

Abstract
The blood-brain barrier (BBB), which constitutes the interface between blood and cerebral parenchyma, has been shown to be disrupted during retroviral associated neuromyelopathies. Human T cell leukemia virus (HTLV-1)-associated myelopathy/tropical spastic paraparesis is a slowly progressive neurodegenerative disease, in which evidence of BBB breakdown has been demonstrated by the presence of lymphocytic infiltrates in the CNS and plasma protein leakage through cerebral endothelium. Using an in vitro human BBB model, we investigated the cellular and molecular mechanisms involved in endothelial changes induced by HTLV-1-infected lymphocytes. We demonstrate that coculture with infected lymphocytes induces an increase in paracellular endothelial permeability and transcellular migration, via IL-1α and TNF-α secretion. This disruption is associated with tight junction disorganization between endothelial cells, and alterations in the expression pattern of tight junction proteins such as zonula occludens 1. These changes could be prevented by inhibition of the NF-κB pathway or of myosin light chain kinase activity. Such disorganization was confirmed in histological sections of spinal cord from an HTLV-1-associated myelopathy/tropical spastic paraparesis patient. Based on this BBB model, the present data indicate that HTLV-1-infected lymphocytes can induce BBB breakdown and may be responsible for the CNS infiltration that occurs in the early steps of retroviral-associated neuromyelopathies.
The blood-brain barrier (BBB)3 is a dynamic physiological structure that is responsible for maintaining the CNS fluid homeostasis. It protects the neural tissue from toxins, infectious agents, and buffers variations in blood composition. The BBB is composed of three major cell types: endothelial cells, pericytes, and astrocytes (1). A major anatomical feature regulating solute entry into the CNS is the presence of closely associated areas called tight junctions (TJs) between opposing endothelial cells (2). Through these complex structures, endothelial cell membranes are able to join together to constitute an impermeable barrier that prevents the passage of cells, molecules, and ions. TJs are formed by transcellular proteins, such as claudins and occludin, joining the cytoskeleton via cytoplasmic proteins, such as zonula occludens 1 (ZO-1). Regulation of TJ protein expression and/or subcellular distribution thus plays a key role in the physiology of the BBB.
BBB breakdown has been shown to occur in retroviral-associated diseases, such as HIV-associated dementia (3, 4). BBB dysfunction has also been reported during human T cell leukemia virus (HTLV)-associated myelopathy/tropical spastic paraparesis (HAM/TSP), a slowly progressive neurodegenerative disease associated with HTLV-1 infection (5, 6). HTLV-1-infected lymphocytes cross the BBB in HAM/TSP patients (7) and massive lymphocytic infiltration has been demonstrated in the CNS parenchyma of a HAM/TSP patient, accompanied by plasma protein leakage (fibrinogen, IgG) through the CNS endothelium (8). The molecular mechanisms that govern BBB breakdown remain unknown (9, 10).
Recently, a human immortalized endothelial cell line (hCMEC/D3) was proposed as a unique model of the human BBB: it demonstrates BBB characteristics, with a high restriction of permeability, and expresses chemokine receptors and up-regulated adhesion molecules in response to inflammatory cytokines (11). We applied this cellular model to the investigation of the mechanisms associated with retroviral-induced BBB disruption, both in terms of permeability to solutes as well as transendothelial migration of lymphocytes. We report here that retroviral-infected lymphocytes induce BBB disruption, as assessed by paracellular permeability changes and increased lymphocyte migration, via the secretion of the proinflammatory cytokines IL-1α and TNF-α. Disruption was shown to be mediated by NF-κB activation and myosin light chain (MLC) phosphorylation induced by cytokines. Such events lead to TJs disorganization and ZO-1 degradation in human cerebral endothelialium, as assessed on cultured human endothelial cells (as well as on spinal cord sections from a HAM/TSP patient).
Materials and Methods
Cells and tissues
The human cerebral microvascular endothelial cell line, hCMEC/D3, was immortalized after transduction with lentiviral vectors encoding the catalytic subunit of human telomerase hTERT and SV40 T Ag, as described previously (11). hCMEC/D3 cells were grown in endothelial growth medium-2 (EGM-2MV; Clonetics; Cambrex Biosciences) without hydrocortisone on Biocoat tissue culture flasks (BD Biosciences).
HUT-102, a T cell line established from neoplasic lymph node cells (12), and C81-66, a T cell line derived from human umbilical cord blood T cells (13) were used as HTLV-1-infected T cell lines. Both cell lines express the viral transactivator Tax-1, although C81-66 is not producing viral particles. CEM, a human HTLV-1-negative T cell line, derived from an acute lymphoblastic leukemia, was used as control cell line. Nonadherent cell lines were grown in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with glutamine (1 mM) and 10% FCS.
PBMCs were obtained from two healthy donors or from two HTLV-1-infected patients with HAM/TSP (referred to as patients A and B). Briefly, thawed PBMCs were cultured for 24 h in RPMI 1640 medium (Invitrogen Life Technologies) supplemented with glutamine (1 mM) and 20% FCS and IL-2 as previously described (14).
Tissue sections were obtained from thoracic spinal cord of a HAM/TSP patient or from an uninfected patient, whose cases have been previously reported (8). Tissues were cryostat-sectioned (6–10 μm thick), air-dried, and fixed in methanol for 20 min before processing for immunofluorescence.
Soluble factors and inhibitors
Purified human TNF-α and IL-1α were obtained from Sanofi Aventis. Monoclonal mouse anti-human (h)TNFα Ab was purchased from HyCult Biotechnologies. Monoclonal murine anti-neurofilament protein (Chemicon) was used as an irrelevant Ab for control experiments. Recombinant vascular endothelial growth factor receptor 2 (VEGF-R2) was obtained from R&D Systems. IL-1 receptor antagonist (IL-1Ra) was obtained from Synergen.
