Seasonal Influenza: The challenges of vaccine strain selection
New measures of influenza virus fitness could improve vaccine strain selection through more accurate forecasts of the evolution of the virus.
- Division of International Epidemiology and Population Studies, Fogarty International Center, National Institutes of Health, United States
Scientists have known since the 1940s that influenza vaccines that perform well one year can be rendered ineffective after the influenza virus mutates. However, despite decades of investment in global surveillance, pathogen sequencing technologies and basic research (Figure 1), vaccines for seasonal influenza have the lowest and most variable performance of any vaccine licensed for use in the United States (CDC, 2016). Now, in eLife, John Huddleston of the Fred Hutchinson Cancer Research Center (FHCRC) and the University of Washington, Trevor Bedford of the FHCRC, and colleagues in the United States, United Kingdom, Japan, Australia and Switzerland present an open-source framework that synthesizes a decade’s worth of innovations in bioinformatics and technology to advance data-driven vaccine design (Huddleston et al., 2020).
Figure 1
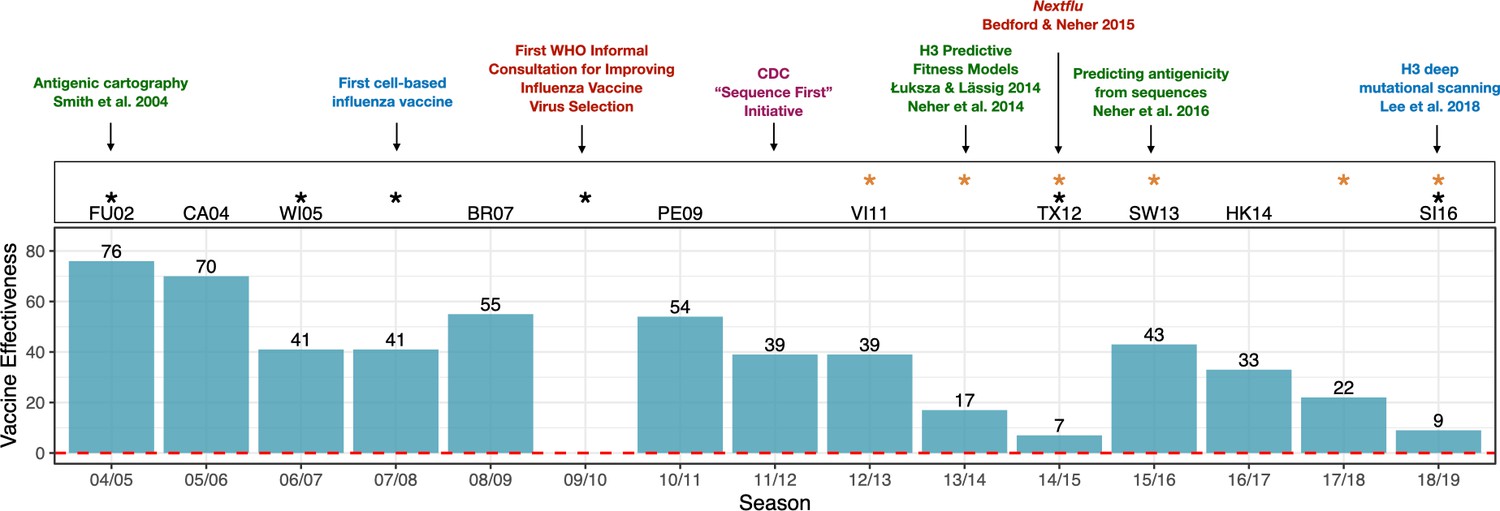
Advances in influenza research and vaccine effectiveness (for A/H3N2) from the 2004/05 flu season onwards.
The effectiveness of vaccines for seasonal influenza (A/H3N2) is highly variable and has been less than 10% in some years (teal bars). The H3N2 vaccine strain is shown for seasons when it was changed from the previous season. Black stars indicate seasons where the vaccine strain mismatched circulating H3N2 viruses (https://www.cdc.gov/flu/season/past-flu-seasons.htm); yellow stars indicate seasons in which H3N2 vaccine strains acquired mutations during passage in eggs. Research advances are listed at the top of the figures and are color coded as follows: surveillance in red; experimental approaches in blue; sequencing approaches in purple; computational approaches in green. Advances in understanding the structure of hemagglutinin (Knossow et al., 1984; Wiley and Skehel, 1987) and predicting the evolution of H3 (Bush et al., 1999) occurred before the period shown in the figure. Point estimates of vaccine effectiveness are taken from the following references: Skowronski et al., 2005 (04/05); Skowronski et al., 2007 (05/06); Skowronski et al., 2009 (06/07); Belongia et al., 2011 (07/08); Skowronski et al., 2010 (08/09); Treanor et al., 2012 (10/11); Ohmit et al., 2014 (11/12); McLean et al., 2015 (12/13);; Gaglani et al., 2016 (13/14); Flannery et al., 2016 (14/15); Jackson et al., 2017 (15/16); Flannery et al., 2019 (16/17); Rolfes et al., 2019 (17/18); Flannery et al., 2020 (18/19); estimates were not available during the 2009/10 A/H1N1 pandemic.
