Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy
- University of Geneva, Switzerland
Abstract
Seed dormancy is an adaptive trait preventing premature germination out of season. In a previous report (Piskurewicz et al., 2016) we showed that dormancy levels are maternally inherited through the preferential maternal allele expression in the seed endosperm of ALLANTOINASE (ALN), a negative regulator of dormancy. Here we show that suppression of ALN paternal allele expression is imposed by non-canonical RNA-directed DNA methylation (RdDM) of the paternal ALN allele promoter. Dormancy levels are further enhanced by cold during seed development. We show that DNA methylation of the ALN promoter is stimulated by cold in a tissue-specific manner through non-canonical RdDM, involving RDR6 and AGO6. This leads to suppression of ALN expression and further promotion of seed dormancy. Our results suggest that tissue-specific and cold-induced RdDM is superimposed to parental allele imprints to deposit in the seed progeny a transient memory of environmental conditions experienced by the mother plant.
https://doi.org/10.7554/eLife.37434.001Introduction
Newly produced seeds exhibit dormancy, a trait whereby germination is blocked upon imbibition under otherwise favorable germination conditions. Dormancy prevents germination out of season and facilitates final dispersion of the plant embryo. The Arabidopsis seed consists of an embryo surrounded by a single cell layer of endosperm and an external seed coat. The endosperm is essential to repress dormant seed germination by releasing the phytohormone abscisic acid (ABA), which blocks embryonic growth (Lee et al., 2010; Chahtane et al., 2017). Dry seeds gradually lose dormancy over a period of time, referred as ‘after-ripening’, thus acquiring the capacity to germinate. After-ripening time is therefore a measure of dormancy levels, which vary across Arabidopsis accessions.
In a previous report, we observed that hybrid seeds arising from reciprocal crosses between high and low dormancy accessions could inherit dormancy levels more akin to the maternal ecotype. We identified a set of genes exhibiting preferential maternal allele expression (genomic imprinting) in the endosperm of dormant seeds, which included seed germination regulators (Piskurewicz et al., 2016). We showed that maternal inheritance of dormancy levels is implemented through the preferential maternal allele endosperm expression of ALLANTOINASE (ALN), a negative regulator of seed dormancy. We also reported that cold temperatures during seed development, which markedly increases dormancy levels in Arabidopsis seeds (Donohue et al., 2008; Penfield and MacGregor, 2017), suppressed the expression of numerous maternally expressed genes (MEGs), including ALN, upon seed imbibition (Piskurewicz et al., 2016).
Genomic imprinting requires ‘imprints’ distinguishing parental alleles. In Arabidopsis, DNA methylation serves as a primary imprint for numerous MEGs (Rodrigues and Zilberman, 2015). The DNA demethylase DEMETER (DME) removes methylated cytosines at specific loci of the central cell maternal genome, resulting, after fertilization, in different DNA methylation levels between maternal and paternal genomes (Choi et al., 2002; Choi et al., 2004; Gehring et al., 2006; Morales-Ruiz et al., 2006). DNA METHYLTRANSFERASE 1 (MET1) is also required for imprinting by maintaining paternal allele CG methylation during DNA replication (Jullien, 2006). In addition to this mechanism, canonical RNA-directed DNA methylation (RdDM) can contribute to paternal allele silencing in some imprinted genes (Vu et al., 2013). Canonical RdDM involves RNA polymerase IV and V (Pol IV, Pol V). Pol IV-dependent RNA transcripts are transcribed by RNA-dependent RNA polymerase 2 (RDR2) to produce double-stranded RNAs (dsRNAs). dsRNAs are cleaved by DICER-LIKE 3 (DCL3) into 24-nt small interfering RNAs (siRNAs, which are then loaded into ARGONAUTE 4 (AGO4). The transcripts emerging from Pol V are recognized by siRNAs bound to AGO4 through sequence complementarity. At this stage, DOMAIN REARRANGED METHYLTRANSFERASE 2 (DRM2) is recruited and methylates cytosine in all sequence contexts (CG, CHG, and CHH; H stands for A, C, or T) (Castel and Martienssen, 2013; Matzke et al., 2015). Non-canonical RdDM pathways, that is pathways not involving Pol IV-RDR2-DCL3, have recently been identified but are less well characterized (Cuerda-Gil and Slotkin, 2016).
Our previous report left unanswered the question of the nature of the mechanism leading to genomic imprinting in the endosperm of mature seeds. Furthermore, the mechanisms allowing mature seeds to ‘remember’ past cold temperatures in order to further regulate imprinted gene expression was not investigated. Here we addressed these questions by focusing on the case of ALN.
Results
ALN imprinting is maintained in met1 mutants and lost in drm1drm2 mutants
To investigate whether ALN paternal allele suppression requires DNA methylation, we pollinated WT Cvi plants with pollen from methyltransferase mutants, met1 (Col ecotype) and drm1drm2, hereafter referred as drm mutants (Ws), lacking functional DRM1 and DRM2.
Preferential maternal ALN allele expression was retained in WT x met1 hybrid seed endosperm but not in that of WT x drm (Figure 1A). We also pollinated WT Cvi plants with nrpd1 (Col-0) pollen, deficient in the largest subunit of Pol IV. Interestingly, WT x nrpd1 hybrid seed endosperm retained preferential maternal ALN allele expression (Figure 1A).
Figure 1 with 1 supplement see all
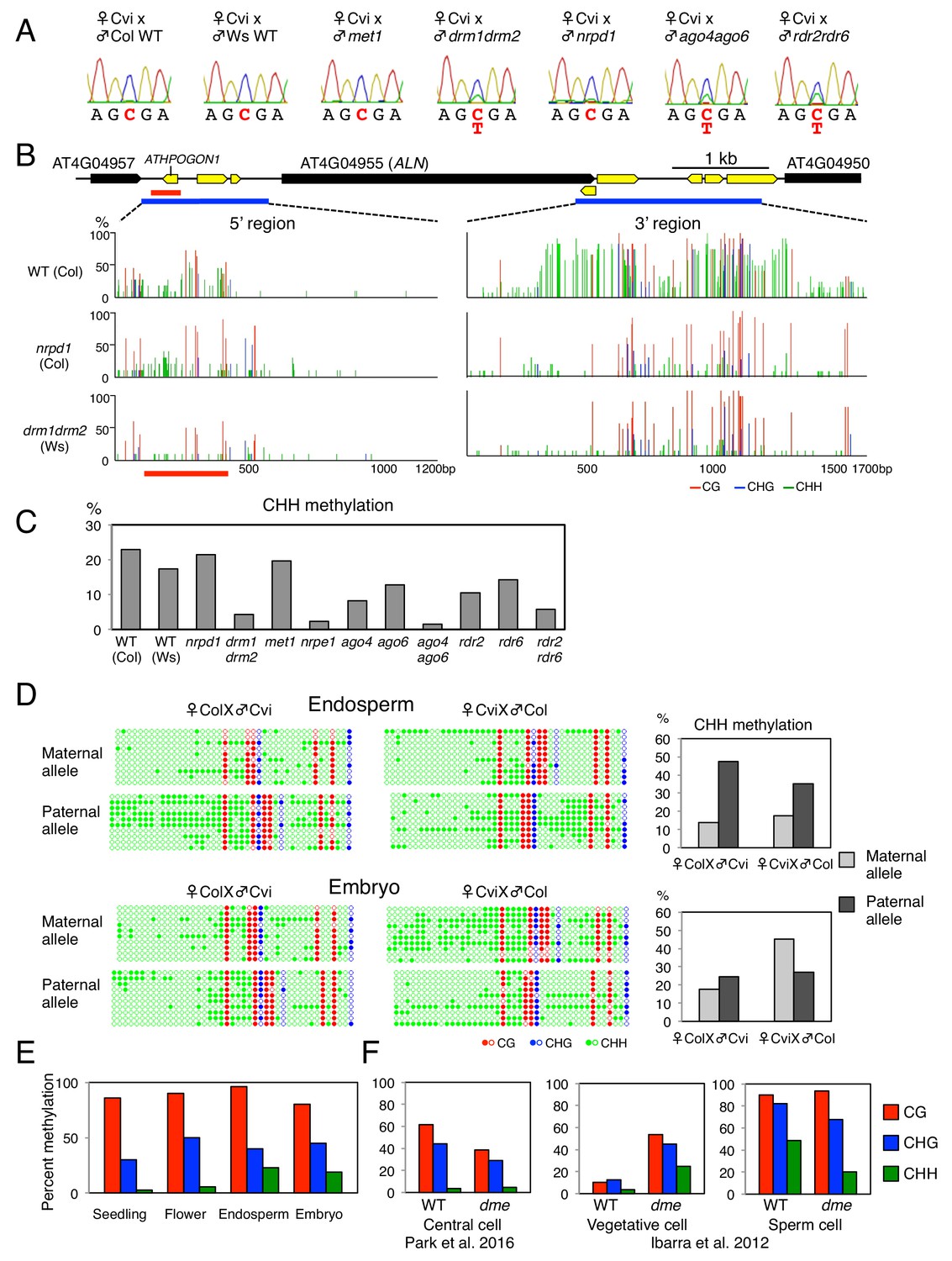
ALN imprinting necessitates non-canonical RdDM.
