
Abstract
We compared genetic variations in the VP1 gene of foot-and-mouth disease viruses (FMDVs) isolated since 2000 from various region of the world. We analyzed relative synonymous codon usage (RSCU) and phylogenetic relationship between geographical regions, and calculated the genetic substitution patterns between Korean isolate and those from other countries. We calculated the ratios of synonymously substituted codons (SSC) to all observed substitutions and developed a new analytical parameter, EMC (the ratio of exact matching codons within each synonymous substitution group) to investigate more detailed substitution patterns within each synonymous codon group. We observed that FMDVs showed distinct RSCU patterns according to phylogenetic relationships in the same serotype (serotype O). Moreover, while the SSC and EMC values of FMDVs decreased according to phylogenetic distance, G + C composition at the third codon position was strictly conserved. Although there was little variation among the SSC values of 18 amino acids, more dynamic differences were observed in EMC values. The EMC values of 4- and 6-fold degenerate amino acids showed significantly lower values while most 2-fold degenerate amino acids showed no significant difference. Our findings suggest that different EMC patterns among the 18 amino acids might be an important factor in determining the direction of evolution in FMDV.
Similar content being viewed by others

Introduction
Korea had been free of the foot-and-mouth disease virus (FMDV) since 1934 until an outbreak in 2000. The isolate was found to be closely related to a FMDV (serotype O) that was isolated from Taiwan in 1999 and known to be included in the Pan Asian clade (Joo et al., 2002; Shin et al., 2003; Oem et al., 2004). The Korean isolate in 2000 was thought to have originated in the Indian subcontinent in the early 1990s, but the reason why FMD had re-emerged after 66 yr was not clear (Kanno et al., 2002). Additionally, there was a further FMD outbreak on January 8 2010, identified as a serotype A (FAO World Reference Laboratory, 2010). Since April 2010, however, most of the FMD cases in Korea have changed from serotype A to O (FAO World Reference Laboratory, 2011), and over 3.5 million animals have been slaughtered.
FMD is an acute systemic disease of clovenhoofed animal species caused by the RNA virus, FMDV. FMDV is a single-stranded, positive-sense RNA virus of the family Picornaviridae. Seven serotypes of FMDV have been identified: A, O, C, Asia 1, and South African Territories 1, 2, and 3. Outbreaks have occurred in every livestock-containing region of the world except New Zealand, and the disease is currently enzootic in all continents except Australia and North America, which have not had an outbreak since 1870 and 1952, respectively (Samuel and Knowles, 2001; Domingo et al., 2002; Grubaman and Baxt, 2004). FMDV comprises four capsid proteins: VP1, VP2, VP3, and VP4. VP1-VP3 genes encode proteins exposed on the capsid surface. VP1 contains the major antigenic determinants for infection, resulting in seven distinct genetic lineages that correlate with serotype (Fox et al., 1989; Samuel and Knowles, 2001).
Many analytical parameters such as effective number of codons (ENC) and relative synonymous codon usage (RSCU) have been used for examining codon-usage bias (Sharp and Li, 1986; Levin and Whittome, 2000; Jenkins and Holmes, 2003; Gu et al., 2004; Basak and Ghosh, 2006; Roychoudhury and Mukherjee, 2010; Zhou et al., 2010b). ENC is a codon-usage parameter that can take values from 20, in the case of extreme bias where one codon is exclusively used for each amino acid, to 61 where the use of alternative synonymous codons is equally likely (Wright, 1990). An ENC value can be calculated for each target nucleotide sequence, showing the average pattern of codon-usage bias regardless of the characteristics of each amino acid group. RSCU is the observed frequency of a codon in a target sequence divided by the frequency expected, minimizing the bias from amino acid composition. In this study, we analyzed RSCU values and phylogenetic relationships between regional isolates, and calculated genetic substitution patterns between those from Korea and those isolated from other countries. We also developed a new analytical parameter to compare the codon substitution patterns in more depth.