SN50, a peptide that specifically inhibits NF-κB in endothelial cells, and the mutated peptide SN50M used as control were used as proposed by the manufacturer (Calbiochem). ML7 is a specific inhibitor for myosin light chain kinase (MLCK) (Calbiochem).
Endothelial permeability assays
Permeability of hCMEC/D3 cell monolayers was measured using a method adapted from Dehouck et al. (15) and Romero et al. (16) on Transwell-Clear filters (polyester; 12 mm diameter; pore size, 0.4 or 3 μm; Costar). Briefly, 105 cells/well were seeded on filters previously coated with rat-tail collagen I (BD Biosciences) and bovine plasma fibronectin (Sigma- Aldrich). At confluence, hydrocortisone was added to EBM-2 medium as recommended by the manufacturer. After 24 h, cells were used for experiments. Coculture experiments were done by addition of 105 lymphocytes to the endothelial monolayer for the indicated period of time in the presence or absence of inhibitors.
For the permeability test, the culture medium was replaced by DMEM without phenol red. FITC-labeled dextran (molecular mass, 70 kDa; Sigma- Aldrich) was added to the upper compartment and inserts were transferred sequentially at 5-min intervals from well to well for 30 min. FITC-dextran that had diffused through the monolayer into the lower compartment at each time point was determined using a fluorescence multiwell plate reader (Wallac Victor 1420; PerkinElmer). The permeability coefficients of the endothelial monolayers (Pe) were then calculated as previously described (16).
Transmission and scanning electron microscopy
For transmission electron microscopy, confluent hCMEC/D3 cell monolayers, grown on filters, were fixed at 6 or 20 h after contact with HTLV-1-infected cells in 2.5% glutaraldehyde and 1% paraformaldehyde in 0.15 M cacodylate buffer supplemented with MgCl2, CaCl2, and sucrose at 0.1 M. After fixation for 2 days at 4°C, the filters were washed in cacodylate buffer and treated with 1% osmium tetroxide solution/1% potassium ferrocyanide for 1 h at room temperature. Cells were dehydrated by serial incubations in increasing ethanol concentrations and embedded in an epoxyresin at 60°C for 48 h. Ultrathin sections were prepared using a Leica ultracut UCT microtome and examined with a JEOL 1200 EX electron microscope operating at 80 kV.
For scanning electron microscopy, hCMEC/D3 cells on filters were washed in PBS and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) overnight at 4°C at 6 or 20 h following coculture with HTLV-1-infected cells or control cells. Cells were washed three times in 0.2 M cacodylate buffer (pH 7.2), postfixed for 1 h in 1% osmium tetroxide in 0.2 M cacodylate buffer (pH 7.2), and then rinsed with distilled water. Cells were then dehydrated, and specimens were sputter-coated twice with carbon, using a Baltec Med010 evaporator. Preparations were then examined with a JEOL JSM 6700F field emission scanning electron microscope operating at 5 kV.
Indirect immunofluorescence
Cultures were washed with PBS and fixed either with methanol (80%) or with 3.5% paraformaldehyde. After permeabilization, cells were preincubated in 5% serum/PBS followed by an incubation with a rabbit anti-ZO-1 Ab (Zymed) for 30 min at 37°C. Samples were then stained with FITC-conjugated goat anti-rabbit IgG (Vector Laboratories) at a 1/100 dilution in 5% milk/PBS for 1 h at 37°C. Some cultures were processed for F-actin staining using Bodipy Phalloidin (Molecular Probes) according to the manufacturer’s instructions. The monolayer was finally washed, mounted with DAPI-containing Vectashield (Vector Laboratories), and visualized with a Zeiss Axiovert apparatus or Leica DMRB.
Tissue sections were processed similarly (with rabbit anti-ZO-1 and anti-occludin Abs from Zymed, mouse anti-PECAM from Sereotec, or goat anti-MLCK from Santa Cruz Biotechnology). In every case, no significant staining was observed after omission of the first Ab (data not shown).
Electroporation of CEM cells
The uninfected CEM cells were transfected with 10 μg of one of the following plasmids: pC-GFP, pC-wild-type Tax-1, pC-M22, pC-M47, bearing genes that code respectively for GFP (control), wild-type Tax-1 protein, M22 mutant of Tax-1 protein (unable to activate the NF-κB pathway), and M47 mutant of Tax-1 protein (unable to activate the CREB pathway) (17). Transfection was performed by electroporation of 107 cells in 300 μl of RPMI 1640 at 250 V and 950 μF using a Gene Pulsor apparatus (Bio-Rad), as described previously (18).
Cells were then transferred into 10 ml of RPMI 1640. Three days later, the percentage of cells expressing Tax-1 (wild type or mutants) or GFP was estimated by FACS. Typically, the percentage of transfected was 30%.
ELISA
Human TNF-α, IL-1α, and IL-1β were detected using Quantikine kits (R&D Systems) according to the manufacturer’s instructions. For nontransfected cell lines, lymphocytes were adjusted to a density of 2 × 105 cells/ml in RPMI 1640 for 6 h. For transfected cells, lymphocytes concentration were adjusted to 6 × 105 cells/ml, to maintain the same density of Tax- or GFP-expressing cells per milliliter.