(A) Sanger sequencing chromatograms at SNPs of ALN. WT plants (Cvi) were pollinated with WT (Col and Ws) and mutant pollen as indicated. RNA was extracted from endosperm of F1 seeds and subjected to RT-PCR followed by Sanger sequencing. Nucleotides at SNP sites are highlighted in red: ‘C’ originates from Cvi and ‘T’ originates from Ws and Col. Representative results are shown from experiments repeated at least 5 times for each genotype. (B) DNA methylation on the ALN 5’ and 3’ flanking regions in endosperm of mature seed. Black box arrows and yellow box arrows show genes and TEs respectively. Blue lines show regions where DNA methylation was studied. Red line (300 bp) shows ALN’s highly methylated 5’ upstream region (referred as ‘POGO region’). All remaining methylation data shown in this figure only correspond to the 300 bp POGO region. Red, blue and green vertical lines represent CG, CHG, and CHH methylation levels, respectively. (C) Percentages of CHH methylation levels corresponding to the POGO region in different RdDM mutants (Materials and methods). drm1drm2 is in Ws background; ago4ago6 is generated after crossing ago4 with ago6 in Ler and C24 background, respectively; all other mutants are in Col-0 background. Percentages of DNA methylation at CG and CHG sites are shown in Figure 1—figure supplement 1. (D) DNA methylation levels in maternal and paternal alleles. DNA extracted from endosperm and embryo of F1 seeds obtained after reciprocally crossing Cvi and Col WT plants was analyzed by sodium bisulfite sequencing. SNPs were used to distinguish maternal and paternal alleles. Filled and open circles represent methylated and unmethylated cytosines, respectively. (E) Percentage of DNA methylation in the POGO region in different tissues. (F) Percentage of DNA methylation in the POGO region in female and male gametes. The data were extracted from published whole genomic DNA methylation data of female (Park et al., 2016) and male (Ibarra et al., 2012) gametes.
-
Figure 1—source data 1
Figure 1B and 2B: DNA methylation in the 5’ and 3’ flanking regions of ALN in the endosperm of mature seeds.
- https://doi.org/10.7554/eLife.37434.004
-
Figure 1—source data 2
Figure 1C: Percentages of DNA methylation levels in the POGO region of different RdDM mutants.
- https://doi.org/10.7554/eLife.37434.005
Altogether, these results show that ALN imprinting requires non-canonical RdDM for paternal allele silencing rather than MET1 or canonical RdDM as previously reported (Gehring, 2013; Vu et al., 2013; Rodrigues and Zilberman, 2015). We therefore hypothesized that ALN imprinting necessitates DNA methylation marks deposited by non-canonical RdDM. Thus, we explored whether genomic DNA neighboring ALN could indeed be targeted by non-canonical RdDM.
ALN upstream sequences are targeted by non-canonical RdDM
The 5’ and 3’ flanking regions of ALN contain transposable elements (TEs), which are often associated with DNA methylation (Figure 1B). We analyzed DNA methylation levels in a region of about 1.3 kbp spanning −1500 to −200 bp relative to ALN’s transcription start site (TSS), and in a region of about 1.7 kbp spanning −200 to +1500 bp relative to ALN’s transcription termination site (TTS) (Figure 1B). We identified a heavily methylated region of about one kbp in the 3’ flanking region and a moderately methylated region of about 300 bp located about 1.0 kbp upstream of ALN’s TSS (Figure 1B). Downstream of ALN’s TTS the levels of CHH methylation, which can be mediated by RdDM, were markedly reduced in drm mutants and nrpd1 mutants (Figure 1B). In contrast, in the methylated region upstream of ALN’s TSS, CHH methylation levels in nrpd1 were similar to WT levels unlike those in drm, which were markedly lower (Figure 1B). We also analyzed DNA methylation upstream of ALN’s TSS in nrpe1, deficient in the largest subunit of Pol V, and met1 mutants. Overall CHH methylation levels in met1 were similar to WT levels whereas they were markedly lower in nrpe1 mutants (Figure 1C, and Figure 1—figure supplement 1).
Interestingly, the methylated region upstream of ALN’s TSS contains a TE belonging to the AthPOGON1 family previously reported to be a target of non-canonical RdDM mediated by RDR6 and AGO6 (referred as RDR6-RdDM) (Nuthikattu et al., 2013; McCue et al., 2015; Cuerda-Gil and Slotkin, 2016). This methylated region is hereafter referred as ‘POGO region’. Indeed, in ago6 and rdr6 mutants the POGO region CHH methylation levels were lower than those of WT, although not as low as those found in drm or nrpe1 mutants (Figure 1B, and Figure 1—figure supplement 1). CHH methylation levels were also decreased in rdr2 and ago4 mutants and further decreased in rdr2rdr6 and ago4ago6 double mutants (Figure 1C, and Figure 1—figure supplement 1).
Altogether, these results show that the POGO region is methylated through a form of RdDM not involving Pol IV, that is a non-canonical form, and involving RDR6 and AGO6 in addition to RDR2 and AGO4.
To further investigate whether CHH methylation in the POGO region affects ALN imprinting, we pollinated WT plants with pollen from rdr2rdr6 and ago4ago6 double mutants. Preferential maternal ALN allele expression was lost both in WT x rdr2rdr6 and WT x ago4ago6 hybrid seeds (Figure 1A). These results consolidate the notion that non-canonical RdDM is required for ALN imprinting. This could involve CHH methylation of the POGO region upstream of ALN’s TSS.