Influenza A and influenza B viruses cause seasonal epidemics every winter. Seasonal influenza A viruses include two different subtypes, H1N1 and H3N2, where H and N (short for hemagglutinin and neuraminidase) are proteins found on the surface of the virus. The human immune system protects the body against influenza infection by producing antibodies that can recognize these proteins. However, the influenza virus mutates frequently, including at sites that affect the immune system's ability to detect the virus. This process – called 'antigenic drift' – helps the virus infect new hosts and spread in populations that previously had immunity to influenza. Indeed, antigenic drift can lead to new strains of the virus that completely displace the currently circulating strains in a matter of months.
To keep pace with antigenic drift, the composition of influenza vaccines must be updated continually. Influenza vaccines contain three or four components that protect against various strains representing the different subtypes. Scientists convene twice a year at the World Health Organization (WHO) to predict which strains will have the highest fitness and therefore dominate the next year's flu season. H3N2 viruses evolve particularly fast and unpredictably compared to other seasonal flu viruses. Because the composition of the vaccine has to be decided a year in advance to allow doses to be manufactured, H3N2 vaccine strains have failed to match naturally circulating strains in six of the past fifteen flu seasons (Figure 1).
For decades, vaccine strain selection has been primarily informed by data from 1950s-era serological assays, which provide a phenotypic measure of how immune systems exposed to recently circulating viruses would see a novel strain. However, the assays have certain disadvantages – they are labor intensive, inconsistent across labs, not publicly available, and difficult to interpret or scale up. This means that these phenotypic measures are only available for a small subset of viruses. To remedy this issue, Huddleston et al. use a phylogenetic model (which includes available serological data and sequence data as inputs) to make predictions for the thousands of strains for which serological information is not available (Bedford et al., 2014; Neher et al., 2016; Smith et al., 2004).
Huddleston et al. compare how antigenic phenotypes from serological assays perform against five newer measures of virus fitness in forecasting future H3N2 virus populations, and find that two of their models provide better forecasts than WHO vaccine strain selections. Moreover, they have now integrated their forecasts for H3N2 into nextstrain.org, an open-source platform that scientists and policymakers use to track the real-time evolution of a wide range of pathogens (Hadfield et al., 2018; Neher and Bedford, 2015). Nextstrain provides a platform to make influenza vaccine strain selection more data-driven, systematic and transparent, and to allow new forecasting methods to be integrated as they show promise.
How does one predict the fitness of an influenza virus? Most mutations are harmful for influenza viruses, except for a subset of beneficial mutations that lead to antigenic drift. For decades researchers have relied on a list of sites in the genome where seemingly beneficial mutations occur to measure antigenic drift and viral fitness (Bedford et al., 2014; Bush et al., 1999; Shih et al., 2007). However, Huddleston et al. find that serological assays (Neher et al., 2016) continue to be more useful than sequence-onlybased measures when making forecasts of future virus populations. Measures of viral fitness based on genetic sequences could not accurately predict H3N2 evolution in recent years due to the emergence of multiple co-circulating strains and the sudden decline of a dominant strain in 2019. While no method predicts the right vaccine strain every time, serology-based methods appear to outperform other approaches.
Over time, alternative approaches to measuring virus fitness will continue to be refined and may become integrated into vaccine strain selection. For example, Huddleston et al. could not include a new serological assay based on virus neutralization in their framework as data from this assay were only available over a short period of time, but it could be integrated as data accrue. Other incremental improvements could be beneficial when used in combination with serological data. For example, how fast a strain is spreading globally can be measured from branching patterns in the phylogenetic tree (Neher et al., 2014). 'Mutational load' (that is, the total number of mutations in sites unrelated to immune detection) provides a simple inverse measure of viral fitness (Luksza and Lässig, 2014), while a technique called deep mutational scanning measures whether experimentally induced mutations have beneficial or harmful effects (Lee et al., 2018), However, as with other sequence-based approaches, the fact that mutations have different effects in different genetic backgrounds may be a disadvantage.
Going forward, the COVID-19 pandemic could disrupt the ecology of flu viruses in the years ahead, and it will be interesting to observe how predictive models fare in a highly perturbed system with no historical precedent. SARS-CoV-2 viruses may also experience post-pandemic strain turnover that requires periodic updates to any COVID-19 vaccine, and it should be possible to adapt platforms built for influenza forecasting to make forecasts for SARS-CoV-2 and other pathogens.
Note
Disclaimer: The conclusions of this study do not necessarily represent the views of the NIH or the US government.
