

Abstract
Neutrophilic airway inflammation is a hallmark of patients with severe asthma. Although we have reported that both IL-33 and IL-17A contributed to IgE-mediated neutrophilic inflammation in mice, the relationship remains unclear. In this article, we examined how IL-17A modifies IL-33–induced neutrophilic inflammation and airway hyperresponsiveness (AHR). IL-33 was intratracheally administered to BALB/c mice on days 0–2; furthermore, on day 7, the effect of the combination of IL-33 and IL-17A was evaluated. Compared with IL-33 or IL-17A alone, the combination exacerbated neutrophilic inflammation and AHR, associated with more increased levels of lung glutamic acid-leucine-arginine+ CXC chemokines, including CXCL1, CXCL2, and CXCL5, and infiltration by alveolar macrophages expressing CXCR2. Treatment with anti-CXCR2 mAb or depletion of alveolar macrophages repressed neutrophilic inflammation and AHR; in addition, depletion of neutrophils suppressed AHR. These findings prompted us to examine the role of CXCR2 in IgE-sensitized mice; a single treatment with anti-CXCR2 mAb in the seventh Ag challenge inhibited late-phase airway obstruction, AHR, and neutrophilic inflammation. In addition to inhibition, multiple treatments during the fourth to seventh challenge attenuated early-phase airway obstruction, eosinophilic inflammation, and goblet cell hyperplasia associated with the reduction of Th2 cytokine production, including IL-4, IL-5, and IL-13. Collectively, IL-33 cooperated with IL-17A to exacerbate AHR by enhancing neutrophilic inflammation via CXCR2 signaling; furthermore, CXCR2 signaling derived Th2 responses. We thus suggest the underlying mechanisms of IL-33 and IL-17A in allergic asthma and CXCR2 as potential therapeutic targets for the disease.
Introduction
Allergic asthma is a chronic inflammatory disorder of the airway associated with an increased level of allergen-specific IgE (1, 2). Although the unifying pathophysiological features of asthma are allergen-induced early- and late-phase asthmatic responses and airway hyperresponsiveness (AHR), which leads to airflow obstruction (2–4), the inflammatory status is complex and heterogeneous. For example, the airway granulocytic infiltrate of most asthmatics in the lungs is dominated by eosinophils (2, 3, 5); however, severe asthma is characterized by predominantly neutrophilic lung inflammation (6, 7). Currently, allergic models of asthma suggest that infiltration by eosinophils arises through the production of CD4+ Th2-type cytokines, such as IL-4, IL-5, and IL-13 in mice (8–10). Furthermore, IL-33 polarizes naive CD4+ T cells into a population of T cells that produce IL-5 (11) and induces the secretion of IL-5 and IL-13 from Th2 cells (12, 13), suggesting that IL-33 promotes a number of key inflammatory pathways that have the potential to initiate and propagate allergic inflammation. However, the Th2 paradigm alone does not seem to explain the full spectrum of asthma, because severe disease is not associated with an exclusive bias toward the production of Th2 cytokines (14, 15).
People with severe asthma have not only severe AHR and robust neutrophils, but also produce IL-17A (14, 15), and experimental mouse models of asthma suggest that IL-17A causes neutrophil recruitment and AHR (16, 17); therefore, it has been postulated that IL-17A production may drive severe forms of this disease. Meanwhile, glutamic acid-leucine-arginine (ELR)+ CXC chemokines contribute to neutrophil migration, and seven human ELR+ CXC chemokines have been identified: growth-related oncogene-α (CXCL1), -β (CXCL2), and -γ (CXCL3); epithelium-derived neutrophil-activating peptide 78 (CXCL5); granulocyte chemotactic protein 2 (CXCL6); neutrophil-activating peptide 2 (CXCL7); and IL-8 (CXCL8) (18). Among these chemokines, CXCL8 is the most potent neutrophil chemoattractant in humans, and its analogs in mice include CXCL1 (keratinocyte-derived chemokine), CXCL2 (MIP-2), and CXCL5 (LPS-induced CXC chemokine); these chemokines, which act through the receptor CXCR2, appear to confer selectivity for promoting neutrophil migration (18–22). In addition, administration of IL-17A to the lungs promotes neutrophilic lung inflammation, as well as the expression of ELR+ CXC chemokines such as CXCL1 and CXCL2 in mice (23, 24). More interestingly, there is a positive correlation between increased expression of ELR+ CXC chemokines such as CXCL8 in humans and the presence of neutrophils in the lungs of severe asthmatics (25, 26), and a clinical study has reported the possibility of CXCR2 as a potential therapeutic target in the disease (27); however, the functional significance and the pathways that regulate the expression of their chemokines in the disease are not fully defined.
We have previously reported that BALB/c mice passively sensitized with an i.p. injection of OVA-specific IgE mAb showed early- and late-phase increases in airway resistance and AHR by repeated intratracheal OVA challenges (28). In our previous (29, 30) and present studies, we demonstrated that the late-phase increase in airway resistance and AHR were closely associated with neutrophilic airway inflammation through IL-33 and IL-17A production. However, the precise relationship between IL-33 and IL-17A remains unclear. Therefore, we investigated how IL-17A modifies IL-33–induced neutrophilic inflammation and AHR, and found that repeated intratracheal administration of IL-33 induced neutrophilic inflammation and AHR, which were enhanced by the presence of IL-17A related to more increased levels of lung CXCL1, CXCL2, and CXCL5 and alveolar macrophages expressing CXCR2 in normal mice. Based on the data, we examined the roles of CXCR2 signaling in neutrophilic inflammation and AHR enhanced by the collaborative action of IL-33 and IL-17A in normal mice. In addition, in IgE-sensitized mice, the contribution of CXCR2 signaling to airway inflammatory responses was investigated.
Materials and Methods
Animals
Seven-week-old male BALB/c mice obtained from Japan SLC (Hamamatsu, Japan) were used in this study to remove confounding effects of menstrual cycle–dependent changes on endogenous female sex hormones. These mice were maintained in a temperature-controlled environment with free access to standard rodent chow and water. All of the experimental procedures were approved by the Experimental Animal Research Committee at Kobe Pharmaceutical University.
Coadministration of IL-33 and IL-17A and treatment with mAbs, dexamethasone, or liposomal clodronate
As shown Fig. 1A, we assessed the effect of coadministration of IL-33 and IL-17A (Biolegend, San Diego, CA) on the accumulation of inflammatory cells in bronchoalveolar lavage fluid (BALF) and AHR. IL-33 in solution (200 ng/mouse) was administered intratracheally to normal mice on days 0, 1, and 2 under anesthesia with isoflurane (Wako Pure Chemicals, Osaka, Japan) in a volume of 20 μl as reported previously (29). Furthermore, on day 7, IL-33 (200 ng/mouse), IL-17A (40 ng/mouse), or IL-33+IL-17A (200 + 40 ng/mouse) was administrated intratracheally, and the accumulation of inflammatory cells in BALF and AHR 24 h after the last instillation was evaluated on day 8. To examine the effect of initial IL-33 treatments, we administered IL-33 (200 ng/mouse), IL-17A (40 ng/mouse), or IL-33+IL-17A (200 + 40 ng/mouse) intratracheally after pretreatment with PBS instead of IL-33.