We previously showed that seed dormancy levels can be regulated by maternal ALN allele expression. Indeed, we showed that aln x WT hybrid F1 seeds obtained after pollinating aln plants with WT pollen have higher dormancy than WT seeds (Piskurewicz et al., 2016). To further assess the role of RdDM in maternal regulation of seed dormancy, we pollinated aln mutant plants with WT and nrpe1 pollen. Dormancy levels in aln x nrpe1 hybrid F1 seeds were lower relative to aln x WT hybrid seeds, further indicating that maternal regulation of seed dormancy requires suppression of ALN paternal allele by RdDM (Figure 1—figure supplement 1C).
The paternal POGO region has higher CHH methylation
To assess whether CHH methylation of the POGO region could be involved in suppressing ALN’s paternal allele, we measured allele-specific methylation in the POGO region of hybrid seeds obtained after reciprocally crossing Col and Cvi plants. In embryos, CG methylation levels were similar between parental alleles whereas CHH methylation levels were higher in Cvi alleles (Figure 1D and Figure 1—figure supplement 1). This bias likely reflects accession differences in DNA methylation levels in the POGO region.
In the endosperm, CG DNA methylation levels were also similar between parental alleles. However, CHH methylation levels were markedly higher in the paternal allele relative to the maternal alleles (Figure 1D and Figure 1—figure supplement 1).
Altogether these results support the notion that preferential maternal ALN allele expression involves ALN paternal allele suppression mediated by methylation of the POGO region through non-canonical RdDM.
ALN expression is not silenced in seedlings and flowers (Schmid et al., 2005; Watanabe et al., 2014). We observed that seedlings and flowers had similar CG methylation levels in the POGO region relative to the mature endosperm or embryo. In contrast, they had markedly lower CHH methylation levels (Figure 1E).
This further suggests that methylation of CHH sites in the POGO region, mediated by non-canonical RdDM, rather than CG sites, which are present at low density in the POGO region, are involved in suppressing ALN paternal allele expression.
Preferential paternal POGO region methylation likely originates in the male germ line
Available genomic methylation data revealed that CHH methylation levels in the POGO region are low (about 4%) in the central cell (Figure 1F and Figure 1—figure supplement 1) (Calarco et al., 2012; Ibarra et al., 2012; Hsieh et al., 2016; Park et al., 2016). DME is active in the central cell where it establishes imprinted gene expression in numerous imprinted genes after removing DNA methylation (Choi et al., 2002; Park et al., 2016). In dme mutants CHH methylation levels in the central cell remain low and similar to those in WT plants (Figure 1F). Thus, in the case of ALN, higher paternal CHH methylation in the POGO region does not result from DME-dependent CHH methylation removal in the central cell.
In the case of sperm cells, different POGO region methylation levels are reported by three different groups but they are systematically higher (48%, 34% and 9%) than those reported in the central cell (Figure 1F and Figure 1—figure supplement 1) (Calarco et al., 2012; Ibarra et al., 2012; Hsieh et al., 2016). In addition to the central cell, DME is also active in the vegetative cell of pollen (Schoft et al., 2011; Ibarra et al., 2012). In WT, CHH methylation levels are low in the vegetative cell (about 3.5%), but they increase markedly (about 25%) in dme mutants, suggesting that DME promotes low CHH methylation in the vegetative cell (Figure 1F). In dme mutants, CHH methylation levels in sperm cells are reduced but they remain higher (>20%) relative to those in the central cell (Figure 1F) (Ibarra et al., 2012).
Altogether, these data suggest that preferential paternal ALN allele POGO region CHH methylation is a phenomenon specific to the male germ cell lineage, which is further maintained in the endosperm after fertilization. In turn, this leads to paternal allele suppression of ALN expression in the endosperm upon imbibition of mature dormant seeds.
Cold temperatures during seed development increase methylation of the POGO region through RDR6-RdDM
Seed development under cold temperatures leads to increased seed dormancy levels, which is associated with low ALN expression upon seed imbibition (Piskurewicz et al., 2016; Penfield and MacGregor, 2017). Interestingly, ALN imprinting was lost in seeds produced at 10 ˚C suggesting that cold perturbs the DNA methylation imprints associated with ALN (Figure 2A). In seeds produced at 10 ˚C, we observed that the length of the methylated POGO region expanded by 200 bp and CHH methylation levels markedly increased while the DNA methylation levels downstream of ALN’s TTS were similar to those observed in seeds produced at 22 ˚C (Figure 2B).
Figure 2 with 1 supplement see all
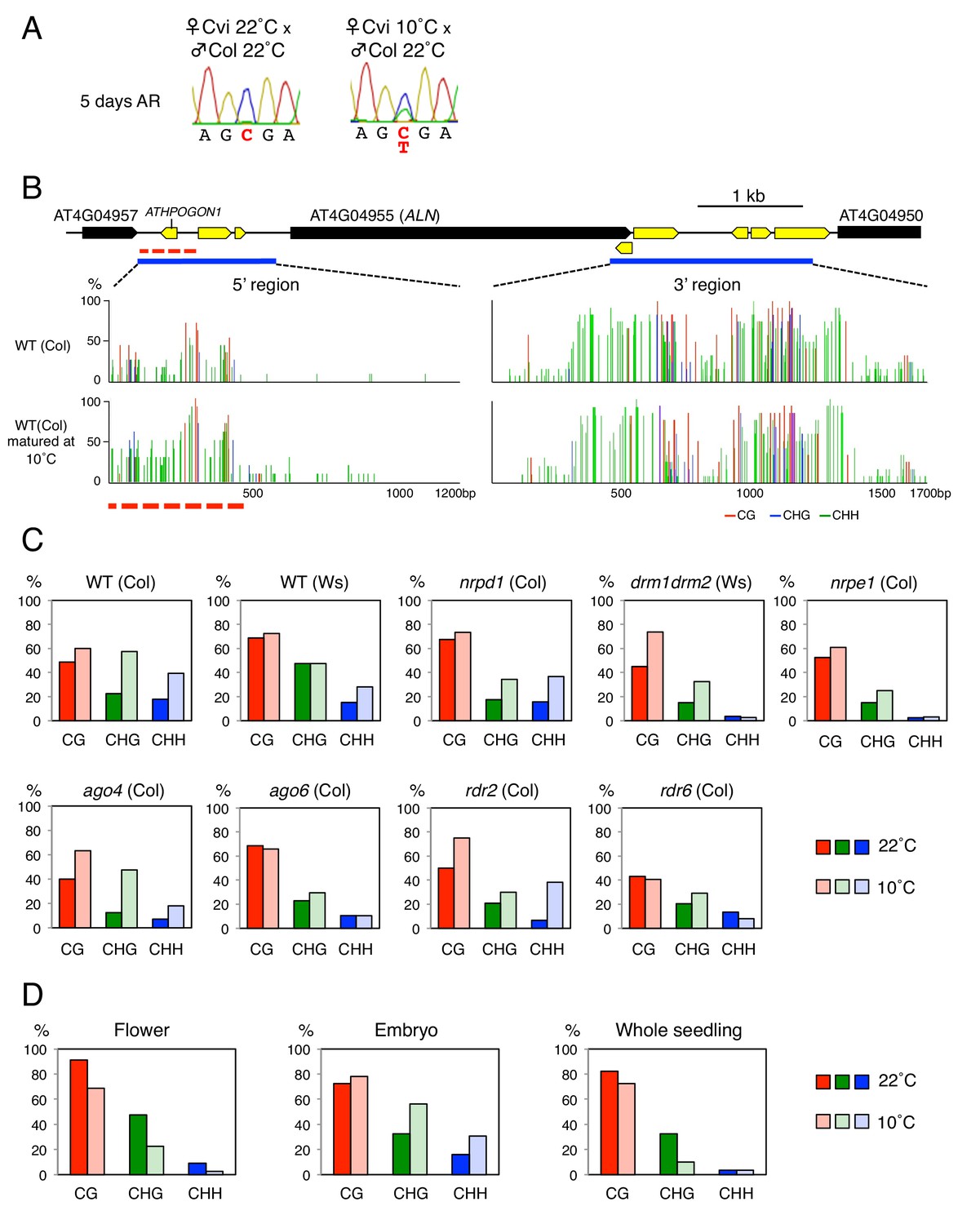
Cold temperatures increase methylation levels and expand the length of the ATHPOGON1-containing region subjected to methylation through RDR6-RdDM.