Immunoblot analyses
TJ proteins expression in hCMEC/D3 cells was investigated by immunoblot analysis. After treatment, cells were washed twice with PBS, lysed in appropriate buffer (50 mM Tris-HCl (pH 7.4), 120 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40, 0.2 mM Na3VO4, 1 mM DTT, 1 mM PMSF) in the presence of a mixture of protease inhibitors (Roche Applied Science), and incubated on ice. Protein concentration was determined by the Bradford method (Bio-Rad). Samples were loaded into 4–20% Tris/Gly gels (NOVEX Invitrogen Life Technologies), subjected to SDS-PAGE, and transferred onto a nitrocellulose membrane (Immobilon-P; Millipore). Following incubation with specific Abs (rabbit anti ZO-1 and occludin from Zymed or mouse anti-MLCK or rabbit anti P-MLC from Sigma-Aldrich) and extensive washing in PBS-Tween (0.05%), membranes were incubated with HRP-conjugated secondary Abs and developed using either the SuperSignal West Pico or SuperSignal West Femto Chemiluminescent substrate kit (Pierce).
To ensure equal amount of protein loaded per well, membranes were stripped with the Re-blot Plus kit (Chemicon International) and reprobed with a specific anti-β-tubulin Ab (Santa Cruz Biotechnology). Normalized quantification was performed with a Fuji IRLas-1000 system.
T lymphocyte migration through brain endothelial monolayers
Lymphocytes were labeled with CellTracker Green BODIPY (Molecular Probes) according to manufacturer’s instructions. Labeled lymphocytes (5 × 105) were added to the upper chamber of Transwell-Clear insert filters (polyester; 12 mm diameter; pore size, 3 μm; Costar) containing confluent hCMEC/D3 monolayers. After 24 h at 37°C, the monolayer was extensively washed with PBS/EDTA, to collect lymphocytes adherent to each side of the membrane. Cells were lysed using water. Fluorescence intensity was determined using fluorescence multiwell plate reader.
Results
HTLV-1-infected lymphocytes increase cerebral endothelial monolayer permeability
To assess changes in the integrity of human cerebral microvascular hCMEC/D3 endothelial cells, we determined the paracellular permeability of cell monolayers to 70-kDa-FITC dextran following coculture with HTLV-1-infected lymphocytes.
The paracellular permeability of control monolayers was low (Pe = 1.9 × 10−5 cm/min), in accordance to that reported previously (11, 19). Similar basal permeability levels were observed whether cells were grown on inserts with pore sizes of 0.4 or of 3 μm (data not shown). In addition, permeability was not modified when hCMEC/D3 monolayers were exposed to uninfected CEM T lymphocytes. In contrast, when hCMEC/D3 cells were cocultured with HTLV-1-infected HUT-102 or C81-66 cells, the permeability coefficient increased by 6 h to Pe = 6.2 × 10−5 and 6.1 × 10−5 cm/min, respectively. An increased permeability coefficient was also observed at 20 h following coculture (Fig. 1 A).
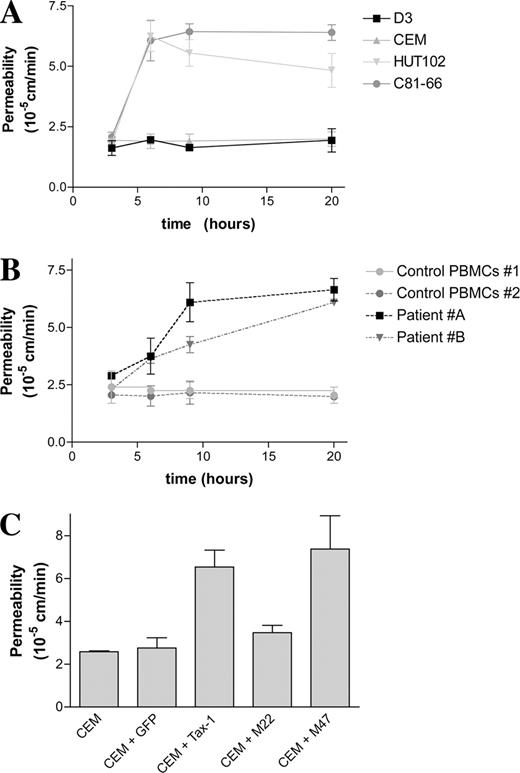
Effect of HTLV-1-infected lymphocytes on BBB permeability. Permeability was assessed by the clearance of 70-kDa-FITC-dextran through hCMEC/D3 monolayers grown on Transwell inserts. A, hCMEC/D3 monolayers were left alone or cocultured with uninfected CEM cells or with HTLV-1-infected HUT-102 or C81-66. Results are from triplicate cultures from one experiment representative of three. B, hCMEC/D3 monolayers were cultured with primary PBMCs from HTLV-1-infected patients (patients A and B) or from two uninfected individuals (control PBMCs). Results are from triplicate cultures from one representative experiment. C, Uninfected CEM cell lines were electroporated with plasmids encoding either GFP, Tax-1, or Tax-1 mutants (M22 and M47, which cannot activate NF-κB or CREB pathways, respectively). A total of 105 Tax-1 expressing cells was cocultured with hCMEC/D3 monolayers for 6 h, and the paracellular permeability of the endothelial cell monolayer was estimated as previously. Results are for triplicate cultures from one experiment representative of three.
Effect of HTLV-1-infected lymphocytes on BBB permeability. Permeability was assessed by the clearance of 70-kDa-FITC-dextran through hCMEC/D3 monolayers grown on Transwell inserts. A, hCMEC/D3 monolayers were left alone or cocultured with uninfected CEM cells or with HTLV-1-infected HUT-102 or C81-66. Results are from triplicate cultures from one experiment representative of three. B, hCMEC/D3 monolayers were cultured with primary PBMCs from HTLV-1-infected patients (patients A and B) or from two uninfected individuals (control PBMCs). Results are from triplicate cultures from one representative experiment. C, Uninfected CEM cell lines were electroporated with plasmids encoding either GFP, Tax-1, or Tax-1 mutants (M22 and M47, which cannot activate NF-κB or CREB pathways, respectively). A total of 105 Tax-1 expressing cells was cocultured with hCMEC/D3 monolayers for 6 h, and the paracellular permeability of the endothelial cell monolayer was estimated as previously. Results are for triplicate cultures from one experiment representative of three.
