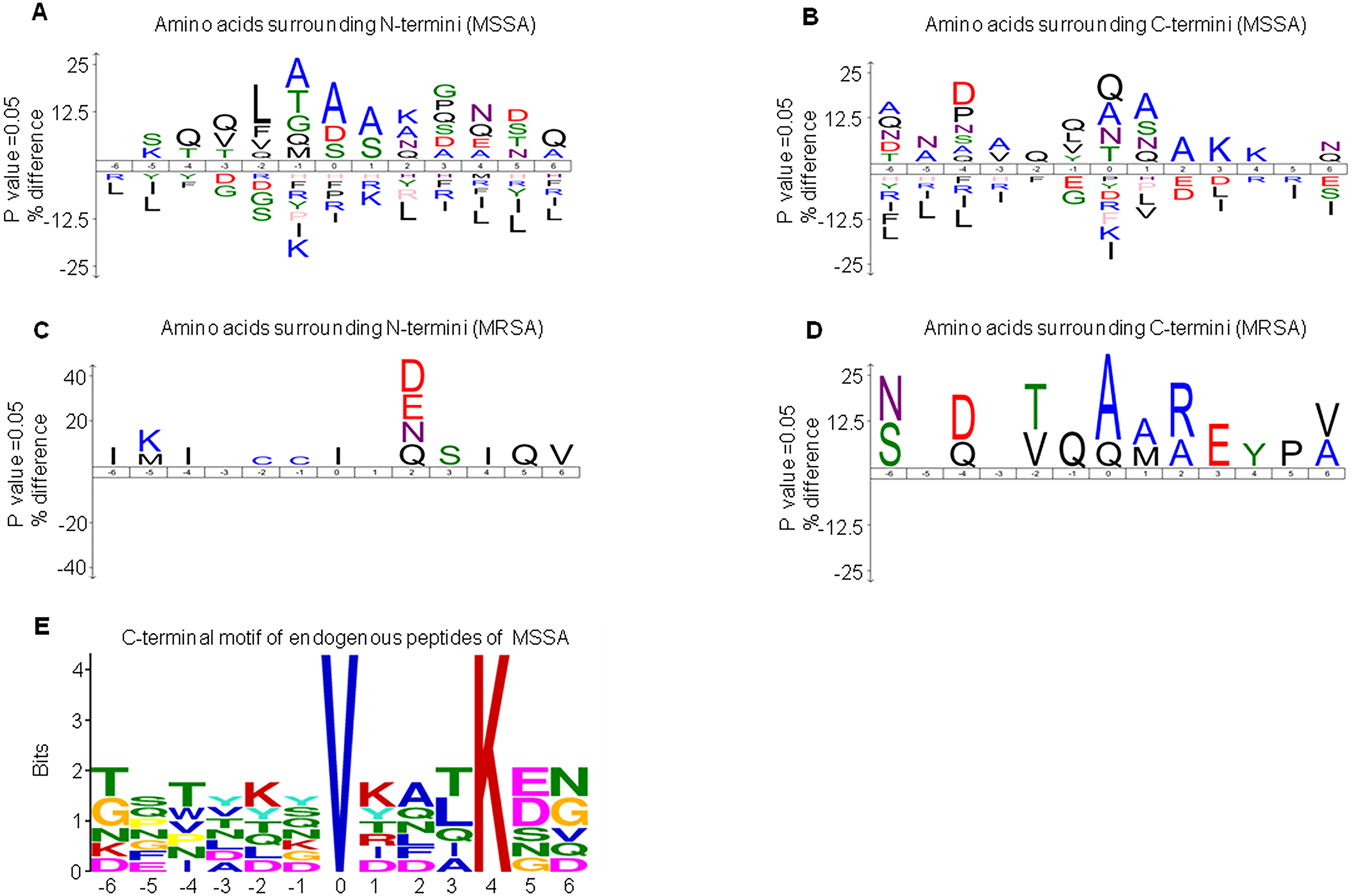{kind=link}
Given that the above sequence logo analysis may be subject to background amino acid distribution, we used motif-x to examine the enrichment of amino acid residues. In-depth motif analysis could help identify how proteolysis generates these peptides. We identified the N-terminus and C-terminus of MSSA-AEP and MRSA-AEP and found that only the C-terminus of the MSSA-AEP had an enriched motif (“VXXXK”) (Fig. 3E, Table S3). There was a valine at P0 and lysine at P4. Seven peptides in MSSA contained this motif; among them, the precursor protein corresponding to the sequence “TFTVDGVRFTKGQ” was Q2FVV8 (a putative transcriptional regulator), and the corresponding precursor protein of the sequence “DSNYTSVKDLKDN” was Q2FVX4 (molybdenum ABC transporter, periplasmic molybdate-binding protein). No enrichment of the proteolytic motif was observed at the end of MRSA-AEP. Therefore, these DE peptides may be produced by differential protease activities between MSSA and MRSA.