(A) Sanger sequencing chromatograms at SNPs of ALN. WT (Cvi) plants cultivated at 22 ˚C or 10 ˚C were pollinated with WT (Col) pollen. RNA was extracted from endosperm of F1 seeds and subjected to RT-PCR followed by Sanger sequencing. Nucleotides at SNP sites are highlighted in red: ‘C’ originates from Cvi and ‘T’ originates from Col. Representative results are shown from experiments repeated at least five times for each genotype. (B) DNA methylation in the ALN 5’ and 3’ flanking regions of the endosperm of mature seeds. Blues lines show regions where DNA methylation was studied. Dashed red line (500 bp) shows the region analyzed for DNA methylation levels. All remaining methylation data shown in this figure only correspond to this 500 bp region. Red, blue and green vertical lines represent CG, CHG, and CHH methylation levels, respectively. (C) Histograms show percentage of DNA methylation in the 500 bp region. DNA extracted from endosperm of WT seeds and RdDM mutant seeds produced at 22 ˚C or 10 ˚C was analyzed by sodium bisulfite sequencing. (D) Cold-induced and seed-specific DNA methylation. Seeds produced at 22 ˚C or 10 ˚C were sown and the resulting seedlings cultivated at 22 ˚C for three weeks prior to DNA extraction and sodium bisulfite sequencing analysis (Whole seedling).
-
Figure 2—source data 1
Figure 2C: Percentages of DNA methylation levels in the POGO region.
- https://doi.org/10.7554/eLife.37434.008
Cold-induced CHH methylation was observed in nrpd1 but not in drm or nrpe1 mutants (Figure 2C and Figure 2—figure supplement 1). This strengthens the notion that the POGO region is a target of non-canonical RdDM. Indeed, cold-induced CHH methylation was dependent on RDR6 and AGO6 but not on RDR2 or AGO4 (Figure 2C and Figure 2—figure supplement 1). These results therefore show that non-canonical RDR6-RdDM is required for cold-induced CHH methylation of the POGO region.
Cold-dependent and tissue-specific stimulation of AGO6 expression likely drives cold-induced methylation of the POGO region in seeds
Increased CHH methylation in the POGO region was observed both in the endosperm and embryo from seeds produced at 10 ˚C, but not in the flowers from plants cultivated at 10 ˚C (Figure 2D). This indicated that cold-induced CHH methylation in mature seeds takes place in gametes or in fertilization tissues or both.
Previous reports showed that AGO6 accumulates in gamete precursor cells (McCue et al., 2015). Furthermore, published microarray data show that AGO6 expression is rather tissue-specific, being expressed in shoot apex, mature pollen and developing seeds (Schmid et al., 2005). In contrast, RDR2, RDR6, and AGO4 expressions are more ubiquitous and higher relative to that of AGO6 (Schmid et al., 2005).
We therefore hypothesized that cold-induced POGO region CHH methylation in mature seeds results from cold-induced stimulation of AGO6 expression in gametes or in fertilization tissues or both. To test the hypothesis, we analyzed AGO6-GFP protein localization in developing seeds using an endogenous AGO6 promoter reporter line (pAGO6:AGO6-GFP) (McCue et al., 2015). Consistent with microarray data, we observed AGO6-GFP fluorescence in seeds developing at 22 ˚C. Strikingly, cold (10 ˚C) markedly increased the AGO6-GFP fluorescence in the pre-fertilization ovule and in developing seeds relative to warm temperatures (22 ˚C) (Figure 3A). Consistent with microarray data, at 22 ˚C AGO6 mRNA expression is low in pre-fertilization ovules relative to stamens (Figure 3B). At 10 ˚C, AGO6 mRNA expression is induced in pre-fertilization ovules (Figure 3B). This suggests that methylation of the POGO region could be stimulated by cold in the female gamete prior to fertilization. Furthermore, we found that AGO6 expression was higher in stamen and early developing seeds in plants cultivated at 10 ˚C relative to plants cultivated at 22 ˚C (Figure 3B). Expression levels of AGO4 were also increased in seeds developing at 10 ˚C relative to those developing at 22 ˚C (Figure 3B). Expression levels of RDR2 and RDR6 were not significantly changed in response to cold (Figure 3A and B).
Figure 3
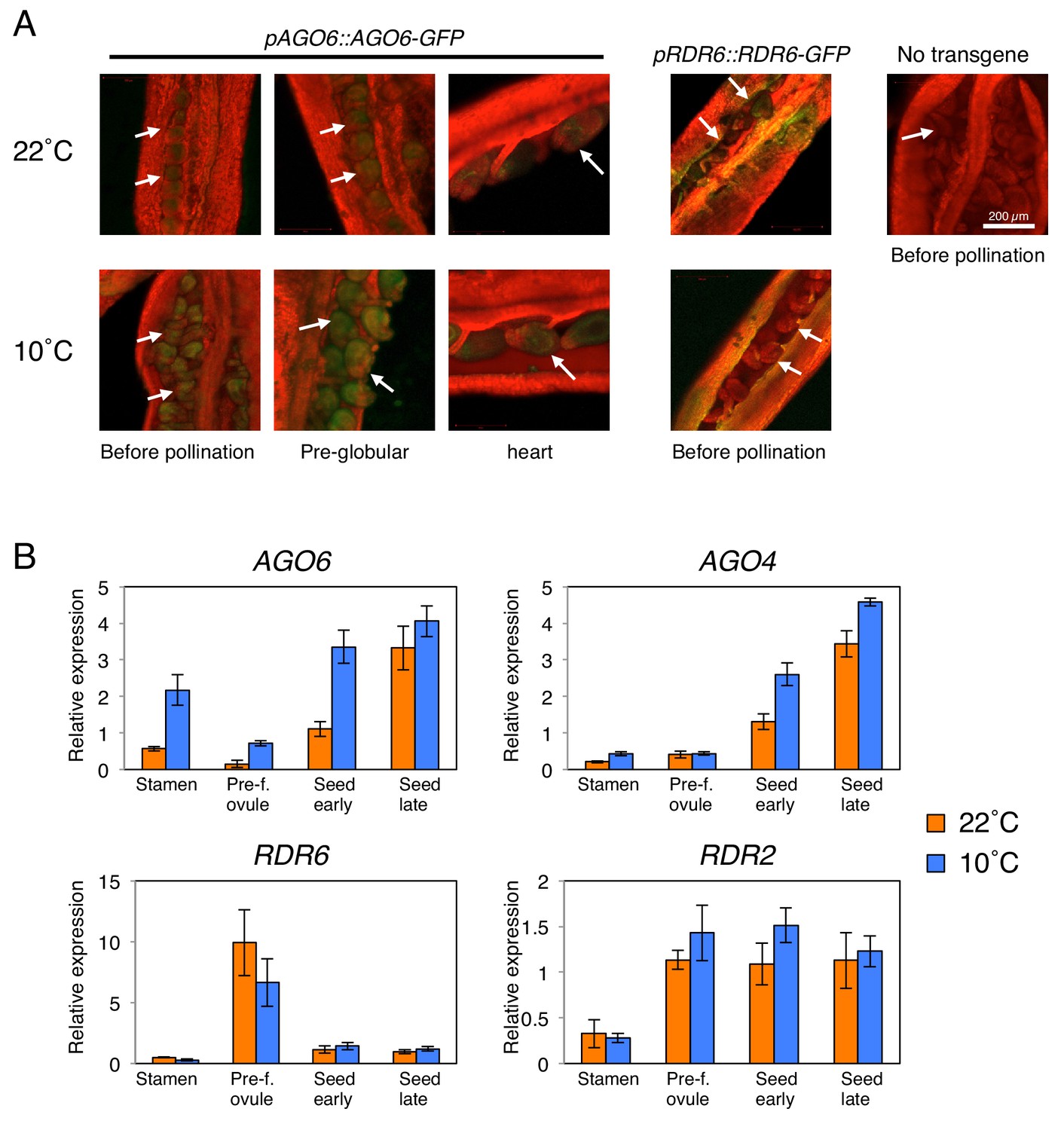
AGO6 expression is tissue-specific and is stimulated by cold.