Similarly, an increase in hCMEC/D3 permeability was observed following coculture with two PBMCs isolated from patients with HAM/TSP (patients A and B), but not with PBMCs from healthy donors (Fig. 1 B). However, BBB disruption by infected PBMCs appeared to be delayed when compared with the effects induced by chronically infected cell lines.
Because C81-66 cells express only the viral transactivator Tax-1 and are able to disrupt hCMEC/D3 cell monolayers, we wondered whether Tax-1 expression in lymphocytes was itself sufficient to mediate this effect. We investigated the capacity of CEM cells expressing Tax-1 (or GFP as control) to affect endothelial permeability. Although GFP-expressing cells did not induce any change in permeability, CEM cells expressing Tax-1 increased the permeability of hCMEC/D3 monolayers at 6 h (Fig. 1,C). Interestingly, M47-expressing-CEM cells (a Tax mutant that is not able to activate the CREB pathway), but not M22-expressing-CEM cells (a Tax mutant that is not able to activate the NF-κB pathway), induced monolayer disruption at 6 h (Fig. 1 C), indicating that infected lymphocyte-mediated BBB disruption requires NF-κB activation by Tax-1.
IL-1α and TNF-α secretion by infected lymphocytes mediates BBB disruption
Because the NF-κΒ pathway is known to mediate proinflammatory cytokines synthesis and secretion, we determined by ELISA the levels of IL-1α, IL-1β, and TNF-α secreted by the different lymphocyte cell lines. Lymphocytes that were able to disrupt the BBB model (HUT-102, C81-66, Tax-1, and M47-expressing CEM cells) secreted higher levels of IL-1α and/or TNF-α than cells that did not increase permeability (Table I). Cytokine secretion levels of PBMCs from patients A and B were slightly lower than those detected by chronically infected cell lines, which could explain the slower BBB disruption kinetics observed with PBMCs. No IL-1β could be detected in the supernatant of most of these cell types.
Secretion levels of IL-1 and TNFαa
| IL-1α | IL-1β | TNFα | |
|---|---|---|---|
| CEM | 0 | 0 | 0 |
| HUT-102 | 88 ± 3 | 0 | 105 ± 5 |
| C81-66 | 117 ± 4 | 0 | 85 ± 2 |
| CEM-GFP | 0 | 0 | 10 ± 2 |
| CEM-Tax1 | 97 ± 7 | 5 ± 4 | 120 ± 12 |
| CEM-M47 | 94 ± 9 | 5 ± 2 | 100 ± 9 |
| CEM-M22 | 30 ± 5 | 0 | 50 ± 7 |
| Control 1 | 0 | 0 | 0 |
| Patient A | 90 ± 6 | ND | 80 ± 4 |
| Patient B | 75 ± 8 | ND | 82 ± 4 |
| IL-1α | IL-1β | TNFα | |
|---|---|---|---|
| CEM | 0 | 0 | 0 |
| HUT-102 | 88 ± 3 | 0 | 105 ± 5 |
| C81-66 | 117 ± 4 | 0 | 85 ± 2 |
| CEM-GFP | 0 | 0 | 10 ± 2 |
| CEM-Tax1 | 97 ± 7 | 5 ± 4 | 120 ± 12 |
| CEM-M47 | 94 ± 9 | 5 ± 2 | 100 ± 9 |
| CEM-M22 | 30 ± 5 | 0 | 50 ± 7 |
| Control 1 | 0 | 0 | 0 |
| Patient A | 90 ± 6 | ND | 80 ± 4 |
| Patient B | 75 ± 8 | ND | 82 ± 4 |
Secretion of IL-1α, IL-1β, and TNFα in HTLV-1-infected cells, uninfected cells, or Tax-1 (and Tax-1 mutants)-transfected cells. The concentration (and SD) of each cytokine (in picograms per milliliter) in the lymphocyte culture medium for 6 h was measured by ELISA. ND, Not done; 0, undetectable levels. CEM, Uninfected cell line; HUT-102 and C81-66, HTLV-1-infected cell lines; CEM-GFP, -Tax-1, -M47, and -M22, uninfected CEM cells transfected with GFP, Tax-1, and mutated M47 and M22 Tax-1 proteins genes, respectively. Control 1, PBMCs from an uninfected individual (control). A and B, PBMCs from HTLV-1-infected patients.
We then analyzed the effect of IL-1α and/or TNF-α added at the concentration determined previously (100 pg/ml) on hCMEC/D3 cell permeability. When added separately, neither IL-1α nor TNF-α were able to induce important increases in endothelial permeability (Fig. 2 A). By contrast, simultaneous addition of both cytokines induced an increase of permeability similar to that observed following coculture with infected cell lines. Of note, at higher concentrations (1–10 ng/ml), addition of each cytokine alone was sufficient to enhance the cell monolayer permeability (data not shown).