IL-17A enhances IL-33–induced neutrophilic inflammation and AHR associated with more increased levels of CXCL1, CXCL2, and CXCL5. (A) Experimental protocol for intratracheal administration of IL-33 and IL-17A in BALB/c mice. IL-33 was administered intratracheally to normal mice on days 0, 1, and 2; furthermore, on day 7, PBS, IL-33, IL-17A, or IL-33+IL-17A was administrated intratracheally. (B) Changes in inflammatory cell numbers in BALF 24 h after the coadministration of IL-33 and IL-17A. (C) Changes in airway responsiveness to MCh 24 h after the coadministration of IL-33 and IL-17A. (D) Changes in levels of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 3 and 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
IL-17A enhances IL-33–induced neutrophilic inflammation and AHR associated with more increased levels of CXCL1, CXCL2, and CXCL5. (A) Experimental protocol for intratracheal administration of IL-33 and IL-17A in BALB/c mice. IL-33 was administered intratracheally to normal mice on days 0, 1, and 2; furthermore, on day 7, PBS, IL-33, IL-17A, or IL-33+IL-17A was administrated intratracheally. (B) Changes in inflammatory cell numbers in BALF 24 h after the coadministration of IL-33 and IL-17A. (C) Changes in airway responsiveness to MCh 24 h after the coadministration of IL-33 and IL-17A. (D) Changes in levels of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 3 and 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
To evaluate the effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, or anti-CXCL5 mAb (R&D Systems, Minneapolis, MN) on the coadministration of IL-33– and IL-17–induced neutrophilic inflammation and AHR, we administered anti-CXCR2 mAb, anti-CXCL1 mAb, or anti-CXCL2 mAb (20 μg/mouse) intratracheally in a volume of 20 μl 30 min before coadministration; anti-CXCL5 mAb (60 μg/mouse) was administered intratracheally 3 h after coadministration, because CXCL5 production was not observed for at least 3 h after coadministration (Fig. 1). Control mice were given the same amount of rat IgG or rat IgG2b. In addition, a dose (20 μg/mouse) of anti–IL-6 mAb (Biolegend) or rat IgG1 was administered intratracheally 30 min before coadministration. Furthermore, in accordance with a previously reported method (29, 31), an i.p. administered dose (150 μg/mouse) of anti–Gr-1 mAb (Biolegend) or rat IgG2b was given 18 h before coadministration to deplete neutrophils; the dose of anti–Gr-1 mAb has been reported to fail to reduce the accumulation of other cells such as eosinophils and monocytes in BALF in a murine model of asthma (31).
Dexamethasone (3 mg/10 ml/kg; dexamethasone 21-phosphate disodium salt; Sigma-Aldrich, St. Louis, MO) was administered i.p. 2 h before coadministration. In addition, to evaluate the role of alveolar macrophages, we used a clodrosome macrophage depletion kit (Encapsula Nano Sciences, Nashville, TN). Liposomal clodronate suspension (500 μg/mouse) was administered intratracheally in a volume of 100 μl 3 h after the coadministration of IL-33 and IL-17A. Control mice were given the same amount of control liposome suspension.
Passive sensitization with OVA-specific IgE mAb and treatment with mAbs
OVA-specific IgE mAb (OE-1) was derived from a B cell hybridoma producing murine IgE, as described previously (28). The hybridoma was grown in CELLine CL1000 with BD-Cell-MAb medium (BD Biosciences, San Diego, CA) supplemented with 20% heat-inactivated FBS, 1% l-glutamine, and 1% penicillin-streptomycin. OE-1 levels in culture supernatants of hybridoma were assayed by ELISA (28). OE-1 levels were calculated by comparison with mouse IgE standards (Southern Biotech, Birmingham, AL).
Passive sensitization with OE-1 was performed with the protocol described previously (30). As shown in Fig. 6A, BALB/c mice were passively sensitized with repeated i.p. injections of a hybridoma supernatant containing OE-1 (100 μg/mouse) on days 0, 1, and 2. Nonsensitized mice were injected with a culture supernatant of the parental myeloma cell line. Both the sensitized and nonsensitized mice were challenged on days 1, 2, 3, 8, 9, 10, and 15 under isoflurane anesthesia with 1% OVA (grade V; Sigma-Aldrich) in a volume of 20 μl by intratracheal administration. In addition, we have reported that purified OE-1 induced allergic airway inflammation by repeated Ag challenges (29).
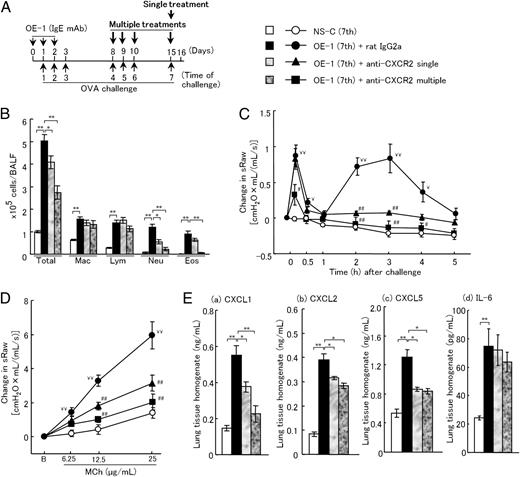
Treatment with anti-CXCR2 mAb inhibits IgE-mediated airway inflammatory responses. (A) Experimental protocol for sensitization with OE-1 and challenge with Ag, and treatment with anti-CXCR2 mAb. A single treatment with anti-CXCR2 mAb was intratracheally administered on day 15 to IgE-sensitized mice. In addition, multiple treatments with anti-CXCR2 mAb were intratracheally administered on days 8, 9, 10, and 15. Negative and positive controls were nonsensitized challenged and IgE-sensitized challenged, control rat IgG2a mAb-treated mice, respectively. (B–E) Effects of treatment with a single dose or multiple doses of anti-CXCR2 mAb on inflammatory cells in BALF (B), biphasic increase in airway resistance (C), AHR (D), and CXCL1 (a), CXCL2 (b), CXCL5 (c), or IL-6 production (d) in the lung tissue homogenates (E). Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01; ¥p < 0.05, ¥¥p < 0.01; or #p < 0.05, ##p < 0.01 compared with the NS-C (seventh) or OE-1 (seventh)+rat IgG2a group, respectively. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti-CXCR2 mAb inhibits IgE-mediated airway inflammatory responses. (A) Experimental protocol for sensitization with OE-1 and challenge with Ag, and treatment with anti-CXCR2 mAb. A single treatment with anti-CXCR2 mAb was intratracheally administered on day 15 to IgE-sensitized mice. In addition, multiple treatments with anti-CXCR2 mAb were intratracheally administered on days 8, 9, 10, and 15. Negative and positive controls were nonsensitized challenged and IgE-sensitized challenged, control rat IgG2a mAb-treated mice, respectively. (B–E) Effects of treatment with a single dose or multiple doses of anti-CXCR2 mAb on inflammatory cells in BALF (B), biphasic increase in airway resistance (C), AHR (D), and CXCL1 (a), CXCL2 (b), CXCL5 (c), or IL-6 production (d) in the lung tissue homogenates (E). Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01; ¥p < 0.05, ¥¥p < 0.01; or #p < 0.05, ##p < 0.01 compared with the NS-C (seventh) or OE-1 (seventh)+rat IgG2a group, respectively. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
A dose (20 μg/mouse) of anti-CXCR2 mAb or anti–IL-6 mAb was administered intratracheally 30 min before the seventh Ag challenge in OE-1–sensitized mice (Fig. 6A, single treatment). Furthermore, on days 8, 9, 10, and 15, a dose (20 μg/mouse) of anti-CXCR2 mAb or anti–IL-6 mAb was administered intratracheally 30 min before each of the Ag challenges to mice sensitized with OE-1 (Fig. 6A, multiple treatments). Control mice were given the same amount of rat IgG2a, rat IgG1, or rat IgG on days 8, 9, 10, and 15.