Results
Phylogenetic relationships between isolates of FMDVs from Korea and those from other countries
To compare overall genetic variation, we created a Neighbor-Joining tree, bootstrapping re-sampling was performed 1,000 times with a total of 68 VP1 sequences of FMDVs isolated from 34 countries (Figure 1). All taxa were classified according to their regions of isolation; Asia, Middle East, Africa, Europe, and South America. The Korean isolate (AF428246) in Group 1-1 showed few distinctive correlations to other nearby Asian countries, such as Hong Kong and the Philippines in Group 3-2, but was correlated with Group 5 and Group 1-2 viruses. Group 5 consists of European countries, including Ireland and the United Kingdom, while Group 1-2 included many Middle Eastern and Southeast Asian countries, including Israel, Saudi Arabia, Turkey, Indonesia, and Laos. Groups 2-1 and 2-2 were composed of many Asian countries adjacent to India (Pakistan, Afghanistan, Bhutan and Nepal), and nearby Arab countries (Iran and United Arab Emirates, Bahrain and Oman). Isolates from South American countries, such as Argentina, Brazil, and Paraguay (Group 3-1) showed the largest variation compared to those from Korea. Viruses from African countries (Ethiopia and Kenya) fell into Group 4-1.

Phylogenetic tree based on the VP1 genes of FMDVs (serotype O) isolated from 34 countries. Branch lengths are drawn to scale, with the bar indicating 0.1 nucleotide replacements per site, and bootstrap values (%) based on the Neighbor-Joining method of 1,000 re-sampled datasets that are not 100% are shown in each node. VP1 sequences which were selected for the visualization of the codon substitution patterns in Figure 3 are marked in bold italic.
Comparison of the RSCU patterns among geographical regions
To determine synonymous codon usage variation among FMDVs worldwide, we calculated RSCU values in 68 VP1 genes (using the same as the taxa sequences in Figure 1). In Figure 2, a scatter plot of the correspondence analysis using RSCU values of target sequences is shown. The countries of FMDV isolation were grouped into 5 geographical regions; Asia, Europe, Middle East, Africa, and South America.

Scatter plots of the correspondence analysis using relative synonymous codon usage (RSCU) values of the VP1 genes of FMDVs. Dim1 and Dim2 represent the values of the first- and second-dimensional factors of each sequence. The percentage in each parenthesis indicates the percent inertia of that axis given by correspondence analysis.
On the basis of the DIM1, the Korean isolate showed RSCU pattern similar to most of the Arab countries in the Middle East and also many Asian countries. Isolates from the United Kingdom also resided in Group 1. Among Asian FMDV isolates, those from Vietnam (DQ165031) showed a similar RSCU pattern to the Korean isolate. Groups 2-5 revealed opposite RSCU patterns to those of Group 1 along DIM1. Group 3, including viruses isolated from the Philippines, Hong Kong, and Vietnam showed the pattern most distinct from Group 1, with an average distance 2.7 times longer than that between Groups 1 and 2, which also clustered into Group 3 in the phylogenetic tree (Figure 1). FMDVs isolated from the South American region (Group 5) revealed opposite RSCU patterns from Group 1 in both DIM1 and DIM2, and African isolates were located between Groups 2 and 5.
Comparison of the codon transition probability matrices (CTMs) of 4 different regions
Figure 3 presents 4 samples of CTMs to visualize substitution patterns between Korea and four other countries; Israel, Malaysia, Ethiopia, and Brazil. These 4 countries represent the four geographical regions of the Middle East, South Eastern Asia, Africa, and South America, respectively. Each row in the Y-axis represents a codon of the VP1 gene from a Korean isolate, and each column in X-axis is a codon of this gene in the virus from the target country. Codon names with their encoded amino acids for each cell in the X- and Y-axes are shown in Supplemental Data Table S2.

2D plots of the codon transition matrices (CTMs) of VP1 genes differed from FMDVs isolated from Korea versus four other countries. Each row in Y-axis represents the seed codon isolated in Korea, and that of X-axis represents the substituted codon in the four target FMDVs isolated from (A) Israel, (B) Malaysia, (C) Ethiopia, and (D) Brazil, representing the Middle East, Asia, Africa and South America, respectively. All the codon names in each axis are shown in Table S2.
In accordance with the phylogenetic result in Figure 1, substitutions between Korea and Israeli isolates showed a relatively simple pattern, resulting in a clear diagonal line (Figure 3A). As the phylogenetic distance increased, however, the substitutions became more and more complex. In the case of FMDVs originating from Malaysia and Ethiopia (Figure 3B and 3C), primarily included in Groups 4-2 and 4-1 in the phylogenetic tree (Figure 1), respectively, mosaic-like substitution patterns in each synonymous codon group were observed, especially in 4-fold degenerate amino acids (alanine, glycine, proline, threonine, and valine). Between FMDV isolates from Korea and Brazil, codon substitution showed a more dispersed pattern to other comparisons, suggesting that non-synonymous codon substitutions were increased between these two countries.