Central role of IL-1α and TNF-α in BBB disruption. A, Effect of IL-1α and TNF-α on BBB permeability. Cytokines were added to hCMEC/D3 monolayers cultured on Transwell inserts for 6 h at 100 pg/ml. Permeability of hCMEC/D3 monolayers was determined by FITC clearance. Results are for triplicate filters from one experiment representative of three. B, Role of soluble factors and NF-κB in HTLV-1-induced HCMEC/D3 monolayer permeability. Cells were treated or not for 1 h with SN50 or control mutant. Inhibitors (anti-TNFα, Ab against TNF-α; soluble VEGF-R2) or specific controls (irrelevant anti-neurofilament Ab, or heated IL-1-Ra and VEGF-R2) were added together with C81-66 lymphocytes, and permeability for FITC dextran was estimated at 6 h of coculture. C, Effect of inhibitors on HUT-102 migration through hCMEC/D3 monolayers. Inhibitors (anti-TNFα, Ab against TNF-α; soluble VEGF-R2) or specific controls (irrelevant anti-neurofilament Ab, or heated VEGF-R2) were added together with HUT-102 lymphocytes. Lymphocytes were initially labeled with the fluorescent marker CellTracker Green BODIPY and added to confluent hCMEC/D3 cells grown on filters. At 24 h, cell migration was estimated by determining the fluorescence in the lower chamber. The migration rate is expressed as ratio (percentage) of fluorescence intensity in the lower compartment vs total fluorescence added to the upper chamber. Results are for triplicate filters from one experiment representative of three. D, Bystander effect of HTLV-1-infected lymphocytes (or cytokines) on migration of uninfected lymphocyte through the barrier. Cytokines, inhibitors, or HUT-102 cells were added together with labeled CEM cells to confluent hCMEC/D3 cells grown on filters. The CEM migration was estimated at 24 h of culture as previously. The migration rate of CEM cells is expressed as ratio (percentage) of fluorescence intensity in the lower compartment vs total fluorescence added to the upper chamber. Results are for triplicate filters from one experiment representative of three.
Central role of IL-1α and TNF-α in BBB disruption. A, Effect of IL-1α and TNF-α on BBB permeability. Cytokines were added to hCMEC/D3 monolayers cultured on Transwell inserts for 6 h at 100 pg/ml. Permeability of hCMEC/D3 monolayers was determined by FITC clearance. Results are for triplicate filters from one experiment representative of three. B, Role of soluble factors and NF-κB in HTLV-1-induced HCMEC/D3 monolayer permeability. Cells were treated or not for 1 h with SN50 or control mutant. Inhibitors (anti-TNFα, Ab against TNF-α; soluble VEGF-R2) or specific controls (irrelevant anti-neurofilament Ab, or heated IL-1-Ra and VEGF-R2) were added together with C81-66 lymphocytes, and permeability for FITC dextran was estimated at 6 h of coculture. C, Effect of inhibitors on HUT-102 migration through hCMEC/D3 monolayers. Inhibitors (anti-TNFα, Ab against TNF-α; soluble VEGF-R2) or specific controls (irrelevant anti-neurofilament Ab, or heated VEGF-R2) were added together with HUT-102 lymphocytes. Lymphocytes were initially labeled with the fluorescent marker CellTracker Green BODIPY and added to confluent hCMEC/D3 cells grown on filters. At 24 h, cell migration was estimated by determining the fluorescence in the lower chamber. The migration rate is expressed as ratio (percentage) of fluorescence intensity in the lower compartment vs total fluorescence added to the upper chamber. Results are for triplicate filters from one experiment representative of three. D, Bystander effect of HTLV-1-infected lymphocytes (or cytokines) on migration of uninfected lymphocyte through the barrier. Cytokines, inhibitors, or HUT-102 cells were added together with labeled CEM cells to confluent hCMEC/D3 cells grown on filters. The CEM migration was estimated at 24 h of culture as previously. The migration rate of CEM cells is expressed as ratio (percentage) of fluorescence intensity in the lower compartment vs total fluorescence added to the upper chamber. Results are for triplicate filters from one experiment representative of three.
The role of TNF-α and IL-1α on BBB disruption was confirmed by treatment of monolayers cocultured with C81-66 cells with specific inhibitors and/or antagonists (Fig. 2 B). A mAb against human TNF-α (anti-TNF-α) was able to restore the permeability coefficient to control level. Similarly, addition of IL-1Ra was able to prevent the increase of the permeability, indicating a role for IL-1 in barrier disruption. Finally, inhibition of the NF-κB pathway in endothelial cells, known to be activated by these cytokines, was sufficient to prevent BBB disruption. By contrast, soluble VEGF-R2 was unable to prevent endothelial permeability changes induced by infected cell lines.
Besides BBB permeability alteration by infected lymphocytes through IL-1α and TNF-α, we also used lymphocyte migration as a criterion of BBB disruption. After 20 h of coculture, 28% of C81-66 and 29.2% of HUT-102 cells had migrated through the hCMEC/D3 monolayer as assessed by fluorometry. A 50% decrease in migration could be achieved by addition to the culture medium of specific inhibitors of IL-1 (IL-1Ra) or TNF-α (specific antibody) (Fig. 2,C). Similarly, addition of IL-1α and TNF-α (100 pg/ml) to the culture medium enhanced the migration of CEM uninfected cells from 7 to 16%. Coculture with HTLV-1-infected lymphocytes was also able to increase the migration of labeled uninfected cells (18%), reflecting a bystander effect of infected lymphocytes (Fig. 2,D). Interestingly, VEGF inhibition, which did not prevent the permeability increase, decreased the migration rate of infected lymphocytes (Fig. 2, B and C), suggesting an additional mechanism to the one described for the increase of BBB permeability.
BBB disruption is associated with TJ disorganization
Scanning electron microscopy of hCMEC/D3 cells showed a confluent cell monolayer composed of closely apposed flat spindle-shaped cells (not shown). This morphology was conserved in hCMEC/D3 cells cocultured with CEM cells (Fig. 3 A). Occasionally, a few scattered CEM lymphocytes adhered to the endothelial monolayer could be observed.