In accordance with a previously reported method (29, 30), a dose (5 μg/mouse) of anti–IL-33 mAb (R&D Systems) or rat IgG2a was administered intratracheally 30 min before each of the challenges on days 8, 9, 10, and 15 in mice sensitized with OE-1. Furthermore, a dose (150 μg/mouse) of anti–IL-17 mAb (R&D Systems) or rat IgG2a was administered i.p. 30 min before the seventh challenge.
Analysis of cells recovered by BAL
To evaluate airway cellular inflammation, we examined the accumulation of inflammatory cells in BALF as detailed previously (32). Animals were killed with diethyl ether. The trachea was cannulated and the left bronchi were tied. The right air lumen was washed twice with 0.5 ml HBSS containing 2% FBS. The cell pellet was suspended in a defined volume (200 μl/sample) of HBSS containing 2% FBS. The total leukocyte count in the lavage fluid was determined by staining with Turk’s solution. For differential cell counts, BAL cells were stained with Diff-Quik solution (Sysmex International Reagents, Kobe, Japan).
Measurement of AHR to methacholine
To estimate AHR to methacholine (MCh), we measured specific airway resistance (sRaw; cmH2O × mL/[mL/s]) before and after the intratracheal instillation of an MCh using a two-chambered, double-flow plethysmograph system (Pulmos-I; M.I.P.S., Osaka, Japan) according to the method by Pennock et al. (33). In brief, increasingly higher doses of MCh (6.25, 12.5, and 25 μg/ml) in solution were administered consecutively via the intratracheal route to mice under isoflurane-induced anesthesia at 30-min intervals. sRaw was measured 2 min after the respective instillations of the three doses of MCh solution.
Measurement of airway resistance
To evaluate the degree of early- and late-phase increases in airway resistance, we measured sRaw before and 10 min to 5 h after the seventh challenge in IgE-sensitized mice.
Measurement of cytokines in lung tissue supernatants
The frozen right lobe (after BAL) was homogenized in 1 ml T-PER (Thermo Scientific, Rockford, IL) containing a Complete Mini Protease Inhibitor Cocktail tablet (Roche, Mannheim, Germany; 1 tablet/10 ml T-PER stock reagent). Lung homogenates were centrifuged at 9000 × g for 10 min at 4°C. CXCL1, CXCL2, CXCL5, IL-4, IL-5, IL-13 (R&D Systems), IL-17A, IL-33, and IL-6 (Biolegend) in supernatants of lung homogenates were measured using quantitative colormetric sandwich ELISA kits. The sensitivity of these assays was 2 pg/ml (CXCL1), 1.5 pg/ml (CXCL2), 0.347 pg/ml (CXCL5), 2 pg/ml (IL-4), 7 pg/ml (IL-5), 1.5 pg/ml (IL-13), 2.7 pg/ml (IL-17A), 15.3 pg/ml (IL-33), and 2 pg/ml (IL-6).
Histological analysis
The left lungs were fixed in 10% neutral-buffered formalin, then dissected, embedded in paraffin, and cut 4 μm thick. Sections were stained with H&E and periodic acid–Schiff (PAS). Scoring for each section was evaluated on a scale of 0 to 4 with increments of 0.5 by a blinded observer for inflammation (H&E) and goblet cell hyperplasia (PAS), as previously reported (32).
Flow cytometry analysis
The whole lung was isolated, cut into 1-mm3 pieces in digestion buffer (RPMI 1640 containing 150 U/ml collagenase [Wako Pure Chemicals], 30 μg/ml DNase I [Sigma-Aldrich], and 10 mM HEPES), and incubated at 37°C for 1 h, as previously reported (30). The resulting single-cell suspension was washed by centrifugation with PBS supplemented with 2% FBS, and cell numbers were determined using trypan blue staining after treatment with ACK lysis buffer to remove erythrocytes.
Leukocytes recovered from collagenase/DNase I–digested lung tissue were incubated with anti-mouse FcγRII/III mAb followed by PerCP-Cy5.5–labeled anti-CXCR2 mAb, FITC-labeled anti-CD11c mAb, and PE-labeled anti-F4/80 mAb (Biolegend). After washing, the stained cells were fixed with 4% paraformaldehyde and then analyzed using FACSCalibur (BD Biosciences) and Cell Quest software (version 3.3; BD Biosciences). For detecting CXCR2+ cells in lung tissues, the cells were gated using side scatter and CXCR2 expression; furthermore, in the cells, alveolar macrophages were sorted based on their expression of F4/80 and CD11c, as shown in a previous report (34). In addition, to measure the absolute number of CXCR2+F4/80+CD11c+ cells in the lungs, the percentage of CXCR2+F4/80+CD11c+ cells was multiplied by the number of total cells in the lungs.
Measurement of OVA-specific IgE Ab in serum
OVA-specific IgE Ab in serum was measured by ELISA, as described previously (28). OVA-specific IgE Ab in serum was detected using plates coated with anti-mouse IgE Ab and adding biotin-labeled OVA. Streptavidin-HRP was added, the plate was developed with 3,3′,5,5′-tetramethylbenzidine solution, and measurements were made at 450 nm using a microplate reader after stopping the reaction with sulfuric acid. Values for serum OVA-specific IgE (1:5) were expressed as absorbance units.
Statistical analyses
Data are shown as the mean ± SEM. Statistical analyses between the two groups were performed using Student t test (two-tailed). To compare more than two groups, we used Dunnett’s test after conducting one-way ANOVA. A p value <0.05 was considered significant.
Results
IL-17A enhances IL-33–induced neutrophilic inflammation and AHR associated with more increased levels of CXCL1, CXCL2, and CXCL5
We have found that in IgE-sensitized mice, not only anti–IL-33 mAb but also anti–IL-17A mAb decreased neutrophilic airway inflammation and AHR; anti–IL-33 mAb failed to inhibit IL-17A production in the lungs (29, 30) (Supplemental Fig. 1). In addition, repeated intratracheal administration of IL-33 alone on days 0, 1, 2, and 7 did not induce IL-17A production in the lungs on day 8 (PBS versus IL-33: 91.7 ± 4.1 versus 97.8 ± 4.9 pg/ml; n = 5). Meanwhile, the production of IL-33 in the lungs of IgE-sensitized mice was significant 24 h after the first and third challenges in comparison with that in nonsensitized mice; a higher level was recognized 24 h after the fourth challenge. In addition, IL-17A production was significant 24 h after the fourth challenge in comparison with that in nonsensitized mice, although no production was seen 24 h after the first and third challenges (Supplemental Fig. 2). Based on these data, we hypothesized that IL-33–induced neutrophilic inflammation and AHR could be enhanced by the presence of IL-17A; furthermore, the condition of IL-33–induced airway inflammation may need to be considered in the interaction with IL-33 and IL-17A. Therefore, before the coadministration of IL-33 and IL-17, IL-33 alone was administered repeatedly; as a result, repeated intratracheal administration of IL-33 induced neutrophilic inflammation and AHR, which were enhanced in combination with IL-17A compared with IL-33 or IL-17A alone–treated mice (Fig. 1B, 1C); infiltration by alveolar macrophages and lymphocytes in IL-33+IL-17A–treated mice was increased in comparison with PBS-treated mice. In addition, sRaw values before the administration of MCh on day 8 were 2.258 ± 0.077 in normal, 2.256 ± 0.083 in PBS-treated, 2.217 ± 0.093 in IL-33–treated, 2.135 ± 0.068 in IL-17A–treated, and 2.272 ± 0.094 in IL-33+IL-17A–treated groups. Throughout the earlier experiment, no significant differences in sRaw before administration were found among the groups. Meanwhile, there were no differences in neutrophilic inflammation between IL-33 alone, IL-17A alone, or IL-33+IL-17A with pretreatment with PBS instead of IL-33 groups; in addition, neutrophilic inflammation by IL-33+IL-17A without IL-33 pretreatment was very weak compared with that by coadministration with IL-33 pretreatment (Table I).