Detailed codon substitution patterns of VP1 genes in each amino acid
To analyze the substitution patterns more intensively, we calculated two parameters: the ratio of synonymously substituted codons (SSC) to all observed substitutions and the proportion of exact matching codons (EMC) within synonymously substituted codons between isolates from Korea and 33 other countries. Table 1 shows the SSC values in 18 amino acids, excluding methionine and tryptophan, which are each encoded by a sole codon. SSC values ranged from 89.7% (serine) to 100.0% (phenylalanine), suggesting that most codon substitutions of FMDVs occurred within the synonymous codon groups. To examine statistical differences in the SSCs among the 18 synonymous codon groups, we performed one-way ANOVA test using Tukey's studentized range test method. Using the SSC value of phenylalanine (SSC = 100.0%) as a baseline, alanine, glycine, threonine, and serine revealed significant differences (P < 0.05). Considering the 4-fold degenerate amino acids, three-fifths showed significantly lower SSC values, and of the 6-fold degenerate amino acids, only serine revealed a significantly lower SSC (89.7%).
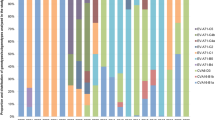
In Figure 4, we present EMC patterns according to both the amino acids and isolated regions (Europe, Middle East Asia, Africa, and South America). In comparison with the SSC patterns, EMC can detect more detailed substitutions within synonymous codon groups before this changes the phenotype of viral genes. In fact, EMC values in synonymous codon groups showed more dramatic differences than those of SSC patterns. On the basis of the amino acid groups, three 2-fold degenerate amino acids, asparagine, cysteine, and glutamine, and all 4-fold and 6-fold degenerate amino acids showed significantly lower EMC values when compared with histidine (EMC = 96.3%), which had the highest EMC value (P < 0.05). The cysteine codon group showed the lowest EMC value (55.4%) followed by alanine (65.0%). Additionally, we also analyzed EMC patterns between FMDVs isolated from Korea and other countries according to their geographical regions (Figure 4B). This showed that as the phylogenetic distance increased, EMC values were gradually decreased, and with countries located in South America demonstrating the lowest EMC values (P < 0.05).

Bar graphs of the exactly matched codon % (EMC) in (A) each synonymous codon group, and in (B) each geographical regions of isolation, such as Europe, Middle East, Asia, Africa, and South America, are shown. All the regional information is presented in Table S1. Each bar represents the % mean, and the error bar indicates the standard deviation. The asterisk means that the value was significantly differed from the value of (A) HIS, or (B) Europe when using Tukey's studentized range test (P < 0.05).
Discussion
In the initial step of our study, we estimated the phylogeny of the VP1 genes among 34 countries by the Neighbor-Joining method with a 1,000 times bootstrapping process. Here, the phylogenetic result indicated that the Korean isolate from 2000 was distinct from isolates from Hong Kong and the Philippines, but closely related to isolates from the United Kingdom and Ireland. South American and East African isolates indicated relatively long phylogenetic distances from the Korean isolate (Figure 1), consistent with other published results (Oem et al., 2004; Yoon et al., 2011). Regarding the origin of FMDVs in Korea, Joo et al. hypothesized that yellow dust spreading from China, and the illegal introduction of traditional Chinese medicines, including deer antlers and other crude materials may have been possible origin of Korean FMD outbreak (Brownlie, 2001; Joo et al., 2002). However, this would not explain the close relationships between the isolates from Korea and the United Kingdom.
In 2010, Zhou et al. analyzed RSCU and codon usage bias (CUB) values using 85 samples of seven serotypes of FMDV (Zhou et al., 2010a). CUB is the absolute deviation between calculated and unbiased RSCU values. Using CUB values, they compared absolute deviation patterns of RSCU values among the seven serotypes of FMDVs; serotype SAT3 had the highest bias while serotype C had the lowest. Serotype O gave intermediate values between serotype A and SAT3 along with other serotypes (Asia 1, A, SAT1). The authors suggested that these results might indicate that differences in evolutionary bias may exist among the seven serotypes, and be primarily determined by codon usage bias in each serotype. In the present study, we computed the RSCU values of 204 VP1 genes of serotype O FMDVs according to the countries from which they were isolated, and observed that FMDVs isolated in each geographical region also had their own RSCU patterns within serotype O (Figure 2). FMDVs were separated into 5 different groups, and these corresponded to the phylogenetic groups in Figure 1. Our findings also indicate that FMDVs in each geographical region have evolved by maintaining their synonymous codon usage patterns. In 2000, Levin and Whittome also reported that there was significant variation in codon usage of genes within the same virus genome, and codon usage bias between viruses was closely related phylogenetically in nucleopolyhedroviruses (Levin and Whittome, 2000). Our research showed a similar trend in FMDVs.