(A) pAGO6::AGO6-GFP and pRDR6::RDR6-GFP transgenic lines were used to image AGO6-GFP and RDR6-GFP fluorescence, respectively, at various stages of ovule and seed development using confocal laser-scanning microscopy. Plants lacking the transgene are shown as a negative control (No transgene). Chlorophyll auto-fluorescence appears in red. Arrows indicate pre- and after-fertilization ovules. (B) qRT-PCR analysis of AGO6, AGO4, RDR2, and RDR6 mRNA accumulation in plants cultivated at 22˚C and 10˚C. RNA was extracted from stamens (Stamen), pre-fertilization ovules (Pre-f. ovule), developing seeds at the heart stage (Seed early) and developing seeds at the mature green stage (Seed late). Expression levels were normalized to those of PP2A. Error bars indicate SD from three technical replicates. Experiments were repeated two times giving similar results.
-
Figure 3—source data 1
Figure 3B: qRT-PCR expression analysis of AGO6, AGO4, RDR2, and RDR6.
- https://doi.org/10.7554/eLife.37434.010
Taken together these results suggest that cold temperatures could stimulate AGO6 expression and product accumulation in gametes and fertilization tissues. They support the hypothesis that cold stimulates RDR6-RdDM in male and female reproductive tissues or early developing seeds or both, thus leading to increased CHH methylation of the POGO region in mature seeds (Figure 4—figure supplement 3). In turn, increased methylation of ALN maternal allele would lead to loss of imprinting.
Furthermore, seedlings arising from seeds produced at 10 ˚C or at 22 ˚C had similar methylation levels in the POGO region (Figure 2D). This strongly suggests that after germination cold-induced CHH methylation is lost as the combined result of cell division and absence of AGO6 expression in seedlings (McCue et al., 2015).
Methylation changes in the POGO region correlate with changes in ALN expression and dormancy levels
We investigated whether changes in the POGO region CHH methylation are associated with changes in ALN endosperm expression levels. ALN endosperm expression upon seed imbibition was higher in WT seeds produced at 22 ˚C relative to seeds produced at 10 ˚C, as previously shown (Figure 4A) (Piskurewicz et al., 2016). In nrpd1 mutant seeds produced at 10 ˚C, in which CHH methylation is increased by cold, ALN expression was suppressed similarly to WT (Figure 4A). In contrast in drm mutants produced at 10 ˚C, ALN expression was comparable to that found in drm seeds produced at 22 ˚C.
Figure 4 with 3 supplements see all
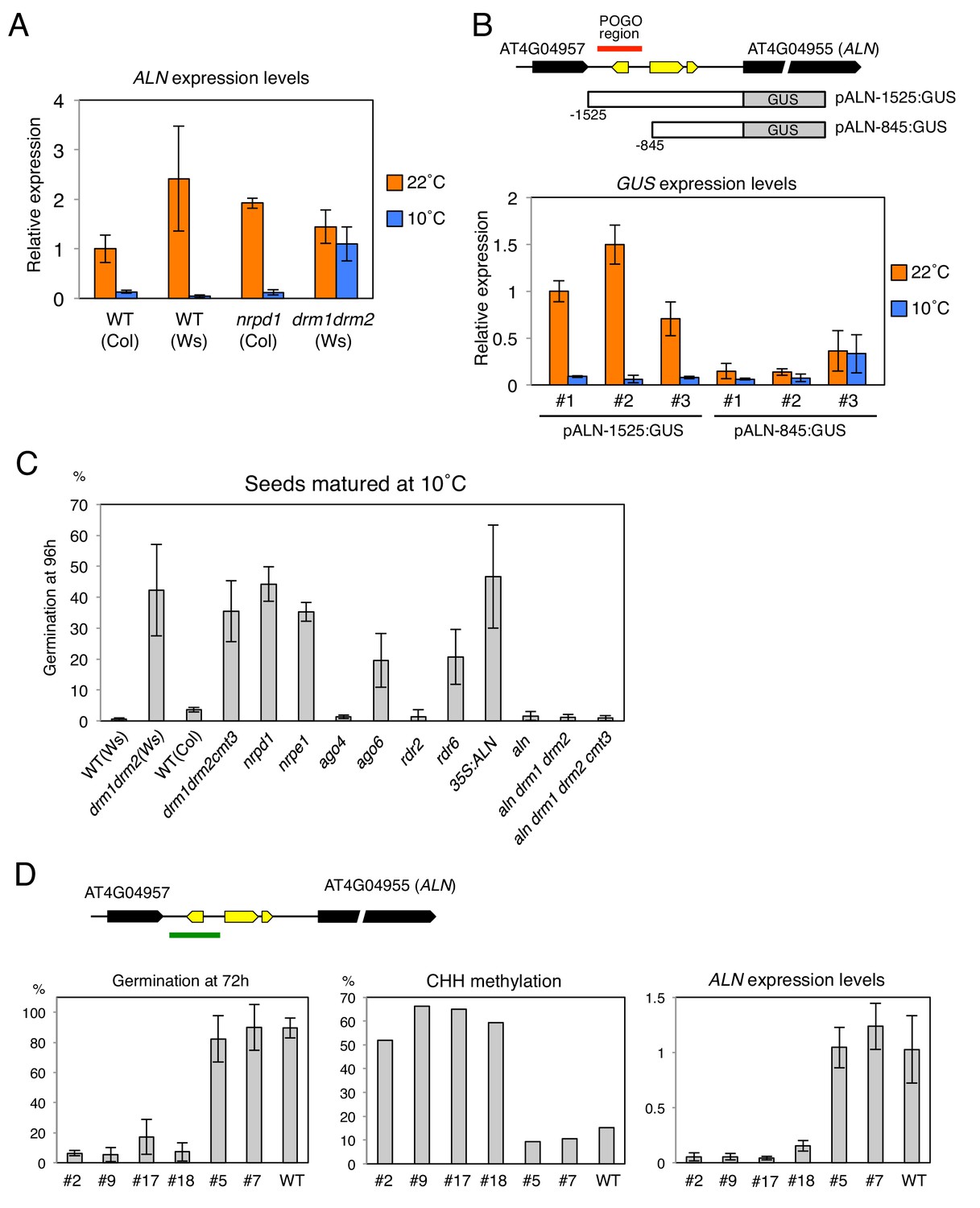
Changes in the POGO region methylation levels correlate with changes in ALN expression and dormancy levels.