| No. of Cells (×105 Cells/BALF) | ||
|---|---|---|
| Total Cells | Neutrophils | |
| PBS alone | 1.65 ± 0.06 | 0.06 ± 0.02 |
| IL-33 (with PBS) | 2.88 ± 0.12 | 0.29 ± 0.07 |
| IL-17A (with PBS) | 2.97 ± 0.31 | 0.13 ± 0.07 |
| IL-33+IL-17A (with PBS) | 2.98 ± 0.25 | 0.33 ± 0.09 |
| IL-33+IL-17A (with IL-33) | 16.72 ± 0.57* | 8.54 ± 0.20* |
| No. of Cells (×105 Cells/BALF) | ||
|---|---|---|
| Total Cells | Neutrophils | |
| PBS alone | 1.65 ± 0.06 | 0.06 ± 0.02 |
| IL-33 (with PBS) | 2.88 ± 0.12 | 0.29 ± 0.07 |
| IL-17A (with PBS) | 2.97 ± 0.31 | 0.13 ± 0.07 |
| IL-33+IL-17A (with PBS) | 2.98 ± 0.25 | 0.33 ± 0.09 |
| IL-33+IL-17A (with IL-33) | 16.72 ± 0.57* | 8.54 ± 0.20* |
Each value is the mean ± SEM of four animals.
*p < 0.01 compared with the IL-33+IL-17A (with PBS) group.
ELR+ CXC chemokines, including CXCL1, CXCL2, and CXCL5 in mice, are a major subclass of chemokines involved in the recruitment of neutrophils (19–22). Therefore, we focused on the roles of these chemokines in AHR and neutrophilic inflammation induced by coadministration of IL-33 and IL-17A. Meanwhile, it has been reported that CXCL1 and CXCL2 chemokine production in the lungs peaked 3 h after the Ag challenge in a murine model of asthma (22); in contrast, the level of CXCL5 in BALF peaked 24 h after the Escherichia coli challenge in mice (35). Therefore, we examined whether coadministration of IL-33 and IL-17A exacerbated the production of these chemokines at 3 and 24 h in comparison with IL-33 or IL-17A alone–treated mice. Significantly higher levels of CXCL1, CXCL2, and CXCL5 were observed 24 h after the coadministration of IL-33 and IL-17A compared with IL-33 or IL-17A alone–treated mice, although levels of these chemokines 3 h after administration showed no difference among the groups (Fig. 1D).
Treatment with anti-CXCR2 mAb inhibits neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A
The results shown in Fig. 1 indicate that IL-17A enhances IL-33–induced neutrophilic inflammation and AHR associated with more increased production of ELR+ CXC chemokines. Therefore, we investigated the role of CXCR2 signaling in these IL-33 and IL-17A coadministration-induced responses. The development of neutrophilic inflammation and AHR induced by coadministration were strongly inhibited by anti-CXCR2 mAb; in addition, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb partially reduced them (Fig. 2A, 2B, 2D, 2E). Furthermore, anti-CXCR2 mAb suppressed CXCL1, CXCL2, and CXCL5 production in the lungs; in addition, anti-CXCL1 mAb decreased CXCL2 and CXCL5 production, although anti-CXCL2 mAb or anti-CXCL5 mAb failed to inhibit CXCL1 and CXCL5 or CXCL1 and CXCL2, respectively (Fig. 2C, 2F). In addition, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb significantly decreased CXCL1, CXCL2, and CXCL5 levels in the lungs, respectively (Fig. 2C, 2F).

Treatment with anti-CXCR2 mAb inhibits neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A. (A, B, D, and E) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on neutrophilic inflammation (A and D) and AHR (B and E) 24 h after the coadministration of IL-33 and IL-17A. (C and F) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on the production of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG or rat IgG2b group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti-CXCR2 mAb inhibits neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A. (A, B, D, and E) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on neutrophilic inflammation (A and D) and AHR (B and E) 24 h after the coadministration of IL-33 and IL-17A. (C and F) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on the production of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 5–12 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG or rat IgG2b group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti–IL-6 mAb inhibits neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A
It has been reported that IL-6 regulates neutrophil trafficking during inflammatory responses (36). Therefore, we examined whether IL-6 contributes to neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A. Although coadministration of IL-33 and IL-17A induced IL-6 production in the lungs, the level was similar to IL-33 or IL-17A alone–treated mice (Fig. 3A). However, IL-6 production was suppressed by anti-CXCR2 mAb and anti-CXCL1 mAb, but not anti-CXCL2 mAb and anti-CXCL5 mAb (Fig. 3B). Furthermore, treatment with anti–IL-6 mAb suppressed AHR and neutrophilic inflammation (Fig. 3C, 3D) although CXCL1, CXCL2, and CXCL5 production in the lungs was not decreased (Fig. 3E).