Annotations of the precursor proteins of MSSA-AEP and MRSA-AEP
To better understand the potential functions of DE endogenous peptides, we used STRING to analyze protein–protein interactions between precursor proteins of MSSA-AEP and MRSA-AEP, and the precursors showed complex interactions (Fig. 4). The peptides from ribosomal-related precursor proteins, which show strong interactions, were highly expressed. These peptides might regulate the functions of precursor proteins and participate in the regulation of translation activities.
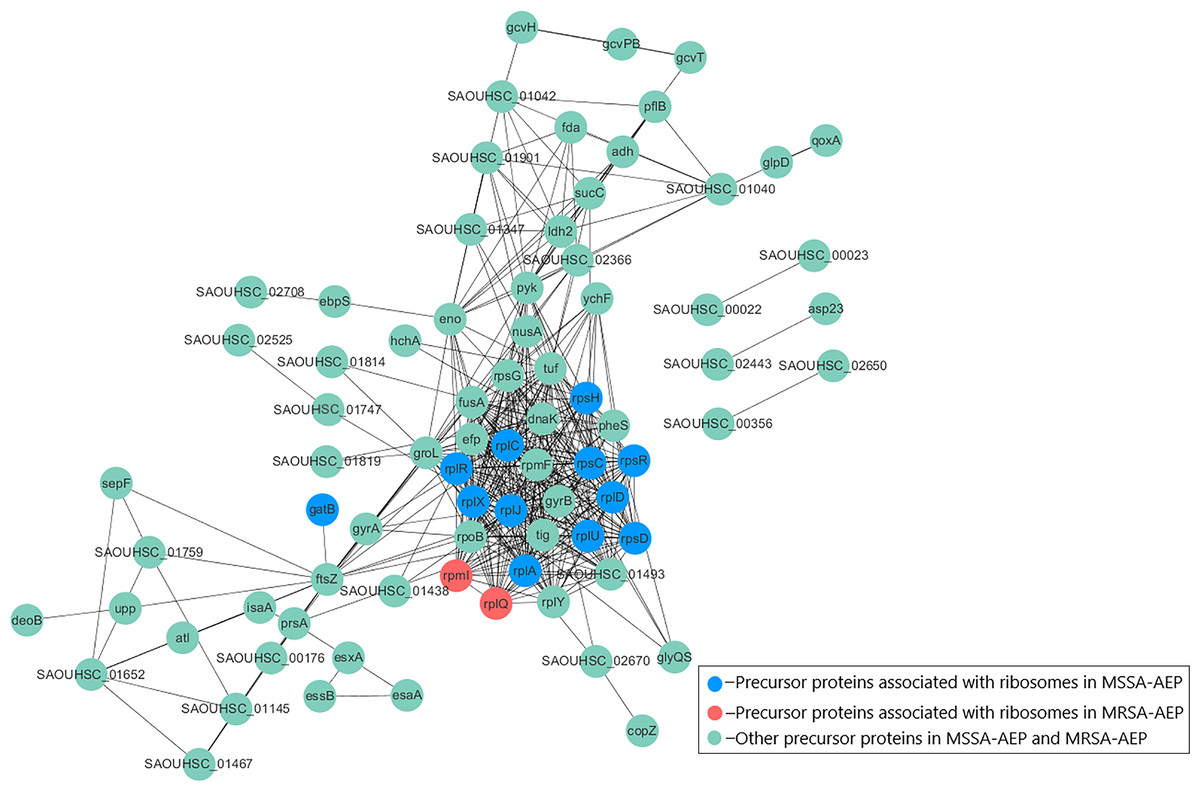
Figure 4: Protein–protein interaction network annotations of DE endogenous peptides between MSSA and MRSA strains by the STRING database.