(A) qRT-PCR analysis of ALN mRNA accumulation in WT, nrpd1 and drm1drm2 endosperm. RNA was extracted from endosperm of seeds produced at 22˚C and 10˚C and imbibed for 48 hr. ALN expression levels were normalized to those of PP2A. Error bars indicate SD of results from three technical replicates. Experiments were repeated three times giving similar results. (B) Analysis of reporter expression driven by ALN upstream sequences. Two different lengths of ALN upstream sequences (open box) were fused to GUS gene (filled box) and transformed into WT plants (Ws background). RNA was extracted from endosperm of seeds produced at 22˚C and 10˚C and imbibed for 48 hr. GUS expression levels were normalized to those of PP2A. Analysis was performed on three independent transgenic lines. Error bars indicate SD of results from three technical replicates. (C) Histograms show percent germination in populations of seeds produced 10 ˚C. Germination was scored 96 hr after seed imbibition (three biological replicates, n = 80–120). drm1drm2 is in Ws background; all other mutants are in Col-0 background. (D) Inducing DNA methylation of the AthPOGON1-containing region upstream of ALN. A DNA fragment covering AthPOGON1 (indicated by a green line) was cloned into a RNAi vector to produce double stranded inverted repeats (Materials and methods) and transformed into WT plants (Ws background). Dormancy levels, CHH methylation levels of endogenous ALN upstream sequences (Materials and methods) and ALN expression levels were analyzed for independent transgenic lines as shown. For the analysis of dormancy levels, seeds were after-ripened for 10 days, and germination was scored 72 hr upon seed imbibition (three replicates, n = 100). RNA was extracted from endosperm 48 hr after imbibition. ALN expression levels were normalized to those of PP2A. Error bars indicate SD of results from three technical replicates.
-
Figure 4—source data 1
Figure 4A, 4B, 4C and 4D: qRT-PCR expression analysis of ALN, GUS. Germination percentages of populations of seeds of different genotypes and produced at 10˚C.
POGO region DNA methylation levels and germination percentages of different transgenic lines
- https://doi.org/10.7554/eLife.37434.015
To further investigate the role of DNA methylation to regulate ALN gene expression, we generated transgenic lines where the expression of the GUS reporter gene is controlled by sequences upstream the ALN’s TSS that either lack (pALN-845) or contain (pALN-1525) the highly methylated POGO region (Figure 4B). In three independent pALN-1525 transgenic lines, harboring the POGO region, GUS expression was suppressed in seeds produced at 10 ˚C relative to that in seeds produced at 22 ˚C (Figure 4B). In contrast in three independent pALN-845 lines, lacking the POGO region, GUS expression levels were similar between seeds produced at 22 ˚C and those produced at 10 ˚C (Figure 4B).
These results suggest that the POGO region indeed contains regulatory elements necessary to suppress ALN transcription in response to cold during seed development. They support the notion that cold-induced CHH methylation suppresses ALN expression upon seed imbibition.
ALN is a negative regulator of seed dormancy and ALN expression levels affect seed dormancy levels (Piskurewicz et al., 2016). We analyzed seed dormancy levels in various RdDM mutant seeds produced at 22˚C and 10˚C. WT and RdDM mutant seeds produced at 22 ˚C had no detectable dormancy after a few days of after-ripening (Figure 4—figure supplement 1). ago4 and rdr2 mutant seeds produced at 10 ˚C increased their dormancy similarly to that of WT seeds. However, cold-produced drm, nrpe1, ago6, and rdr6 mutant seeds, unable to increase POGO region methylation, were less dormant compared to WT (Figure 4C and Figure 4—figure supplement 1).
These results establish a strong positive correlation between POGO region methylation and dormancy levels in seeds produced at 10 ˚C. However, we also observed that nrpd1 seeds produced at 10 ˚C were less dormant than WT seeds even though they increased CHH methylation (Figure 4C and Figure 4—figure supplement 1). One possible explanation for this apparent discrepancy is if Pol IV promotes dormancy irrespective of temperature during seed development. To better reveal differences in dormancy levels of seeds produced at 22 ˚C, we used suboptimal germination conditions (De Giorgi et al., 2015). After one week of after-ripening, nrpd1 seeds were able to germinate unlike WT and drm seeds, indeed suggesting Pol IV promotes seed dormancy irrespective of the temperature (Figure 4—figure supplement 1).
As expected, seeds produced at 10 ˚C from plants overexpressing ALN had low dormancy levels (Figure 4C and Figure 4—figure supplement 1). Furthermore, seeds produced at 10 ˚C from aln drm1 drm2 and aln drm1 drm2 cmt3 mutants had high dormancy levels similar to WT seeds (Figure 4C and Figure 4—figure supplement 1). These observations strongly suggest that high ALN expression in drm mutants contributes to their lower seed dormancy levels.
It is reported that RNAi targeted to specific loci can induce de novo DNA methylation (Mette et al., 1999; Mette et al., 2000; Matzke et al., 2004). To further study the role of POGO region methylation in cold-dependent ALN expression suppression and regulation of dormancy, we generated transgenic lines expressing an inverted repeat construct to be transcribed into dsRNA in order to generate siRNAs targeting the POGO region. In the T2 generation of transgenic plants, different dormancy levels were observed among different transgenic lines cultivated at 22 ˚C (Figure 4D and Figure 4—figure supplement 2). Among 22 transgenic lines, only four lines produced seeds with dormancy levels similar to WT seeds. The remaining 18 lines had higher dormancy levels compared to WT seeds. We selected four lines producing seeds with the highest dormancy levels and two lines producing seeds with dormancy levels similar to WT seeds. In the low dormancy lines endogenous POGO region CHH methylation and ALN expression levels were similar to those of WT plants (Figure 4D and Figure 4—figure supplement 2). In contrast, in the high dormancy lines endogenous POGO region CHH methylation levels were markedly increased relative to those in WT plants and ALN expression was suppressed (Figure 4D and Figure 4—figure supplement 2).
Altogether, these results support a model where cold-induced and RDR6-RdDM-dependent ALN promoter CHH methylation promotes seed dormancy by repressing ALN expression in seeds (Figure 4—figure supplement 3).
Discussion
ALN imprinting is implemented by non-canonical RdDM
Here we showed that ALN preferential maternal allele expression depends on DNA methylation of the paternal allele through non-canonical RdDM rather than MET1 as previously proposed for numerous MEGs (Gehring, 2013; Rodrigues and Zilberman, 2015). An association between RdDM and imprinted gene expression early upon fertilization has previously being described showing that some imprinted genes require canonical RdDM for paternal allele suppression; however, they also require MET1 activity (Vu et al., 2013).
The low abundance of CG sites in the ALN promoter suggests that CG methylation is not involved in suppressing ALN expression. In contrast, we showed that CHH methylation of POGO region located upstream of ALN’s TSS positively correlate with low ALN expression. This indicates that CHH methylation mediated by RdDM regulates ALN gene expression.