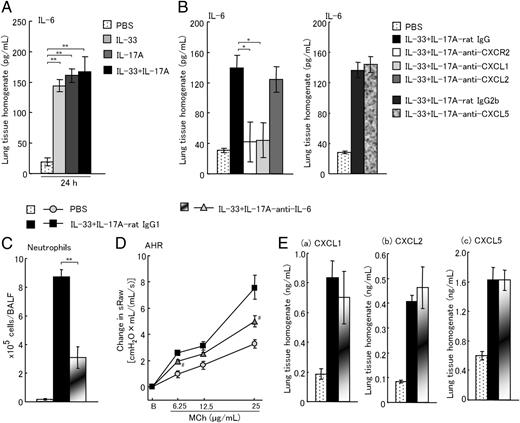
Treatment with anti–IL-6 mAb inhibits neutrophilic inflammation and AHR induced by the coadministration of IL-33 and IL-17A. (A) Change in level of IL-6 in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (B) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on the production of IL-6 in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (C and D) Effects of anti–IL-6 mAb on neutrophilic inflammation (C) and AHR (D) 24 h after the coadministration of IL-33 and IL-17A. (E) Effects of anti–IL-6 mAb on the production of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG1 group. Eos, eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti–IL-6 mAb inhibits neutrophilic inflammation and AHR induced by the coadministration of IL-33 and IL-17A. (A) Change in level of IL-6 in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (B) Effects of anti-CXCR2 mAb, anti-CXCL1 mAb, anti-CXCL2 mAb, and anti-CXCL5 mAb on the production of IL-6 in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (C and D) Effects of anti–IL-6 mAb on neutrophilic inflammation (C) and AHR (D) 24 h after the coadministration of IL-33 and IL-17A. (E) Effects of anti–IL-6 mAb on the production of CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG1 group. Eos, eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Depletion of alveolar macrophages inhibits neutrophilic inflammation and AHR induced by coadministration of IL-33 and IL-17A
As shown earlier, CXCL1, CXCL2, CXCL5, and IL-6 production, which contribute to neutrophil recruitment, were produced via CXCR2 signaling (Figs. 2, 3). It has been reported that high levels of CXCL1 and CXCL2 induced by OVA challenge were observed in alveolar macrophages of mice sensitized with OVA (37). Based on this information, we hypothesized that chemokine and/or cytokine production may be induced via alveolar macrophages expressing CXCR2 increased by coadministration of IL-33 and IL-17A. First, we assessed whether CXCR2 expression was increased on alveolar macrophages in the lungs of IL-33+IL-17A–treated mice. The absolute number of alveolar macrophages (CD11c+F4/80+) expressing CXCR2 was calculated by multiplying by total cells in the lungs and percentages of the alveolar macrophages expressing CXCR2; in addition, the number of total cells in the lungs was 11.75 ± 0.44 × 106 cells/lung in PBS-treated, 13.36 ± 0.74 × 106 cells/lung in IL-33–treated, 14.93 ± 0.75 × 106 cells/lung in IL-17A–treated, and 24.64 ± 2.36 × 106 cells/lung in IL-33+IL-17A–treated groups. Percentages and absolute cell numbers of alveolar macrophages (CD11c+ F4/80+) expressing CXCR2 in the lungs of IL-33 alone–, IL-17A alone–, and IL-33+IL-17A–treated groups were increased in comparison with the PBS-treated group (Fig. 4A). Furthermore, the absolute cell numbers of alveolar macrophages (CD11c+ F4/80+) expressing CXCR2 in the IL-33+IL-17A–treated group were increased compared with the IL-33 or IL-17A alone–treated group, although the percentages did not change among the groups (Fig. 4A). Therefore, we investigated the effects of the depletion of alveolar macrophages by liposomal clodronate on neutrophilic inflammation, AHR, and the production of ELR+ CXC chemokine and IL-6 induced by coadministration of IL-33 and IL-17A. The treatment inhibited neutrophilic inflammation and AHR; furthermore, CXCL1 and CXCL2 production in the lungs was suppressed, but not CXCL5 and IL-6 (Fig. 4B–D). In addition, the number of alveolar macrophages was significantly reduced in the IL-33+IL-17A–clodronate–treated group (2.61 ± 0.41 × 105 cells/BALF, p < 0.05) in comparison with the IL-33+IL-17A vehicle–treated group (4.75 ± 0.36 × 105 cells/BALF).

Depletion of alveolar macrophages inhibits neutrophilic inflammation and AHR induced by the coadministration of IL-33 and IL-17A. (A) Representative dot plots depicting the percentages of alveolar macrophages expressing CXCR2 (CXCR2+CD11c+F4/80+) in the lungs 24 h after the coadministration of IL-33 and IL-17A (a). Changes in the number of CXCR2+CD11c+F4/80+ cells in the lungs of PBS, IL-33, IL-17, and IL-33+IL-17 groups (b). (B and C) Effects of liposomal clodronate on neutrophilic inflammation (B) and AHR (C) 24 h after the coadministration of IL-33 and IL-17A. (D) Effects of liposomal clodronate on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A vehicle group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Depletion of alveolar macrophages inhibits neutrophilic inflammation and AHR induced by the coadministration of IL-33 and IL-17A. (A) Representative dot plots depicting the percentages of alveolar macrophages expressing CXCR2 (CXCR2+CD11c+F4/80+) in the lungs 24 h after the coadministration of IL-33 and IL-17A (a). Changes in the number of CXCR2+CD11c+F4/80+ cells in the lungs of PBS, IL-33, IL-17, and IL-33+IL-17 groups (b). (B and C) Effects of liposomal clodronate on neutrophilic inflammation (B) and AHR (C) 24 h after the coadministration of IL-33 and IL-17A. (D) Effects of liposomal clodronate on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A vehicle group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Depletion of neutrophils inhibits AHR induced by coadministration of IL-33 and IL-17A
Subsequently, we examined the role of neutrophils in AHR and ELR+ CXC chemokine and IL-6 production, because neutrophils highly expressed CXCR2 (38). The depletion of neutrophils by treatment with anti–Gr-1 mAb suppressed AHR in IL-33+IL-17A–treated mice (Fig. 5A, 5B); furthermore, this treatment suppressed IL-6 production in the lungs, but not CXCL1, CXCL2, and CXCL5 (Fig. 5C). Meanwhile, steroids are considered one of the most effective treatments for asthma. Therefore, we assessed the effect of dexamethasone on neutrophilic inflammation and AHR. As shown in Fig. 5D–F, neutrophilic inflammation, AHR, and CXCL1, CXCL2, CXCL5, and IL-6 production were not suppressed by this treatment.

Depletion of neutrophils inhibits AHR and IL-6 production induced by the coadministration of IL-33 and IL-17A. (A and B) Effects of anti–Gr-1 mAb on neutrophilic inflammation (A) and AHR (B) 24 h after the coadministration of IL-33 and IL-17A. (C) Effects of anti–Gr-1 mAb on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (D and E) Effects of dexamethasone on neutrophilic inflammation (D) and AHR (E) 24 h after the coadministration of IL-33 and IL-17A. (F) Effects of dexamethasone on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of four to five animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG2b. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Depletion of neutrophils inhibits AHR and IL-6 production induced by the coadministration of IL-33 and IL-17A. (A and B) Effects of anti–Gr-1 mAb on neutrophilic inflammation (A) and AHR (B) 24 h after the coadministration of IL-33 and IL-17A. (C) Effects of anti–Gr-1 mAb on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. (D and E) Effects of dexamethasone on neutrophilic inflammation (D) and AHR (E) 24 h after the coadministration of IL-33 and IL-17A. (F) Effects of dexamethasone on the production of CXCL1 (a), CXCL2 (b), CXCL5 (c), and IL-6 (d) in the lung tissue homogenates 24 h after the coadministration of IL-33 and IL-17A. Each value is the mean ± SEM of four to five animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01 compared with IL-33+IL-17A rat IgG2b. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti-CXCR2 mAb or anti–IL-6 mAb inhibits IgE-mediated airway inflammatory responses
In IgE-sensitized mice, treatment with anti–IL-33 mAb or anti–IL-17A mAb inhibited CXCL1, CXCL2, and CXCL5 production in the lungs 24 h after the seventh Ag challenge (Table II). Therefore, we examined whether CXCR2 signaling is also important for the development of IgE-mediated airway inflammatory responses. Treatment with a single dose or multiple doses of anti-CXCR2 mAb significantly suppressed neutrophilic accumulation in BALF, the late-phase increase in airway resistance, and AHR; in addition, CXCL1, CXCL2, and CXCL5, but not IL-6, were decreased by this treatment (Fig. 6B–E). Furthermore, multiple treatments inhibited the early-phase increase in airway resistance and eosinophilic inflammation, although the single treatment did not show inhibitory effects on these responses (Fig. 6B, 6C). In addition, throughout the earlier experiment, sRaw values before the seventh challenge were 2.484 ± 0.036 in the nonsensitized challenged, 2.505 ± 0.050 in the IgE-sensitized challenged rat IgG2a-treated, 2.508 ± 0.007 in the IgE-sensitized challenged anti-CXCR2 mAb single-treated, and 2.515 ± 0.024 in the IgE-sensitized challenged anti-CXCR2 mAb multiple-treated groups, indicating that no significant differences in sRaw before the challenge were found among the groups. Meanwhile, treatment with a single dose or multiple doses of anti–IL-6 mAb showed similar inhibitory effects of anti-CXCR2 mAb on the biphasic increase in airway resistance, AHR, and neutrophilic and eosinophilic inflammation (Fig. 7A–C). Although single treatment with anti–IL-6 mAb did not suppress CXCL1, CXCL2, and CXCL5 production, multiple treatments inhibited only CXCL1 production (Fig. 7D).