The protein–protein interactions were annotated from the STRING database and visualized by Cytoscape. The red nodes represent the translation-related proteins and ribosomal components in the precursor proteins of MRSA-AEP, while the blue nodes represent the translation-related proteins and ribosomal components in the precursor proteins of MSSA-AEP. The edges represent protein–protein associations from the STRING database.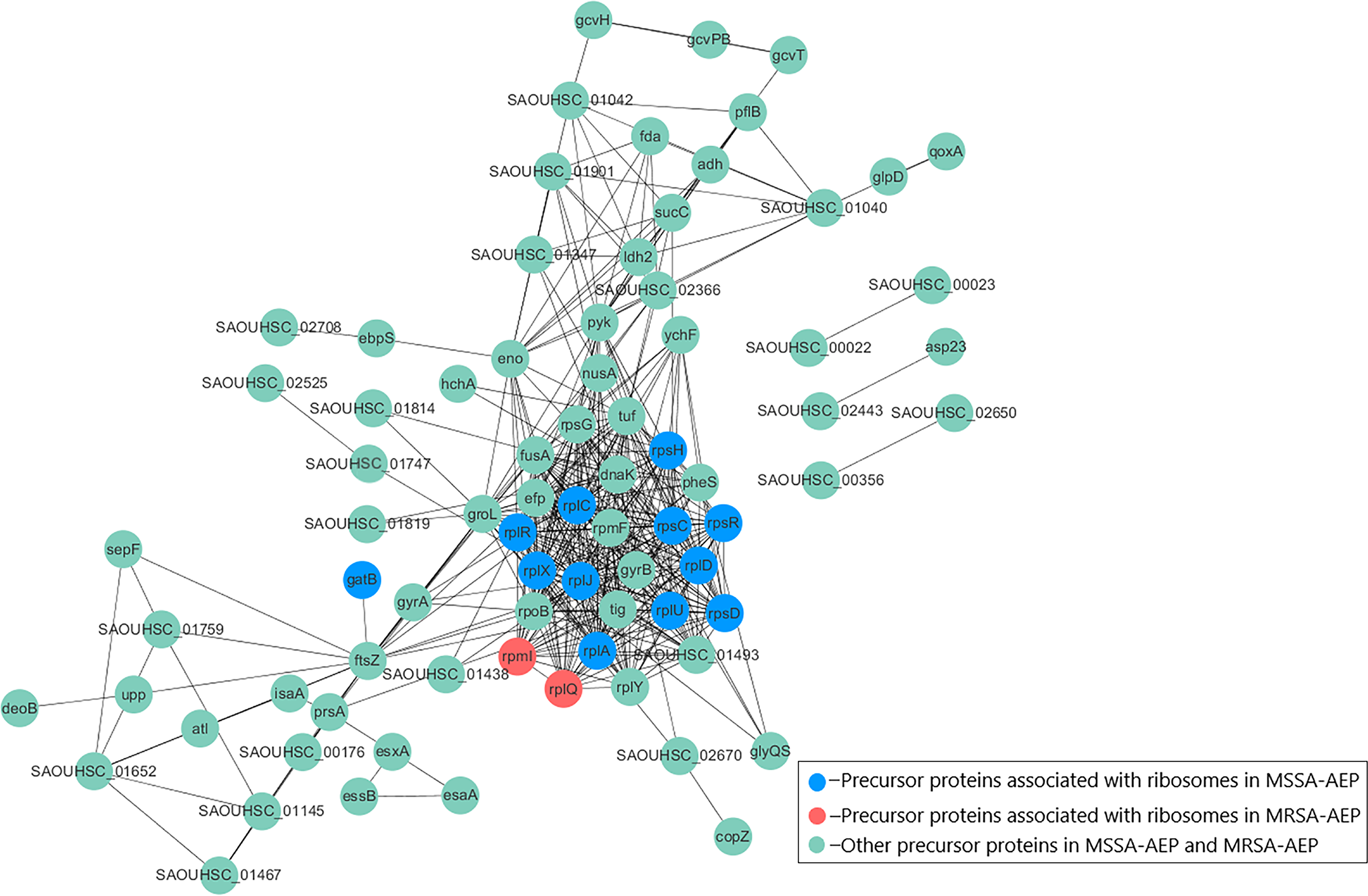{kind=link}
To further annotate their functions, the corresponding precursor proteins of MSSA-AEP and MRSA-AEP were subjected to gene ontology and KEGG analyses (Table S4). Gene ontology annotation revealed significant enrichment of precursor proteins in “translation” (14 peptides), “structural constituent of ribosome” (13 peptides), and “rRNA binding” (9 peptides), indicating that many precursor proteins were involved in ribosome biogenesis or translational regulation (Fig. 5A), which is consistent with the results shown in the protein interaction network diagram of the precursor proteins. rpsR, rpsC, rpsD, rpsH, rplA, rplJ, rplQ, rplR, rplU, rplX, rplC, rplD, and