Developmental origin of the imprinting marks
Our results indicate that ALN paternal allele CHH methylation is established in sperm cells during gametogenesis and maintained during seed development, which may be due to non-canonical RdDM specifically taking place in the male gamete lineage or fertilization tissues or both (Figure 4—figure supplement 3).
However, it remains to be understood how higher CHH methylation levels are maintained in the paternal allele relative to the maternal allele after fertilization (Figure 1C). Guiding siRNAs for RdDM are not expected to discriminate paternal from maternal alleles. Indeed, maternal ALN allele CHH methylation levels are higher in the endosperm relative to those observed in flower and seedling tissues though they remain, in the endosperm, lower relative to those of the paternal ALN allele (Figure 1D and E).
One possible explanation for this discrimination is that RdDM is more efficient on methylated DNA, thus maintaining higher paternal ALN allele methylation over time after fertilization. Consistent with this hypothesis, Pol V is preferentially associated with methylated loci (Wierzbicki et al., 2012). It was shown that the SET domain proteins SUVH2 and SUVH9 bind to methylated DNA and facilitate the recruitment of Pol V to RdDM-targeted loci (Liu et al., 2014). Thus, after DNA replication, Pol V might be preferentially associated with hemi-methylated paternal chromosome, which would then facilitate DRM2-dependent CHH methylation of both strands.
Another possibility is that higher paternal allele CHH methylation levels at the time of fertilization serve as a primary imprint, which can generate a secondary imprint such as histone modification. This secondary imprint would then attract RdDM and thus maintain higher levels of CHH methylation in the paternal ALN allele. In plants, H3K27 trimethylation was shown to serve as a secondary imprint in some paternally expressed genes (Gehring, 2013; Rodrigues and Zilberman, 2015). In these cases, DNA hypomethylation of the maternal allele promotes H3K27 trimethylation, which suppresses maternal allele expression. This scenario could also operate in the case of ALN. Consistent with th possibility that H3K27 trimethylation could serve as a secondary imprint, a recent report showed that H3K27 trimethylation also takes place in hypermethylated paternal alleles (Moreno-Romero et al., 2016).
Cold-dependent RdDM in mature seeds is reset after germination
Previous studies associated environmental cues, including cold temperatures, with changes in CHH methylation levels in Arabidopsis and different plant species (Dubin et al., 2015; Secco et al., 2015; Wibowo et al., 2016; Secco et al., 2017). In most cases changes are transient, which is consistent with the notion that CHH methylation can be a dynamic process contrarily to CG methylation, which is faithfully maintained by MET1 after each cell division (Kawakatsu et al., 2017).
Here we found that CHH methylation levels of the POGO region located upstream of ALN’s TSS are stimulated by cold temperatures during seed development, which represses ALN expression, thus promoting seed dormancy. Interestingly, increased cold- and RDR6-RdDM-dependent stimulation of CHH methylation is observed in seed tissues but not in flowers. This reflects the tissue-specific activity of RDR6-RdDM (Figure 3A and B). Cold-induced CHH methylation is lost in the seedling after germination (Figure 2D).
It has been reported that there are resetting mechanisms of epigenetic changes induced by environmental cues (Choi et al., 2009; Crevillén et al., 2014; Iwasaki and Paszkowski, 2014; Crisp et al., 2016). One notable example is FLC, encoding a repressor of flowering. FLC expression is suppressed by H3K27 trimethylation by prolonged cold (vernalization). H3K27 trimethylation-imposed suppression of FLC expression is maintained after the vernalization period and this epigenetic mark is then removed during late embryo development (Berry and Dean, 2015). This ‘resetting’ mechanism ensures that the plant’s progeny can once again adapt appropriately their flowering time in a new environment.
Similarly, we propose that cold-induced CHH methylation of the POGO region located upstream of ALN’s TSS is a seed-specific mechanism allowing the seed to keep information of past cold temperatures in order to optimize seed germination timing. After germination, the information is erased in the embryo thus allowing optimal gene expression once again in the next generation.
Materials and methods
Plant material
Request a detailed protocolArabidopsis mutants, met1‐1 (Kankel et al., 2003), nrpd1a-3 (Herr et al., 2005), drm1-1 drm2-1 (Cao and Jacobsen, 2002), nrpe1 (SALK_017795), ago4 (SALK_027933), ago6-2 (SALK_031553), ago4-1 ago6-1 (Zheng et al., 2007), rdr2 (SALK_206644), rdr6-11 (Peragine et al., 2004), rdr2-1 rdr6-15 (Garcia-Ruiz et al., 2010) were obtained from The European Arabidopsis Stock Centre (RRID:SCR_004576). pAGO6::AGO6-GFP (McCue et al., 2015) and pRDR6::RDR6-GFP (McCue et al., 2012) transgenic lines were kindly provided by Dr. Keith Slotkin. 35S :ALN (Watanabe et al., 2014) construct was kindly provided by Dr. Atsushi Sakamoto.
Plant growth conditions and germination assays
Request a detailed protocolPlants were grown at 21–23°C, 16 hr/8 h day/night photoperiod, light intensity of 80 μE·m−2·s−1, humidity of 70%. For cold temperature treatment, plants were transferred to growth cabinets at 10°C after bolting. For germination assays, seeds were surface-sterilized (5% Bleach, 0.05% Tween) and sown on a Murashige and Skoog medium containing 0.8% (w/v) Bacto-Agar (Applichem). Between 80 and 150 seeds were examined with a Stemi 2000 (Zeiss) stereomicroscope and photographed with a high-resolution digital camera (Axiocam Zeiss) at different times of seed imbibition.
Plasmid construction and plant transformation
Request a detailed protocolFor the GUS reporter gene construct, a 1525 bp fragment spanning −1525 to −1, and a 845 bp fragments spanning −845 to −1 relative to the TSS of ALN were amplified from WT Col-0 genome by PCR and cloned into BamHI and SalI sites flanking the GUS coding region of pBI101 plasmid. For the RNAi vector, the 601 bp flagment spanning −846 to −1446 bp relative to ALN’s TSS was similarly amplified by PCR and cloned into the pRNAi-GG plasmid following the protocol from Yan et al. (2012). The 35S::ALN plasmid was described previously (Watanabe et al., 2014). The resulting constructs were introduced into Agrobacterium tumefaciens GV3101 strains to transform wild-type Col-0 or Ws plants by the floral‐dip method as previously described (Clough and Bent, 1998). Plants from T2 and T3 generation were used for analysis. Primers used in this study are listed in Supplementary file 1.
DNA methylation analysis
Request a detailed protocolGenomic DNA from embryo and endosperm was extracted as described previously (Piskurewicz and Lopez-Molina, 2011). Genomic DNA from flower and seedling was extracted with DNeasy Plant Mini Kit (Qiagen). Bisulfite DNA conversion was performed using EpiTect Bisulfite Kits (Qiagen). PCR was performed with primers listed in Supplementary file 1. Amplified fragments were cloned into the pGEM-T Easy Vector (Promega) and 10 to 12 independent clones for each sample were sequenced. Experiments were repeated at least twice. Sequencing data were analyzed with Kismeth (http://katahdin.mssm.edu/ kismeth).