| CXCL1 | CXCL2 | CXCL5 | |
|---|---|---|---|
| Anti–IL-33 mAb | |||
| NS-C (7th) | 0.09 ± 0.01 | 0.06 ± 0.01 | 0.62 ± 0.03 |
| OE-1 (7th) + rat IgG2a | 0.78 ± 0.11* | 0.52 ± 0.05* | 2.43 ± 0.20* |
| OE-1 (7th) + anti–IL-33 | 0.50 ± 0.03** | 0.27 ± 0.08** | 1.43 ± 0.13** |
| Anti–IL-17A mAb | |||
| OE-1 (7th) + rat IgG2a | 0.67 ± 0.04 | 0.34 ± 0.04 | 2.23 ± 0.14 |
| OE-1 (7th) + anti–IL-17A | 0.43 ± 0.05** | 0.22 ± 0.03** | 1.39 ± 0.11** |
| CXCL1 | CXCL2 | CXCL5 | |
|---|---|---|---|
| Anti–IL-33 mAb | |||
| NS-C (7th) | 0.09 ± 0.01 | 0.06 ± 0.01 | 0.62 ± 0.03 |
| OE-1 (7th) + rat IgG2a | 0.78 ± 0.11* | 0.52 ± 0.05* | 2.43 ± 0.20* |
| OE-1 (7th) + anti–IL-33 | 0.50 ± 0.03** | 0.27 ± 0.08** | 1.43 ± 0.13** |
| Anti–IL-17A mAb | |||
| OE-1 (7th) + rat IgG2a | 0.67 ± 0.04 | 0.34 ± 0.04 | 2.23 ± 0.14 |
| OE-1 (7th) + anti–IL-17A | 0.43 ± 0.05** | 0.22 ± 0.03** | 1.39 ± 0.11** |
Values are in ng/ml. Anti–IL-33 mAb was intratracheally administered on days 8, 9, 10, and 15 (OE-1 [seventh] + anti–IL-33). Negative and positive controls were nonsensitized challenged (NS-C [seventh]) and OE-1–sensitized challenged, control rat IgG2a mAb-treated (OE-1 [seventh] + rat IgG2a) mice, respectively. Furthermore, anti–IL-17A mAb was i.p. administered on day 15 (OE-1 [seventh] + anti–IL-17A). Positive control was OE-1–sensitized challenged, control rat IgG2a mAb-treated (OE-1 [seventh] + rat IgG2a) mice. Each value is the mean ± SEM of 5–11 animals.
*p < 0.01 compared with the NS-C (seventh) group, **p < 0.05 compared with the OE-1 (7th) + rat IgG2a group.

Treatment with anti–IL-6 mAb inhibits IgE-mediated airway inflammatory responses. (A–D) Effects of treatment with a single dose or multiple doses of anti–IL-6 mAb on inflammatory cells in BALF (A), biphasic increase in airway resistance (B), AHR (C), and CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates (D). Each value is the mean ± SEM of five to six animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01, compared with the OE-1 (seventh)+rat IgG1 group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Treatment with anti–IL-6 mAb inhibits IgE-mediated airway inflammatory responses. (A–D) Effects of treatment with a single dose or multiple doses of anti–IL-6 mAb on inflammatory cells in BALF (A), biphasic increase in airway resistance (B), AHR (C), and CXCL1 (a), CXCL2 (b), and CXCL5 (c) in the lung tissue homogenates (D). Each value is the mean ± SEM of five to six animals. *p < 0.05, **p < 0.01; #p < 0.05, ##p < 0.01, compared with the OE-1 (seventh)+rat IgG1 group. Eos, Eosinophils; Lym, lymphocytes; Mac, macrophages; Neu, neutrophils; total, all cells.
Furthermore, airway inflammation (H&E) and goblet cell hyperplasia (PAS) in the lungs were inhibited by multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb (Fig. 8A, 8B). In addition, multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb suppressed OVA-specific IgE Ab (Fig. 9A), IL-5, and IL-13 (Fig. 9B); anti-CXCR2 mAb, but not anti–IL-6 mAb, inhibited IL-4 production (Fig. 9B).

Multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb inhibit IgE-mediated airway inflammation and goblet cell hyperplasia. (A and B) Effects of multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb on IgE-mediated airway inflammation (H&E) (A) and goblet cell hyperplasia (PAS) (B) in OE-1 (7th)+rat IgG (a), OE-1 (7th)+anti-CXCR2 mAb (b), or OE-1 (7th)+anti–IL-6 mAb groups (c). Scale bars, 100 μm. Histological appearance 24 h after the seventh challenge was scored for inflammation and goblet cell hyperplasia (d). Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01.
Multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb inhibit IgE-mediated airway inflammation and goblet cell hyperplasia. (A and B) Effects of multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb on IgE-mediated airway inflammation (H&E) (A) and goblet cell hyperplasia (PAS) (B) in OE-1 (7th)+rat IgG (a), OE-1 (7th)+anti-CXCR2 mAb (b), or OE-1 (7th)+anti–IL-6 mAb groups (c). Scale bars, 100 μm. Histological appearance 24 h after the seventh challenge was scored for inflammation and goblet cell hyperplasia (d). Each value is the mean ± SEM of 4–10 animals. *p < 0.05, **p < 0.01.

Multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb inhibit IgE-mediated Ag-specific IgE Ab and Th2 cytokine production. (A and B) Effects of multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb on IgE-mediated endogenous Ag-specific IgE Ab production in the serum (A) and Th2 cytokine production including IL-4 (a), IL-5 (b), and IL-13 (c) in the lung tissue homogenates (B). Each value is the mean ± SEM of 4–12 animals. *p < 0.05, **p < 0.01.
Multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb inhibit IgE-mediated Ag-specific IgE Ab and Th2 cytokine production. (A and B) Effects of multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb on IgE-mediated endogenous Ag-specific IgE Ab production in the serum (A) and Th2 cytokine production including IL-4 (a), IL-5 (b), and IL-13 (c) in the lung tissue homogenates (B). Each value is the mean ± SEM of 4–12 animals. *p < 0.05, **p < 0.01.