In Figure 3, we show detailed variations of phylogenetic distances in codon-basis, presenting both synonymous and non-synonymous codon substitution patterns. Interestingly, the G + C composition of FMDVs in the third codon position seemed to be conserved during substitutions, resulting in mosaic-like substitution patterns in each codon group. Regarding the GC-end pattern of codon in FMDV, Zhong et al. (2007) investigated mutational bias on codon usage in all seven serotypes by comparing the G + C content at the first and second codon positions with that at the third codon positions, and concluded that there were correlations between them. They suggested that patterns of G + C composition may be the result of mutation pressure, not natural selection. In our study, we showed that those GC-end codon patterns were strictly conserved when the degree of codon substitutions increased according to phylogenetic distance. In addition, Zhou et al. (2010a) reported that FMDVs preferred to use C- than G-end codons, and hypothesized that these compositional constraints might play a key role in contributing to the pattern of synonymous codon usage.
In 2011, Yoon et al. analyzed phylogenetic relationships and rates of nucleotide substitutions of FMDV using the Bayesian Markov Chain Monte Carlo approach, and observed that the ω (dN/dS) value of serotype O was 0.0685, indicating that it is under strong purifying selection. Lewis-Rogers et al. (2008) also analyzed the capsid proteins of FMDVs and concluded that most amino acids were under purifying selection at the genotypic level as their evidence suggested that less than 29% of residues were identified as being under either selection category. In our study, we also observed that there was less variation among amino acids when we calculate SSC values between Korean isolate and those from 33 other countries. Using the EMC parameter, we investigated substitution patterns within each synonymous codon group in more details, and found that there were dynamic differences among synonymous codon groups as well as isolated regions (Figure 4). Our results indicate that 2-fold degenerate amino acids might have stricter substitution conditions within the synonymous codon group, versus codons encoding 4- and 6-fold degenerate amino acids. Our finding also suggests that different EMC patterns among amino acids might be a key factor in determining the evolutionary direction in FMDV in the long term. In fact, the SSC values of the 4- and 6-fold degenerate amino acids were relatively low compared with those of the other amino acids, indicating that non-synonymous codon substitutions preferentially occurred in those codon groups. Regarding nucleotide substitution rates in FMDV, Haydon et al. (2001) reported that the rates in capsid genes were in the range 0.5-1.5 × 10-2 nucleotide substitutions per site per year, and the ratio of synonymous to non-synonymous changes was variable across data sets, and is sensitive to the interval of time. They mentioned that complete sequences of capsid genes VP1-VP3 for serotypes O, A, and C indicate ratios of synonymous to non-synonymous substitutions of approximately 3:1. We observed that there were differences in the substitution patterns within each synonymous codon group as well as the ratio of synonymous to non-synonymous substitutions, and these differences of EMC pattern among amino acids might have an important role in FMDV evolution.
In conclusion, we determined phylogenetic relationships among 204 VP1 genes of FMDVs originated from 34 countries, and analyzed correlations between phylogenetic distance and codon substitution patterns. Correspondence analysis using RSCU values showed that there was unique synonymous codon usage bias in FMDVs isolated in a specific region, and this corresponded with the phylogenetic distance between them. For more in-depth comparisons, we identified codon substitution patterns by calculating SSC and EMC values, and generated the CTMs between Korean isolate and those from other countries (Figure 3). In general, the abundance of nonsynonymous (dN) and synonymous (dS) pairwise variability in nucleotide sequence and their ratio (dS/dN) have been used to determine the selection power, such as mutation pressure or a natural selection (Bonhoeffer et al., 1995; Nielsen, 1999). If the dN exceeds the natural expectation ratio, this is evidence for positive selection (Kimura, 1983). In this paper, we introduced a new parameter, EMC, to facilitate the comparison of codons within synonymous substitutions to showed more detailed selection patterns within synonymous codon groups. Here, we found that G + C composition of FMDVs in the third codon position was strictly conserved while SSC and EMC values decreased, and those values in 4- and 6-fold degenerate amino acids decreased significantly. Moreover, we observed that the EMC values were inversely proportional to phylogenetic distances, and each amino acid showed a unique EMC pattern, indicating that viral evolution caused by gradual codon substitution might be influenced by differences of EMC patterns among amino acids. It might be possible to develop detailed substitution rules for synonymous substitutions, and the cumulative changes might control final nonsynonymous substitutions.