gatB were the precursor proteins with ribosome function in MSSA-AEP, and rplQ and rpmI were the precursor proteins in MRSA-AEP (Fig. 4). The expression of peptides from rplQ was much lower in MSSA-AEP than in MRSA-AEP; thus, we considered rpIQ to be the precursor protein of MRSA-AEP. These ribosomal endogenous peptides may regulate the translational efficiency (Cotter, Hill & Ross, 2005; Repka et al., 2017) and be responsible for the proteome differences between MSSA and MRSA.
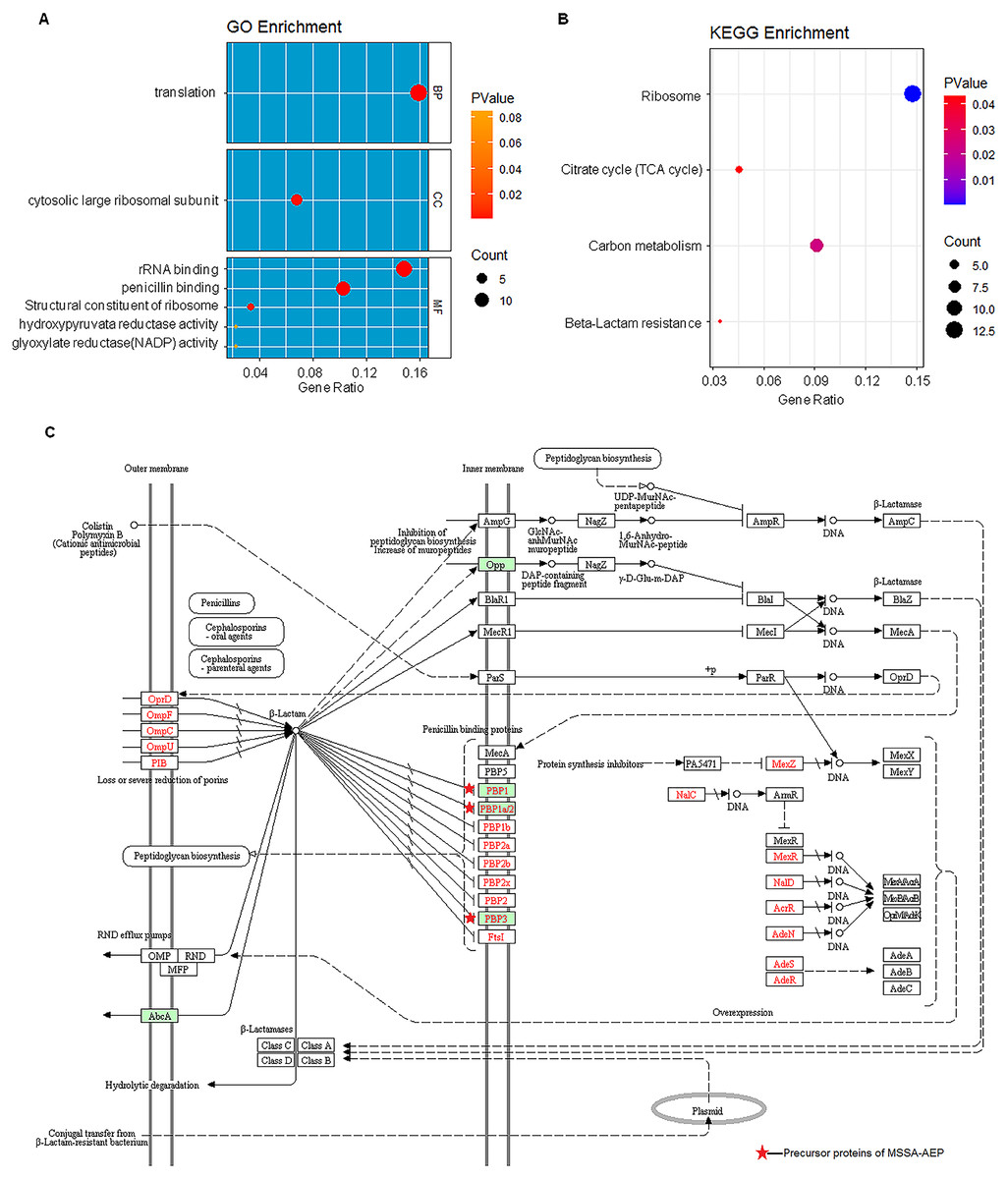
Figure 5: The gene ontology and KEGG enrichment analysis of precursor proteins of MSSA-AEP and MRSA-AEP.
(A) Enriched gene ontology terms for biological process (BP), cellular component (CC), and molecular function (MF) are shown together with the gene ratio. (B) Enriched KEGG pathways. (C) Beta-lactam resistance pathway red stars represent pathway-related precursor proteins in MSSA-AEP.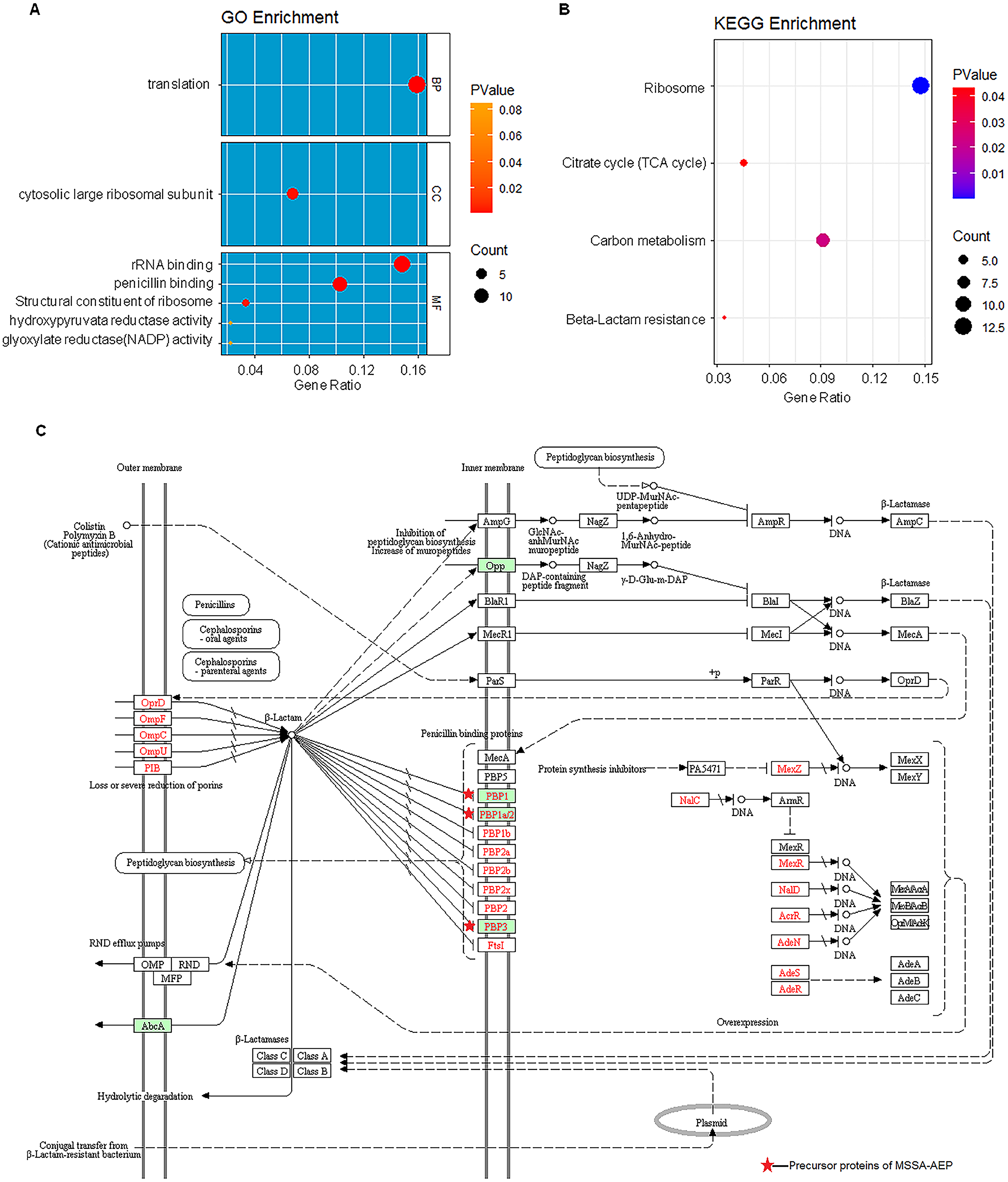{kind=link}