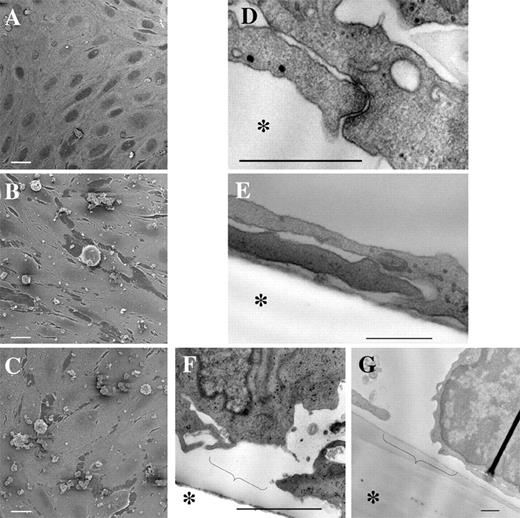
Ultrastructural analysis of hCMEC/D3 cells cocultured with lymphocytes for 6 h. A–C, Scanning electron microscopy of hCMEC/D3 monolayers cultured with CEM (A) or HTLV-1-infected HUT-102 (B), or C81-66 (C) cells. White bar represents 10 μm. D–G, Transmission electron microscopy showing electron-dense TJs in hCMEC/D3 cells cocultured with CEM cells for 6 h (D) or absence of TJs in hCMEC/D3 cells cocultured with HUT-102 (E). Disruption of hCMEC/D3 monolayer after 6 h of coculture with C81-66 (F) or PBMCs from patient A (G). Bar represents 500 nm. Asterisk (∗) indicates the filter location of the Transwell device. Brackets indicate the local cell retraction observed.
Ultrastructural analysis of hCMEC/D3 cells cocultured with lymphocytes for 6 h. A–C, Scanning electron microscopy of hCMEC/D3 monolayers cultured with CEM (A) or HTLV-1-infected HUT-102 (B), or C81-66 (C) cells. White bar represents 10 μm. D–G, Transmission electron microscopy showing electron-dense TJs in hCMEC/D3 cells cocultured with CEM cells for 6 h (D) or absence of TJs in hCMEC/D3 cells cocultured with HUT-102 (E). Disruption of hCMEC/D3 monolayer after 6 h of coculture with C81-66 (F) or PBMCs from patient A (G). Bar represents 500 nm. Asterisk (∗) indicates the filter location of the Transwell device. Brackets indicate the local cell retraction observed.
By contrast, HTLV-1-infected cells (HUT-102 and C81-66 cell lines) strongly adhered to hCMEC/D3 cells (Fig. 3, B and C). In areas close to adherent infected lymphocytes, endothelial cell retraction was observed. Approximately 4% of the total surface of endothelial monolayers was involved in retraction areas, compared with 0.2% in control CEM cocultures, as calculated by NIH ImageJ software analysis performed in 12 different microscopic fields. Such cell retractions are a possible consequence of a decrease in cell-to-cell adhesion.
By transmission electron microscopy analysis, control endothelial cells showed close apposition to each other, with electron-dense areas at points of contact between adjacent cells, likely indicating the presence of adherent and tight junction (TJ) complexes (Fig. 3,D). No changes were observed in cocultures of hCMEC/D3 cells with CEM cells. At 6 h following initiation of coculture with HTLV-1-infected lymphocytic cell lines, a drastic decrease—or even absence in some preparations—in the number of intercellular junctional structures between hCMEC/D3 cells was observed (Fig. 3,E). As seen by scanning electron microscopy, cytopathic effects on hCMEC/D3 cells were more pronounced in the areas of close vicinity with infected lymphocytes (Fig. 3, F and G).
These data are consistent with an increase in the permeability of endothelial monolayer as a consequence of alterations of endothelial junctional integrity.
IL-1α and TNF-α treatment alters the expression of tight junctional proteins in hCMEC/D3 endothelial cells via MLC phosphorylation
To confirm the loss of integrity of TJ, we detected by immunofluorescence the localization of ZO-1 (a cytoplasmic TJ component) and actin filaments (that stabilize TJ structure). They both form a continuous staining along the membranes in control monolayers (hCMEC/D3 cells alone or cocultured with CEM cells) (Fig. 4, A and C). When hCMEC/D3 cells were cocultured with HTLV-1-infected lymphoid cells, the organization of the actin cytoskeleton was modified: the staining of cortical F-actin decreased, whereas the density of stress fibers increased (Fig. 4,B). Moreover, ZO-1 staining at the periphery of the cell became less continuous (Fig. 4 D). Such an alteration could be mostly prevented by addition in the medium of the inhibitors of IL-1 and TNF-α (data not shown).

Changes in ZO-1 and actin expression patterns during HTLV-1 infection. A and B, Detection of F-actin by bodipy-phalloidin staining on endothelial hCMEC/D3 cells cocultured with CEM (A) or with HUT-102 cells (B) for 6 h. Magnification, ×3000. C and D, Detection of ZO-1 by immunofluorescence on endothelial hCMEC/D3 cells cocultured with HUT-102 (D) or with CEM cells (C) for 6 h. Magnification, ×12,000. E, Immunoblot analysis of the expression levels of ZO-1, occludin, MLCK, and phosphorylation of MLC by hCMEC/D3 for 6 h following IL-1α and TNF-α treatment. β-Tubulin was used for normalization. F, Effect of inhibition of MLC phosphorylation on TJ protein levels and endothelial permeability of IL-1α- and TNF-α-treated hCMEC/D3. Cells were treated or not for 6 h with cytokines (100 pg/ml) with or without ML7, a specific inhibitor for MLCK activity. Western blotting was performed for ZO-1 and phosphorylated MLC. Simultaneously, the effect of the inhibitor on the permeability was estimated in hCMEC/D3 monolayers cocultured with C81-66 cells. Results are from triplicate cultures from one experiment representative of three.