Discussion
Allergic asthma has long been considered an eosinophilic bronchitis; however, its symptoms were not alleviated by a marked reduction in eosinophil numbers in blood and the airway by treatment with anti–IL-5 in clinical cases (39, 40). Some asthmatics, particularly those who have severe asthma (6, 40, 41) and are resistant to corticosteroids (42), have raised neutrophil counts in their airways, suggesting neutrophils to be a more valid target than eosinophils in certain stages of the pathogenesis. Meanwhile, although we have found that not only IL-33 but also IL-17A-secreting Th17 cells contributed to IgE-mediated neutrophilic inflammation (29, 30), the relationship between IL-33 and IL-17A has not yet been defined. In this study, we focused on how IL-17A modifies IL-33–induced neutrophilic inflammation and AHR; as a result, repeated intratracheal administration of IL-33 induced neutrophilic inflammation and AHR, which were enhanced in combination with IL-17A associated with more increased levels of lung CXCL1, CXCL2, and CXCL5 and alveolar macrophages expressing CXCR2. Treatment with anti-CXCR2 mAb or depletion of alveolar macrophages suppressed neutrophilic inflammation and AHR; in addition, depletion of neutrophils inhibited AHR, suggesting the importance of CXCR2 signaling to regulate the cooperative action of IL-33– and IL-17A–enhanced AHR via neutrophilic inflammation. Based on these data, we attempted to examine the role of CXCR2 in IgE-sensitized mice; a single treatment with anti-CXCR2 mAb at the seventh Ag challenge inhibited late-phase airway obstruction, AHR, and neutrophilic inflammation. Thus, we propose the mechanism that IL-17A exacerbates IL-33–induced AHR by enhancing neutrophilic inflammation via CXCR2 signaling in allergic asthma.
Treatment with anti–IL-33 mAb or anti–IL-17A mAb inhibited neutrophilic inflammation and AHR in IgE-sensitized mice (29, 30) (Supplemental Fig. 1). Therefore, we hypothesized that the interaction with IL-33 and IL-17A may enhance neutrophilic inflammation and AHR. Furthermore, IL-33 expression in the lungs was observed during the first to fourth Ag challenges, although IL-17 was recognized only in the fourth challenge in IgE-sensitized mice (Supplemental Fig. 2), suggesting that the condition of IL-33–induced airway inflammation may need to be considered in the interaction with IL-33 and IL-17A. Therefore, before the coadministration of IL-33 and IL-17, IL-33 alone was administered repeatedly; as a result, repeated intratracheal administration of IL-33 induced neutrophilic inflammation and AHR, which were strongly enhanced by the presence of IL-17A (Fig. 1). In addition, as expected, there was no enhancement of neutrophilic inflammation by coadministration of IL-33 and IL-17A without IL-33 pretreatments compared with IL-33 or IL-17A alone–treated mice (Table I). In the previous study (30), we found that repeated administration of IL-33 alone induced airway inflammation related to Th2 cytokine production in the lungs, such as IL-4, IL-5, and IL-13. Several cell types, including CD4+ Th2 cells and type 2 innate lymphocyte cells (ILC2s) producing Th2 cytokines, are targets for IL-33; indeed, IL-33–responsive adaptive immune cells, such as allergen-specific Th2 cells, and innate cells, such as ILC2s, cooperate to induce allergic airway inflammation (43, 44). Thus, initial IL-33–responsive Th2 cells and ILC2s may elicit changes that conditioned the mice to a quantitatively different response as a consequence of the coadministration of IL-33 and IL-17A. Meanwhile, consistent with previous data (29), in which the depletion of neutrophils inhibited AHR in IgE-sensitized mice, the development of AHR induced by the coadministration of IL-33 and IL-17A was also inhibited by the depletion, suggesting that the increased numbers of neutrophils in the lungs are associated with the development of AHR. The finding that activated neutrophils release a large array of inflammatory mediators, oxygen radicals, and protease has lent support to the notion of their involvement in the intense inflammation found in severe asthma (5, 45).
It has been reported that ELR+ CXC chemokines such as CXCL1, CXCL2, and CXCL5 mainly contribute to neutrophilic inflammation in mice (19–22). Therefore, we examined the relationship between IL-33 and IL-17A in the production of CXCL1, CXCL2, and CXCL5. Repeated intratracheal administration of IL-33 induced CXCL1, CXCL2, and CXCL5, which were enhanced by the presence of IL-17A (Fig. 1), suggesting the possibility that more increased ELR+ CXC chemokine production induced by IL-33 and IL-17A may enhance the development of AHR via neutrophilic inflammation. Therefore, we investigated the role of CXCR2 in AHR and neutrophilic inflammation. Treatment with anti-CXCR2 mAb suppressed neutrophilic inflammation and AHR in mice treated with IL-33 and IL-17A (Fig. 2). Thus, the cooperative action of IL-33 and IL-17A exacerbates AHR by enhancing neutrophilic inflammation induced by CXCR2 signaling.
Furthermore, we examined the detailed interaction with CXCL1, CXCL2, and CXC5 in neutrophilic inflammation and AHR enhanced by IL-33 and IL-17A. Interestingly, treatment with anti-CXCR2 mAb decreased CXCL1, CXCL2, and CXCL5 production in the lungs; in addition, anti-CXCL1 mAb suppressed AHR and neutrophilic inflammation, as well as CXCL2 and CXCL5 production (Fig. 2). Although anti-CXCL2 mAb or anti-CXCL5 mAb failed to inhibit CXCL1 and CXCL5 or CXCL1 and CXCL2, respectively, both treatments suppressed AHR and neutrophilic inflammation (Fig. 2). Thus, these data suggest that CXCL1-CXCR2 signaling controls CXCL2 and CXCL5 production, which contributes to neutrophilic inflammation and AHR. Meanwhile, it has been reported that IL-6 induces neutrophilic inflammation (36). Consistent with this finding, treatment with anti–IL-6 mAb reduced neutrophilic inflammation and AHR (Fig. 3); furthermore, IL-6 production was inhibited by treatment with anti-CXCR2 mAb or anti-CXCL1 mAb. Thus, IL-6 regulated by CXCL1-CXCR2 signaling also contributed to neutrophilic inflammation and AHR. However, anti–IL-6 mAb had no effect on ELR+ CXC chemokines, including CXCL1, CXCL2, and CXCL5, produced by coadministration of IL-33 and IL-17A; coadministration of IL-33 and IL-17A did not induce greater production of IL-6 in the lungs. It has been reported that IL-6 suppressed neutrophil apoptosis dependent on the increased number of neutrophils (46). Based on these findings, it is suggested that ELR+ CXC chemokine production enhanced by IL-33 and IL-17A strongly induces neutrophilic inflammation, and IL-6 exhibits prolonged survival of neutrophils. Furthermore, in this study, the depletion of neutrophils inhibited IL-6 production (Fig. 5), indicating that neutrophils may inhibit their own apoptosis through IL-6 production.
To define the cells important for coadministration of IL-33– and IL-17A–induced ELR+ CXC chemokine production via CXCR2, we assessed the potential of alveolar macrophages because they are important sources of CXCL1 and CXCL2 (37). In this study, the number of alveolar macrophages expressing CXCR2 was increased after the coadministration of IL-33 and IL-17A in comparison with IL-33 or IL-17A alone–treated mice (Fig. 4), suggesting that IL-17A enhances IL-33–induced alveolar macrophages expressing CXCR2. Furthermore, the depletion of alveolar macrophages by treatment with liposomal clodronate suppressed neutrophilic inflammation and AHR, as well as CXCL1 and CXCL2 production in the lungs (Fig. 4), indicating the possibility that the infiltration of alveolar macrophages expressing CXCR2 after coadministration plays a critical role in neutrophilic inflammation and AHR through CXCL1 and CXCL2 production. However, the inhibitory levels of CXCL1 and CXCL2 in the lungs by the depletion of alveolar macrophages were weak, although neutrophilic inflammation was strongly inhibited. It has been reported that activated alveolar macrophages produce TNF-α and/or IL-1β to express adhesion molecules such as ICAM-1 and E-selectin, which contribute to neutrophil migration through the endothelium (47, 48), suggesting that, in this study, depletion of alveolar macrophages may repress the recruitment of neutrophils through decreased expressions of not only CXCL1 and CXCL2, but also adhesion molecules. Meanwhile, the depletion of alveolar macrophages and neutrophils did not inhibit CXCL5 production (Figs. 4, 5). It has been reported that this chemokine is upregulated in lung epithelial and endothelial cells during inflammation (49), suggesting that epithelial and endothelial cells might contribute to the production of CXCL5 enhanced by the cooperative action of IL-33 and IL-17A.