KEGG pathway analysis also revealed that these DE peptides were significantly enriched in various metabolic pathways, such as carbon metabolism (8 peptides), citric acid cycle (TCA cycle) (4 peptides) (Fig. 5B). In addition, DE endogenous peptides were involved in β-lactam resistance. All relevant precursor proteins were expressed by MSSA-AEP, and PBP1, PBP1a/2, and PBP3 were penicillin-binding proteins (Fig. 5C), which is consistent with “penicillin-binding” in the gene ontology analysis. Thus, MSSA-AEP and MRSA-AEP are related to the differences in metabolism, pathogenicity, and drug resistance between MSSA and MRSA.
Discussion
The regulation of proteins have been studied a lot in S. aureus (Becher et al., 2009; Zhao et al., 2017). Small peptides (e.g., neuropeptides) are able to play important functions (Fridjonsdottir et al., 2018), However, small peptides, especially the endogenous peptides, are less studied in S. aureus. In this study, a proteomic analysis was conducted to characterize the endogenous peptides of S. aureus (Schrader, 2018; Vincent, 2000) and identified 292 MSSA-AEP and 35 MRSA-AEP, indicating different endogenous peptidomes between MRSA and MSSA.
Endogenous peptides can be produced by proteolytic enzymes (Wang et al., 2012). We analyzed the features of peptide sequences near the proteolytic N-terminal and C-terminal sites of MSSA-AEP and MRSA-AEP and observed a non-random distribution of amino acids. Specifically, we found that the amino acid distribution of MSSA-AEP and MRSA-AEP was uneven, and the amino acid enrichment of MRSA-AEP was significantly reduced. The C-terminus of MSSA-AEP was enriched in specific amino acid residues, potentially reflecting protease activity during peptide maturation, while MRSA-AEP did not have an enriched motif because there were fewer peptides identified in MRSA than MSSA. Deeper peptide coverage may help us discover motifs. The motif analysis suggested that endogenous peptides were not randomly broken and were regulated by proteases. However, the exact enzyme recognition motif is still unknown due to the lack of research on the substrate features of proteolytic enzymes. Additional studies to characterize such protease would help explain the differential endogenous peptidome between MSSA and MRSA.