Changes in ZO-1 and actin expression patterns during HTLV-1 infection. A and B, Detection of F-actin by bodipy-phalloidin staining on endothelial hCMEC/D3 cells cocultured with CEM (A) or with HUT-102 cells (B) for 6 h. Magnification, ×3000. C and D, Detection of ZO-1 by immunofluorescence on endothelial hCMEC/D3 cells cocultured with HUT-102 (D) or with CEM cells (C) for 6 h. Magnification, ×12,000. E, Immunoblot analysis of the expression levels of ZO-1, occludin, MLCK, and phosphorylation of MLC by hCMEC/D3 for 6 h following IL-1α and TNF-α treatment. β-Tubulin was used for normalization. F, Effect of inhibition of MLC phosphorylation on TJ protein levels and endothelial permeability of IL-1α- and TNF-α-treated hCMEC/D3. Cells were treated or not for 6 h with cytokines (100 pg/ml) with or without ML7, a specific inhibitor for MLCK activity. Western blotting was performed for ZO-1 and phosphorylated MLC. Simultaneously, the effect of the inhibitor on the permeability was estimated in hCMEC/D3 monolayers cocultured with C81-66 cells. Results are from triplicate cultures from one experiment representative of three.
We then studied the level of expression of ZO-1 and occludin (a transmembrane component of TJ) in hCMEC/D3 cells by immunoblotting. Because HTLV-1-infected lymphocytes themselves express some TJ proteins such as ZO-1 (data not shown), we evaluated the variations in expression of these proteins in hCMEC/D3 cells treated with IL-1α and TNF-α for 6 h, in the concentration ranges we found in the supernatant of infected lymphocytes. The levels of ZO-1 decreased after 5 h of cytokine treatment, reaching 50% of basal levels at 6 h; occludin expression was also partially decreased but to a lesser extent (15% at 6 h of coculture) (Fig. 4 E).
We also observed that treatment of human cerebral endothelial cells induce an increase of expression of MLCK with a maximum of expression at 3 h, followed by an increase in MLC phosphorylation level (Fig. 4,E). We wondered whether this pathway could be involved in TJ disorganization. We then tested the effect of the specific inhibition of MLCK activity with ML7 in endothelial permeability and ZO-1 expression. We observed that the inhibition of MLCK activity largely restored the permeability of endothelial monolayers exposed to infected C81-66 lymphocytes to basal levels (Fig. 4,F). Moreover, this prevented the decrease in ZO-1 levels and increase in MLC phosphorylation induced by proinflammatory cytokines treatment (Fig. 4 F).
Finally, changes in the expression of TJ proteins was investigated by immunofluorescence on sections from thoracic spinal cord from a HAM/TSP patient (8). In control spinal cord section from an uninfected patient, the ZO-1 immunoreactivity exhibited a classical continuous staining along blood vessels (Fig. 5,A). In a section from a HAM/TSP patient, ZO-1 exhibited a discontinuous pattern, ranging from almost conserved areas (Fig. 5,B) to some drastically disorganized (Fig. 5,C); in vivo BBB alteration may occur in some areas and do not concern the wide tissue. In contrast, occludin staining was continuous and almost similar in the different areas (Fig. 5, D–F).
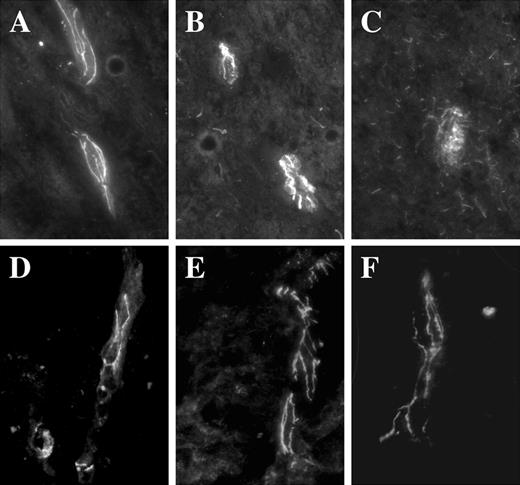
Expression patterns of TJ proteins in spinal cord sections from a HAM/TSP patient. A–C, Detection of ZO-1 by immunofluorescence on cryostat sections from spinal cord of a HAM/TSP patient (B and C) or uninfected patient (A). A more diffused pattern is observed in C when compared with unaffected areas (B), or to the pattern observed on uninfected patient (A). Magnification, ×1000. D–F, Detection of occludin by immunofluorescence on cryostated sections from spinal cords of a HAM/TSP patient (D–F). In all areas, the staining was continuous.
Expression patterns of TJ proteins in spinal cord sections from a HAM/TSP patient. A–C, Detection of ZO-1 by immunofluorescence on cryostat sections from spinal cord of a HAM/TSP patient (B and C) or uninfected patient (A). A more diffused pattern is observed in C when compared with unaffected areas (B), or to the pattern observed on uninfected patient (A). Magnification, ×1000. D–F, Detection of occludin by immunofluorescence on cryostated sections from spinal cords of a HAM/TSP patient (D–F). In all areas, the staining was continuous.
Discussion
BBB disruption induced by infected lymphocytes is an early event of the pathogenesis of retroviral-associated neuromyelopathies. BBB disruption during HIV-associated encephalitis and HIV-associated dementia has been extensively studied in recent years. Particularly, the role of HIV-infected monocytes and the viral Tat protein in altering cell junction expression has been pointed out (for a review, see Ref. 20). Concerning HTLV-1 infection, BBB alteration during HAM/TSP has been suggested by previous reports showing lymphocytic infiltrates (7, 8) and leakage of plasma proteins within the CNS parenchyma (8).
Our study is the first one, to our knowledge, to take advantage of an in vitro model of the human BBB (11), to elucidate the early steps of HAM/TSP pathogenesis.