References
-
Enhanced genetic characterization of influenza A(H3N2) viruses and vaccine effectiveness by genetic group, 2014-2015Journal of Infectious Diseases 214:1010–1019.https://doi.org/10.1093/infdis/jiw181
-
Influenza vaccine effectiveness in the United States during the 2016-2017 seasonClinical Infectious Diseases 68:1798–1806.https://doi.org/10.1093/cid/ciy775
-
Nextstrain: real-time tracking of pathogen evolutionBioinformatics 34:4121–4123.https://doi.org/10.1093/bioinformatics/bty407
-
Influenza vaccine effectiveness in the United States during the 2015-2016 seasonNew England Journal of Medicine 377:534–543.https://doi.org/10.1056/NEJMoa1700153
-
Influenza vaccine effectiveness in the United States during 2012-2013: variable protection by age and virus typeJournal of Infectious Diseases 211:1529–1540.https://doi.org/10.1093/infdis/jiu647
-
Effects of influenza vaccination in the United States during the 2017-2018 influenza seasonClinical Infectious Diseases 69:1845–1853.https://doi.org/10.1093/cid/ciz075
-
Effectiveness of vaccine against medical consultation due to laboratory-confirmed influenza: results from a sentinel physician pilot project in british Columbia, 2004-2005Canada Communicable Disease Report 31:161–168.
-
Component-specific effectiveness of trivalent influenza vaccine as monitored through a sentinel surveillance network in Canada, 2006-2007The Journal of Infectious Diseases 199:168–179.https://doi.org/10.1086/595862
-
The structure and function of the hemagglutinin membrane glycoprotein of influenza virusAnnual Review of Biochemistry 56:365–394.https://doi.org/10.1146/annurev.bi.56.070187.002053
Article and author information
Author details
Publication history
- Version of Record published: October 13, 2020 (version 1)
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 2,315
- views
-
- 182
- downloads
-
- 6
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Seasonal Influenza: The challenges of vaccine strain selection
eLife 9:e62955.
https://doi.org/10.7554/eLife.62955
Further reading
-
- Biochemistry and Chemical Biology
- Evolutionary Biology
Stramenopiles form a clade of diverse eukaryotic organisms, including multicellular algae, the fish and plant pathogenic oomycetes, such as the potato blight Phytophthora, and the human intestinal protozoan Blastocystis. In most eukaryotes, glycolysis is a strictly cytosolic metabolic pathway that converts glucose to pyruvate, resulting in the production of NADH and ATP (Adenosine triphosphate). In contrast, stramenopiles have a branched glycolysis in which the enzymes of the pay-off phase are located in both the cytosol and the mitochondrial matrix. Here, we identify a mitochondrial carrier in Blastocystis that can transport glycolytic intermediates, such as dihydroxyacetone phosphate and glyceraldehyde-3-phosphate, across the mitochondrial inner membrane, linking the cytosolic and mitochondrial branches of glycolysis. Comparative analyses with the phylogenetically related human mitochondrial oxoglutarate carrier (SLC25A11) and dicarboxylate carrier (SLC25A10) show that the glycolytic intermediate carrier has lost its ability to transport the canonical substrates malate and oxoglutarate. Blastocystis lacks several key components of oxidative phosphorylation required for the generation of mitochondrial ATP, such as complexes III and IV, ATP synthase, and ADP/ATP carriers. The presence of the glycolytic pay-off phase in the mitochondrial matrix generates ATP, which powers energy-requiring processes, such as macromolecular synthesis, as well as NADH, used by mitochondrial complex I to generate a proton motive force to drive the import of proteins and molecules. Given its unique substrate specificity and central role in carbon and energy metabolism, the carrier for glycolytic intermediates identified here represents a specific drug and pesticide target against stramenopile pathogens, which are of great economic importance.
-
- Evolutionary Biology
- Genetics and Genomics
A protein’s genetic architecture – the set of causal rules by which its sequence produces its functions – also determines its possible evolutionary trajectories. Prior research has proposed that the genetic architecture of proteins is very complex, with pervasive epistatic interactions that constrain evolution and make function difficult to predict from sequence. Most of this work has analyzed only the direct paths between two proteins of interest – excluding the vast majority of possible genotypes and evolutionary trajectories – and has considered only a single protein function, leaving unaddressed the genetic architecture of functional specificity and its impact on the evolution of new functions. Here, we develop a new method based on ordinal logistic regression to directly characterize the global genetic determinants of multiple protein functions from 20-state combinatorial deep mutational scanning (DMS) experiments. We use it to dissect the genetic architecture and evolution of a transcription factor’s specificity for DNA, using data from a combinatorial DMS of an ancient steroid hormone receptor’s capacity to activate transcription from two biologically relevant DNA elements. We show that the genetic architecture of DNA recognition consists of a dense set of main and pairwise effects that involve virtually every possible amino acid state in the protein-DNA interface, but higher-order epistasis plays only a tiny role. Pairwise interactions enlarge the set of functional sequences and are the primary determinants of specificity for different DNA elements. They also massively expand the number of opportunities for single-residue mutations to switch specificity from one DNA target to another. By bringing variants with different functions close together in sequence space, pairwise epistasis therefore facilitates rather than constrains the evolution of new functions.




{kind=link}