Methods
Nucleotide sequences
VP1 genes (over 500 bp) of FMDV serotype O isolated between 2000 and 2009 (as of January 25, 2010) were obtained from the Taxonomy Browser at the National Center for Biotechnology Information. Sequences that contained one or more ambiguous nucleotides in the initial source data were excluded. The selected sequences were placed into categories including accession number, country isolated from, and year by using JAVA codes. In total, 204 nucleotide sequences of VP1 genes originating from 34 countries were screened and added into a MySQL database to facilitate further computational analyses. All the annotation information of the target sequences is shown in Supplemental Data Table S1.
Phylogenetic analysis
To compare the overall sequence variation in the FMDVs, we randomly selected 1 VP1 sequence per each year (if available) from each country. In total, 68 nucleotide sequences were selected and aligned using ClustalW version 2.0.12 (Larkin et al., 2007) implemented via the Bioworks workflow client (http://bioworks.kisti.re.kr). The annotation information, including year of isolation, country, and accession number, is presented in Figure 1. A phylogram was generated by PAUP* version 4.0 (Swofford, 1999) using the Neighbor-Joining method with 1,000 times bootstrapping process, and the TreeView program (Page, 2002) was used for a graphical representation.
Calculation of relative synonymous codon usage (RSCU) values
RSCU values of VP1 genes of the FMDVs were calculated using C++ codes and the same sequences used in the phylogenetic analysis described above. RSCU is a ratio of the observed frequency of a codon to the frequency expected if all the synonymous codons are used in an unbiased manner. The RSCU values can be calculated as:

where Xij is the frequency of occurrence of the jth e codon for the ith amino acid, and ni is the number of codons for the ith synonymous amino acid group (Sharp and Li, 1986). All 59 codons, except the three stop codons, and the sole codons for methionine (= AUG) and tryptophan (= UGG) were analyzed according to the FMDVs' country of origin.
Calculation of codon substitutions between isolates from Korea and those from other countries
We compared 204 VP1 genes of FMDVs isolated from Korea with isolates from other countries. Detailed annotation information for target sequences is shown in Table S1. This analysis was performed in three steps.
The first step was to conduct multiple sequence alignments between Korean isolate and those from other countries using ClustalW (ver. 2.0.12; Larkin et al., 2007) with strict alignment conditions (40.0 and 0.8 for gap open and gap extension penalties, respectively). After alignment, the FASTA-format aligned results were divided into country of isolation with their gap information. The second step was to count all the possible codon substitutions between Korean sequence and those from other countries at each codon position along the VP1 gene. This generated 33 codon substitution matrices (CSMs) for each pair of countries of isolation, where each cell represents each codon in the Korean isolate or those from other countries (Table S2). Three stop codons, 2 sole codons for methionine and tryptophan, and codons that included one or more gaps in any codon position were excluded from this analysis. In the third and final step, we computed 2 types of codon substitution patterns from CSMs using C++ codes. We calculated the ratio of synonymously substituted codons (SSC) to all observed substitutions from each synonymous codon group of Korean isolate, and developed a new codon substitution parameter, "EMC", the proportion of exact matching codons within each synonymous codon group, to investigate synonymous codon substitutions in more detail. Equations for the two analytical parameters can be defined as:

Where TSa is the total number of codon substitutions, and SSa means the number of codons synonymously substituted from Korean isolate encoding the amino acid "a". In equation 3, MCa is the number of codons substituted to the exact same codon so that from the Korean isolate in "a" amino acid encoding group. SSC is a parameter for determining overall substitution patterns, particularly the synonymous substitution from the Korean isolate, and EMC provides more detailed information about the substitutions within each synonymously substituted codon group. All computational work was conducted using the Tachyon Supercomputer at KISTI Supercomputing Center (http://www.ksc.re.kr).