We first demonstrate that endothelial monolayer permeability increase following coculture with lymphocytes that express NF-κB-activating Tax-1 protein. Interestingly, BBB disruption was an early event that did not require endothelial infection. Indeed, the time course of the barrier disruption was not consistent with the course of viral infection of endothelial cells. Furthermore, the nonproductively infected C81-66 cell line was able to induce the same deleterious effects as virion-producing cell lines.
We also show that proinflammatory cytokines, TNF-α and IL-1α, are together responsible for endothelial permeability increase at physiologically relevant concentrations (i.e., at the concentrations measured in the supernatant of chronically infected cells). There was a correlation between cytokines secretion levels and the kinetic of the disruption; in fact, barrier function impairment was delayed following coculture with PBMCs isolated from HAM/TSP patients when compared with HTLV-1-infected cell lines. The difference of the secretion level of cytokines may be due to a lower Tax-1 expression in PBMCs when compared with cell lines. Moreover, the increased endothelial permeability was prevented by blocking the biological action of either IL-1 or TNF-α. It is worth mentioning that, although IL-1α and TNF-α were previously reported to “open” the BBB, they were used in those studies at much higher concentrations, which did not allow to observe any synergy between these cytokines (19, 21, 22, 23, 24). Consistent with our results, a correlation between TNF-α levels in patient’s sera and progression to HAM/TSP had been previously reported (25). Whether other proinflammatory cytokines could contribute to the alteration of BBB function during HAM/TSP pathogenesis remains to be determined. A good candidate would be IFN-γ, which has been demonstrated to be a deleterious factor in the pathogenesis of other CNS inflammatory disorders, such as multiple sclerosis (26). However, in a rat model of HTLV-1-induced myelopathy, IFN-γ has been shown to play a protective role and to be secreted at a later stage of the disease (27).
Our data show that BBB disruption is associated with TJ disorganization, causing a decrease in endothelial cell-to-cell adhesion (detected by scanning electron microscopy), and loss of electron-dense junctional structures between endothelial cells (detected by transmission electron microscopy). Finally, the dissociation of TJs correlates with actin disorganization and degradation of junctional proteins such as ZO-1, and, to a lesser extent, occludin. This confirms that ZO-1 plays a crucial role in the integrity of TJ structures (28, 29).
Alteration of the blood-cerebrospinal fluid barrier was recently reported in an in vitro model of rat choroid plexus, through a TNF-α/IL-1α-mediated mechanism (30). However, at variance with the present study, the authors demonstrated in that case that the alteration of the barrier was associated with a reduction of the transporter-mediated cerebrospinal fluid-to-blood efflux of organic anions. In this study, we demonstrate that the mechanism of endothelial dysfunction is quite different. It is characterized by the disappearance of electron-dense junctional structures in endothelial cells, a decreased expression, and an altered localization of the TJ protein ZO-1, associated with loss of junctional integrity. Our in vitro observations are reminiscent of HAM/TSP pathogenesis as the disorganization of TJ in blood vessels could be seen in spinal cord sections from a HAM/TSP patient.
The mechanism mediating the cytokine-induced BBB alteration was largely dependent on the activation of MLCK. We report that MLCK was expressed downstream of NF-κB activation, in agreement with the known regulation of MLCK gene promoter by this pathway (31, 32). BBB disruption was prevented either by inhibiting MLCK synthesis (by inhibiting NF-κB pathway) or by inhibiting MLCK activity (by ML7). The role of MLCK in actin reorganization and permeability increase was previously described in several endothelial cell lines (33, 34, 35), but our report is the first one to describe such regulation in human cerebral endothelial cells.
In conclusion, we characterized in the present study the signaling cascades leading from adhesion of HTLV-1-infected lymphocytes on brain endothelial cells to endothelial barrier disruption, using an in vitro model of human BBB. We provided evidence that high level secretion by infected lymphocytes of proinflammatory cytokines (IL-1α and TNF-α) was directly responsible for BBB dysfunction via activation of the MLCK pathways leading to actin cytoskeleton and TJs alterations, and to permeability increase and monocyte transendothelial migration. We propose that this mechanism may mediate the massive lymphocyte infiltration observed in the CNS of HAM/TSP patients. Whether initial BBB disruption is maintained by similar or additional mechanisms at later stages of the disease is currently unknown. However, impairment of astrocytic function after contact with HTLV-1-infected T cells has been demonstrated to induce the secretion of proinflammatory cytokines, such as TNF-α and IL-1α (36, 37). This proinflammatory cytokine secretion by activated astrocytes could play a role in maintaining BBB disruption. However, our results suggest that preventing the elevation of IL-1α and TNF-α plasma concentrations may constitute a potential therapeutic strategy in this severe disease.
Acknowledgments
We thank E. Perret for help with the imaging and A. Mallet and S. Guadagnini for help with the scanning electron microscopy. We thank M. C. Cumont for help in obtaining and preparing tissue sections. We are grateful to J. M. Cavaillon and J. Estaquier for helpful discussions and advice.
Disclosures
The authors have no financial conflict of interest.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This work was supported by a grant from the Association pour la Recherche sur la Sclérose en Plaques and Institut Pasteur (Programme Transversal de Recherche 190). P.V.A. is a grant recipient from the Ministère de la Recherche (France).
Abbreviations used in this paper: BBB, blood-brain barrier; TJ, tight junction; ZO-1, zonula occludens 1; HTLV, human T cell leukemia virus; HAM/TSP, human T cell leukemia virus-associated myelopathy/tropical spastic paraparesis; MLC, myosin light chain; MLCK, myosin light chain kinase; h, human; VEGF, vascular endothelial growth factor; IL-1-Ra, IL-1 receptor antagonist.