It has been reported that Ag-induced AHR, neutrophilic inflammation, and ELR+ CXC chemokine production, such as CXCL1 and CXCL2 in experimental asthmatic models associated with IL-17A using Th17 cell–reconstituted SCID and RORγt-tg mice, were steroid resistant (50, 51). Consistent with these data, we found that dexamethasone failed to inhibit coadministration of IL-33– and IL-17A–induced AHR, neutrophilic inflammation, and ELR+ CXC chemokine production, indicating that AHR and neutrophilic inflammation exacerbated by IL-33 and IL-17A were also steroid resistant. Exposure of epithelial cells to IL-17A in vitro significantly increased glucocorticoid receptor β (GRβ), which is thought to act as a dominant negative inhibitor of GRα, the classical GR (52); indeed, bronchial biopsies from individuals with asthma showed increased expression of IL-17A and decreased expression of GRα (53), suggesting that IL-17A may directly induce steroid insensitivity. Meanwhile, IL-17A production induced by an Ag challenge was not inhibited by dexamethasone in Th17 cell–reconstituted SCID mice (50); in contrast, we found that IL-33 production in the lungs was inhibited by dexamethasone in OVA+alum-sensitized/OVA-challenged mice (T. Nabe, N. Mizutani, and S. Yoshino, unpublished observations), and pretreatment with IL-33 was needed to induce changes in the interaction with IL-33 and IL-17A, indicating that the reduction of IL-33 production by steroids may regulate responses such as AHR, neutrophilic inflammation, and ELR+ CXC chemokine production in experimental asthmatic models associated with IL-33 and IL-17A.
To clarify the importance of CXCR2 signaling in allergic airway responses, we first investigated the effects of anti–IL-33 mAb or anti–IL-17A mAb on the production of ELR+ CXC chemokines in IgE-sensitized mice; these mAbs suppressed CXCL1, CXCL2, and CXCL5 production in the lungs (Table II), suggesting that IL-33 and IL-17A contributed to IgE-mediated ELR+ CXC chemokine production. Next, we examined the effect of anti-CXCR2 mAb on IgE-mediated airway inflammatory responses. In mice sensitized with IgE, a single treatment with anti-CXCR2 mAb at the seventh challenge inhibited late-phase airway obstruction, AHR, neutrophilic inflammation, and ELR+ CXC chemokine production (Fig. 6), suggesting that CXCR2 signaling contributes to the development of IgE-mediated airway inflammatory responses. Meanwhile, a single treatment with anti–IL-6 mAb also suppressed late-phase airway obstruction, AHR, and neutrophilic inflammation in IgE-sensitized mice (Fig. 7). However, IL-6 production was not inhibited by anti-CXCR2 mAb in IgE-sensitized mice (Fig. 6), although production was inhibited by the mAb in IL-33+IL-17A–treated mice. Thus, the mechanism of IL-6 production in IgE-mediated airway inflammation was not congruent with that in the cooperative action of IL-33 and IL-17A, suggesting that in our IgE-sensitized model, IL-6 production may occur by other mechanisms independent of the CXCR2 signaling induced by IL-33 and IL-17A, because there are more complex mechanisms in IgE-mediated airway inflammation.
It is widely known that eosinophilic inflammation and goblet cell hyperplasia are characteristic features of asthma associated with Th2 responses (8–10, 54, 55). It is of interest to examine whether CXCR2 signaling also contributes to these responses. In this study, we examined the effect of multiple treatments with anti-CXCR2 mAb during the fourth to seventh challenges on eosinophilic inflammation and goblet cell hyperplasia, because eosinophilic inflammation and goblet cell hyperplasia were exacerbated during these challenges (30). Interestingly, the infiltration of eosinophils and goblet cell hyperplasia was suppressed by multiple treatments with anti-CXCR2 mAb in IgE-sensitized mice (Figs. 6, 8). It is known that Th2 cytokines, including IL-5 and IL-13, particularly contribute to eosinophilic inflammation and goblet cell hyperplasia, respectively (8–10); in this study, Th2 cytokine production, including IL-5 and IL-13, were inhibited by the treatment (Fig. 9), suggesting that CXCR2 signaling induced these IgE-mediated responses through Th2 immune responses. Meanwhile, it has been reported that IL-6 contributes to Th2 immune responses (56). In addition to the inhibitory effects of anti-CXCR2 mAb, multiple treatments with anti–IL-6 mAb also inhibited eosinophilic inflammation and goblet cell hyperplasia associated with the reduction of Th2 cytokine production such as IL-5 and IL-13 (Figs. 7–9). However, anti–IL-6 mAb showed a slight inhibition of Th2 cytokine production, especially in IL-13; in contrast, IL-6 directly induced goblet cell hyperplasia (57), indicating that in addition to IL-13, goblet cell hyperplasia in IgE-sensitized mice may be induced directly by IL-6.
Multiple treatments with anti-CXCR2 mAb or anti–IL-6 mAb inhibited early-phase airway obstruction (Figs. 6, 7). Furthermore, both multiple treatments inhibited the production of Ag-specific IgE at the seventh challenge in IgE-sensitized mice (Fig. 9). Our previous report showed that the level of Ag-specific IgE was significantly increased over the injection levels of IgE (OE-1) alone (28). Based on these data, the suppression of the early-phase increase in airway resistance by these treatments likely contributed to the reduction of endogenous Ag-specific IgE Ab in IgE-sensitized mice.
In conclusion, we demonstrated that: 1) IL-17A exacerbates IL-33–induced neutrophilic inflammation and AHR associated with enhanced ELR+ CXC chemokines in the lungs and alveolar macrophages expressing CXCR2; 2) CXCR2 signaling regulates the cooperative action of IL-33– and IL-17A–enhanced AHR via neutrophilic inflammation; and 3) in IgE-sensitized mice, CXCR2 signaling plays essential roles in the development of airway inflammatory responses. Published works have suggested that anti-IgE therapy is effective for some patients with poorly controlled asthma (58–60). On the basis of our results, the importance of CXCR2 signaling by the interaction with IL-33 and IL-17A in IgE-mediated allergic asthma associated with neutrophilic inflammation may provide insight into the underlying pathogenesis of chronic inflammatory disease, in which IgE has an important pathogenic role.
Acknowledgements
We thank Yuki Fujimoto, Kaori Kubota, and Mio Sasaki (Kobe Pharmaceutical University) for assistance with sample measurements.
Footnotes
This work was supported by Grant-in-Aid for Young Scientists (B) 23790164 (to N.M.) and Grant-in-Aid for Scientific Research (C) 23590093 (to T.N.) from the Japan Society for the Promotion of Science.
The online version of this article contains supplemental material.
References
Disclosures
The authors have no financial conflicts of interest.