MRSA is resistant to methicillin, a type of beta-lactam antibiotic that causes bacteria to stop dividing and die (Peacock & Paterson, 2015). The KEGG analysis showed that the beta-lactam resistance pathway was enriched in MSSA-AEP but not MRSA-AEP. In MSSA-AEP, peptides from the penicillin-binding proteins PBP1, 1a/2, and 3, which have high affinities for beta-lactam antibiotics (Fisher & Mobashery, 2021), may be involved in its sensitivity to penicillin.
In addition, analysis of the precursor proteins of MSSA-AEP and MRSA-AEP by gene ontology analysis revealed the enrichment of ribosome or translational regulation, which is consistent with the observed complex interactions among ribosomal proteins in the protein–protein interaction network analysis. We found that there were 21 ribosomal peptides in MSSA-AEP, and only 4 ribosomal peptides in MRSA-AEP. It is widely accepted that ribosomes can regulate cell growth and metabolism (Fu et al., 2013; Jiang et al., 2015). For example, 50S ribosomal protein L1 (rplA) is a ribosomal structural component highly expressed in MSSA-AEP. L1 has dual functions as a ribosomal structural protein that binds rRNA and as a translational repressor that binds its mRNA (Nevskaya et al., 2006) and is related to bacterial growth (Dean & Nomura, 1980). Many antibiotics interfere with protein synthesis and function by binding to ribosomes and thereby inhibiting bacterial reproduction (Foster, 2017). In comparison with MRSA-AEP, MSSA-AEP have stronger ribosome-related functions, and might be involved in the regulation of bacterial growth.
We also observed that the precursor proteins of MRSA-AEP were mainly enriched in metabolism pathways, including the TCA cycle. The activity of the TCA cycle is related to the virulence and survival of pathogens, the production of the substances of biofilm mucus, intercellular adhesin, and persistent infection (Gao & Stewart, 2004; Oscarsson, Tegmark-Wisell & Arvidson, 2006). For example, one peptide among the MRSA-AEP is derived from the branched-chain alpha-keto acid dehydrogenase subunit E2 (SAOUHSC_01042), which is essential in the adhesion of S. aureus to host eukaryotic cells and their survival (Khairon et al., 2016). Aside from proteins in the TCA cycle, we found that MRSA-AEP were also derived from other metabolic processes, such as one peptide from fructose-1,6-diphosphate aldehydes (fda) that is closely related to the viability of pathogenic microorganisms (Yadav et al., 2013). Two MRSA-AEP were derived from CopZ, the excess of which causes bacterial toxicity and affects the growth, vitality, and metabolism of bacteria (Stewart et al., 2020; Xu et al., 2021). The phenotypic changes of MRSA, such as the formation of smaller colonies and the ability to colonize for a longer period, may be related to its metabolic changes. These changes in metabolism pathways of MRSA-AEP are associated with antibiotic resistance and increased toxicity.
By analyzing MSSA-AEP precursor proteins, we also found that that many of them were involved in immunity regulation. For example, S. aureus recombinant alkaline shock protein 23 (Asp23) has immunogenicity and protective effects and is a promising vaccine candidate as a prophylactic therapeutic agent for S. aureus (Francis & Kuyyalil, 2018). Immunodominant staphylococcal antigen A (isaA), which is expressed on the surface of the bacterial cell wall, plays a role in immunogenicity in the cleavage of peptidoglycan and the inhibition of biofilm formation (Valliammai et al., 2020). ESAT-6 secretion accessory factor (esaA) and ESAT-6 secretion machinery protein (essB) are immunogenic antigens, both of which can induce a strong immune response and have protective effects against bacterial infection (Klein et al., 2021; Ma et al., 2020). The peptide derived from ESAT-6, essB, and esaA may also be able to induce immune responses when secreted.
Conclusion
In sum, our characterization of the DE endogenous peptides between MSSA and MRSA revealed their complex regulatory roles in antibiotic resistance, bacterial survival, and immune responses. Additional functional studies of these DE endogenous peptides between MSSA and MRSA are needed to help elucidate the mechanisms underlying their pathogenicity, phenotypes, and differences in antibiotic resistance.