Figure 3 shows four sample CSMs between Korea and other four countries; Israel, Malaysia, Ethiopia, and Brazil, representing the Middle East, Asia, Africa and South America, respectively. To examine codon substitution patterns in each row in more depth, all CSMs were converted into the codon transition probability matrices (CTMs) using the "transition probability" concept of Markov theory (Stewart, 2009). A CTM can be defined as:

where C(i, j) is the counted number of codons in each cell of the CSM, and Sum(i, ) is the total codon substitutions in each row of the CSM. Thus each P(i, j) had a value between 0.0 and 1.0, and the sum total of each row should be 1.0.
Statistical analysis
Correspondence analysis was performed to examine correlations between VP1 genes of the FMDVs. This is a type of compositional method that is able to analyze associations between objects and a set of descriptive characteristics (Hair et al., 1998). We assigned the countries of isolation as objects, and RSCU values for all the amino acid groups as descriptive characteristic factors. The graphs resulting from the correspondence analysis include first and second dimensional representations of the data, in which the distance between two coordinates within the same row or column represent the χ2 distance (Johnson et al., 2002; Perrière and Thioulouse, 2002). Additionally, we used Tukey's studentized range (HSD) test to investigate which FMDVs differed significantly (P < 0.05) from VP1 genes isolated from Korea by comparing the SSC and EMC values (Figure 4). All the data were analyzed to ensure they met the assumptions for the one-way ANOVA model.
Abbreviations
- CSM:
-
codon substitution matrix
- CTM:
-
codon transition probability matrix
- EMC:
-
the ratio of exact matching codons within each synonymous substitution group
- FMDV:
-
foot-and-mouth disease virus
- RSCU:
-
relative synonymous codon usage
- SSC:
-
the ratio of synonymously substituted codons
References
Basak S, Ghosh TC . Temperature adaptation of synonymous codon usage in different functional categories of genes: a comparative study between homologous genes of Methanococcus jannaschii and Methanococcus maripaludis . FEBS Lett 2006 ; 580 : 3895 - 3899
Bonhoeffer S, Holmes EC, Nowak MA . Causes of HIV diversity . Nature 1995 ; 376 : 125 -
Brownlie J . Strategic decisions to evaluate before implementing a vaccine programme in the face of a foot-and-mouth disease (FMD) outbreak . Vet Rec 2001 ; 148 : 358 - 360
Domingo E, Baranowski E, Escarmis C, Sobrino F . Foot-and-mouth disease virus . Comp Immunol Microbiol Infect Dis 2002 ; 25 : 297 - 308
FAO World Reference Laboratory for Foot-and-Mouth Disease . 03/03/2011 . ME Reports : 2010 [online]. Available from http://www.wrlfmd.org/fmd_genotyping/2010.htm [accessed 3 March 2011]
FAO World Reference Laboratory for Foot-and-Mouth Disease . 03/03/2011 . ME Reports : 2011 [online]. Available from http://www.wrlfmd.org/fmd_genotyping/2011.htm [accessed 3 March 2011]
Fox G, Parry NR, Barnett PV, McGinn B, Rowlands DJ, Brown F . The cell attachment site on foot-and-mouth disease virus includes the amino acid sequence RGD (arginine-glycine-aspartic acid) . J Gen Virol 1989 ; 70 : 625 - 637
Grubman MJ, Baxt B . Foot-and-mouth disease . Clin Microbiol Rev 2004 ; 17 : 465 - 493
Gu W, Zhou T, Ma J, Sun X, Lu Z . Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales . Virus Res 2004 ; 101 : 155 - 161
Hair JF, Anderson RE, Tatham RL, Black WC . In Multivariate Data analysis . 1998, Prentice Hall, Upper Saddle River, New Jersey, USA
Haydon DT, Samuel AR, Knowles NJ . The generation and persistence of genetic variation in foot-and-mouth disease virus . Prev Vet Med 2001 ; 51 : 111 - 124
Jenkins GM, Holmes EC . The extent of codon usage bias in human RNA viruses and its evolutionary origin . Virus Res 2003 ; 92 : 1 - 7
Johnson RA, Wichern DW, Holmes EC . In Applied Multivariate Statistical Analysis . 2002, Prentice Hall, Upper Saddle River, New Jersey, USA