RNA extraction and RT-qPCR
Request a detailed protocolTotal RNA was extracted as described previously (Piskurewicz and Lopez-Molina, 2011). Total RNAs were treated with RQ1 RNase-Free DNase (Promega) and reverse-transcribed using ImpromII reverse transcriptase (Promega) and oligo(dT)15 primer (Promega). Quantitative RT–PCR was performed by using the ABI 7900HT fast real-time PCR system (Applied Biosystems) and Power SYBR Green PCR master mix (Applied Biosystems). Relative transcript levels were calculated using the comparative ΔCt method and normalized to the PP2A (AT1G69960) gene transcript levels. Primers used in this study are listed in Supplementary file 1.
Confocal microscopy
Request a detailed protocolThe confocal fluorescent images were obtained by a Zeiss LSM 700 confocal microscope and ZEN microscope and imaging software package (Zeiss). GFP signals were taken with 488 nm laser and 520/35 nm band-pass filter. Chlorophyll autofluorescence signals were taken with 639 nm laser and 640 LP band-pass filter.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
-
NCBI Gene Expression OmnibusID GSE40501. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA.
-
NCBI Gene Expression OmnibusID GSE38935. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes.
-
NCBI Gene Expression OmnibusID GSE89789. DNA demethylation is initiated in the central cells of Arabidopsis and rice.
-
NCBI Gene Expression OmnibusID GSE87170. DNA methylation inheritance across generations.
References
-
Environmental perception and epigenetic memory: mechanistic insight through FLCThe Plant Journal 83:133–148.https://doi.org/10.1111/tpj.12869
-
RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyondNature Reviews Genetics 14:100–112.https://doi.org/10.1038/nrg3355
-
Primary seed dormancy: a temporally multilayered riddle waiting to be unlockedJournal of Experimental Botany 68:857–869.https://doi.org/10.1093/jxb/erw377
-
Non-canonical RNA-directed DNA methylationNature Plants 2:16163.https://doi.org/10.1038/nplants.2016.163
-
Genomic imprinting: insights from plantsAnnual Review of Genetics 47:187–208.https://doi.org/10.1146/annurev-genet-110711-155527
-
Genetic analysis of RNA-mediated transcriptional gene silencingBiochimica Et Biophysica Acta (BBA) - Gene Structure and Expression 1677:129–141.https://doi.org/10.1016/j.bbaexp.2003.10.015
-
RNA-Directed DNA methylation: the evolution of a complex epigenetic pathway in flowering plantsAnnual Review of Plant Biology 66:243–267.https://doi.org/10.1146/annurev-arplant-043014-114633
-
Effects of environmental variation during seed production on seed dormancy and germinationJournal of Experimental Botany 68:819–825.https://doi.org/10.1093/jxb/erw436
-
Isolation of genetic material from arabidopsis seedsMethods in Molecular Biology 773:151–164.https://doi.org/10.1007/978-1-61779-231-1_10
-
Evolution and function of genomic imprinting in plantsGenes & Development 29:2517–2531.https://doi.org/10.1101/gad.269902.115
-
A gene expression map of arabidopsis thaliana developmentNature Genetics 37:501–506.https://doi.org/10.1038/ng1543
-
Nutrient stress-induced chromatin changes in plantsCurrent Opinion in Plant Biology 39:1–7.https://doi.org/10.1016/j.pbi.2017.04.001
Article and author information
Author details
Funding
Swiss National Science Foundation (31003A-152660/1)
- Luis Lopez-Molina
Swiss National Science Foundation (31003A-179472/1)
- Luis Lopez-Molina
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are especially grateful to Keith Slotkin and Olivier Voinnet for generously sharing seed material. We thank Atsushi Sakamoto for providing the 35S::ALN plasmid. We thank Olivier Voinnet for helpful discussions.
We thank all members of the LLM laboratory for discussions. This work was supported by grants from the Swiss National Science Foundation and by the State of Geneva.
Version history
- Received: April 11, 2018
- Accepted: March 9, 2019
- Version of Record published: March 26, 2019 (version 1)
Copyright
© 2019, Iwasaki et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 9,500
- views
-
- 765
- downloads
-
- 46
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Non-canonical RNA-directed DNA methylation participates in maternal and environmental control of seed dormancy
eLife 8:e37434.
https://doi.org/10.7554/eLife.37434
Further reading
-
- Biochemistry and Chemical Biology
- Plant Biology
Metabolism and biological functions of the nitrogen-rich compound guanidine have long been neglected. The discovery of four classes of guanidine-sensing riboswitches and two pathways for guanidine degradation in bacteria hint at widespread sources of unconjugated guanidine in nature. So far, only three enzymes from a narrow range of bacteria and fungi have been shown to produce guanidine, with the ethylene-forming enzyme (EFE) as the most prominent example. Here, we show that a related class of Fe2+- and 2-oxoglutarate-dependent dioxygenases (2-ODD-C23) highly conserved among plants and algae catalyze the hydroxylation of homoarginine at the C6-position. Spontaneous decay of 6-hydroxyhomoarginine yields guanidine and 2-aminoadipate-6-semialdehyde. The latter can be reduced to pipecolate by pyrroline-5-carboxylate reductase but more likely is oxidized to aminoadipate by aldehyde dehydrogenase ALDH7B in vivo. Arabidopsis has three 2-ODD-C23 isoforms, among which Din11 is unusual because it also accepted arginine as substrate, which was not the case for the other 2-ODD-C23 isoforms from Arabidopsis or other plants. In contrast to EFE, none of the three Arabidopsis enzymes produced ethylene. Guanidine contents were typically between 10 and 20 nmol*(g fresh weight)-1 in Arabidopsis but increased to 100 or 300 nmol*(g fresh weight)-1 after homoarginine feeding or treatment with Din11-inducing methyljasmonate, respectively. In 2-ODD-C23 triple mutants, the guanidine content was strongly reduced, whereas it increased in overexpression plants. We discuss the implications of the finding of widespread guanidine-producing enzymes in photosynthetic eukaryotes as a so far underestimated branch of the bio-geochemical nitrogen cycle and propose possible functions of natural guanidine production.
-
- Plant Biology
Root gravitropic bending represents a fundamental aspect of terrestrial plant physiology. Gravity is perceived by sedimentation of starch-rich plastids (statoliths) to the bottom of the central root cap cells. Following gravity perception, intercellular auxin transport is redirected downwards leading to an asymmetric auxin accumulation at the lower root side causing inhibition of cell expansion, ultimately resulting in downwards bending. How gravity-induced statoliths repositioning is translated into asymmetric auxin distribution remains unclear despite PIN auxin efflux carriers and the Negative Gravitropic Response of roots (NGR) proteins polarize along statolith sedimentation, thus providing a plausible mechanism for auxin flow redirection. In this study, using a functional NGR1-GFP construct, we visualized the NGR1 localization on the statolith surface and plasma membrane (PM) domains in close proximity to the statoliths, correlating with their movements. We determined that NGR1 binding to these PM domains is indispensable for NGR1 functionality and relies on cysteine acylation and adjacent polybasic regions as well as on lipid and sterol PM composition. Detailed timing of the early events following graviperception suggested that both NGR1 repolarization and initial auxin asymmetry precede the visible PIN3 polarization. This discrepancy motivated us to unveil a rapid, NGR-dependent translocation of PIN-activating AGCVIII kinase D6PK towards lower PMs of gravity-perceiving cells, thus providing an attractive model for rapid redirection of auxin fluxes following gravistimulation.




{kind=link}
{kind=link}
{kind=link}
{kind=link}