Joo YS, An SH, Kim OK, Lubroth J, Sur JH . Foot-and-mouth disease eradication efforts in the Republic of Korea . Can J Vet Res 2002 ; 66 : 122 - 124
Kanno T, Yamakawa M, Yoshida K, Sakamoto K . The complete nucleotide sequence of the PanAsia strain of foot-and-mouth disease virus isolated in Japan . Virus Genes 2002 ; 25 : 119 - 125
Kimura M . Rare variant alleles in the light of the neutral theory . Mol Biol Evol 1983 ; 1 : 84 - 93
Larkin MA, Blackshields G, Brown NP, Chenn R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DGS . Clustal W and Clustal X version 2.0 . Bioinformatics 2007 ; 23 : 2947 - 2948
Levin DB, Whittome B . Codon usage in nucleopolyhedroviruses . J Gen Virol 2000 ; 81 : 2313 - 2325
Lewis-Rogers N, McClellan DA, Crandall KA . The evolution of foot-and-mouth disease virus: impacts of recombination and selection . Infect Genet Evol 2008 ; 8 : 786 - 798
Nielsen R . Changes in ds/dn in the HIV-1 env gene . Mol Biol Evol 1999 ; 16 : 711 - 714
Oem JK, Lee KN, Cho IS, Kye SJ, Park JH, Joo YS . Comparison and analysis of the complete nucleotide sequence of foot-and-mouth disease viruses from animals in Korea and other PanAsia strains . Virus Genes 2004 ; 29 : 63 - 71
Page RD . Visualizing phylogenetic trees using TreeView . Curr Protoc Bioinformatics 2002 ; Chapter 6:Unit 6.2
Perrière G, Thioulouse J . Use and misuse of correspondence analysis in codon usage studies . Nucleic Acids Res 2002 ; 30 : 4548 - 4555
Roychoudhury S, Mukherjee D . A detailed comparative analysis on the overall codon usage pattern in herpesviruses . Virus Res 2010 ; 148 : 31 - 43
Samuel AR, Knowles NJ . Foot-and-mouth disease type O viruses exhibit genetically and geographically distinct evolutionary lineages (topotypes) . J Gen Virol 2001 ; 82 : 609 - 621
Sharp PM, Li WH . Codon usage in regulatory genes in Escherichia coli does not reflect selection for 'rare' codons . Nucleic Acids Res 1986 ; 14 : 7737 - 7749
Shin JH, Sohn HJ, Choi KS, Kwon BJ, Ko YJ, An DJ, Cha SH, Park JH, Jeong WS, Park JY, Choi CU, Kweon CH, Song JY, Kim JY, An SH, Kim SJ, Joo YS . Molecular epidemiological investigation of foot-and-mouth disease virus in Korea in 2000 . J Vet Med Sci 2003 ; 65 : 9 - 16
Stewart WJ . Probability, Markov Chains, Queues, and Simulation: The Mathematical Basis of Performance Modeling, 2009, Princeton University Press, Princeton (NJ), USA
Swofford DL . PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4, 1999, Sinauer, Sunderland (MA), USA
Wright F . The 'effective number of codons' used in a gene . Gene 1990 ; 87 : 23 - 29
Yoon SH, Lee KN, Park JH, Kim H . Molecular epidemiology of foot-and-mouth disease virus serotypes A and O with emphasis on Korean isolates: temporal and spatial dynamics . Arch Virol 2011 ; 156 : 817 - 826
Zhong J, Li Y, Zhao S, Liu S, Zhang Z . Mutation pressure shapes codon usages in the GC-Rich genome of foot-and-mouth disease virus . Virus Genes 2007 ; 35 : 767 - 776
Zhou JH, Zhang J, Chen HT, Ma LN, Liu YS . Analysis of synonymous codon usage in foot-and-mouth disease virus . Vet Res Commun 2010a ; 34 : 393 - 404
Zhou JH, Zhang J, Ding YZ, Chen HT, Ma LN, Liu YS . Characteristics of codon usage bias in two regions downstream of the initiation codons of foot-and-mouth disease virus . BioSystems 2010b ; 101 : 20 - 28
Acknowledgements
This study was supported by the Korea Institute of Science and Technology Information. We also acknowledge the invaluable contribution of the researchers who have made their data publicly available.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Experimental & Molecular Medicine website
Supplementary information
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ahn, I., Bae, SE. & Son, H. Comparative study of codon substitution patterns in foot-and-mouth disease virus (serotype O). Exp Mol Med 43, 587–595 (2011). https://doi.org/10.3858/emm.2011.43.10.066
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2011.43.10.066
