
Nano-Microparticle Platforms in Developing Next-Generation Vaccines
by
 , ,
, ,
Giuseppe Cappellano
1,2 ,
,
Hugo Abreu
1 ,
,
Chiara Casale
1, 
Umberto Dianzani
1,3,* and
Annalisa Chiocchetti
1,2 1
Dipartimento di Scienze della Salute, Interdisciplinary Research Center of Autoimmune Diseases—IRCAD, Università del Piemonte Orientale, 28100 Novara, Italy
2
Center for Translational Research on Autoimmune and Allergic Disease—CAAD, Università del Piemonte Orientale, 28100 Novara, Italy
3
Laboratorio di Biochimica Clinica, Dipartimento di Scienze della Salute, AOU Maggiore della Carità, Università del Piemonte Orientale, Corso Mazzini 18, 28100 Novara, Italy
*
Author to whom correspondence should be addressed.
Vaccines 2021, 9(6), 606; https://doi.org/10.3390/vaccines9060606
Submission received: 3 May 2021
/
Revised: 3 June 2021
/
Accepted: 3 June 2021
/
Published: 5 June 2021
(This article belongs to the Special Issue Advances in Vaccine Development)
Abstract
:The first vaccines ever made were based on live-attenuated or inactivated pathogens, either whole cells or fragments. Although these vaccines required the co-administration of antigens with adjuvants to induce a strong humoral response, they could only elicit a poor CD8+ T-cell response. In contrast, next-generation nano/microparticle-based vaccines offer several advantages over traditional ones because they can induce a more potent CD8+ T-cell response and, at the same time, are ideal carriers for proteins, adjuvants, and nucleic acids. The fact that these nanocarriers can be loaded with molecules able to modulate the immune response by inducing different effector functions and regulatory activities makes them ideal tools for inverse vaccination, whose goal is to shut down the immune response in autoimmune diseases. Poly (lactic-co-glycolic acid) (PLGA) and liposomes are biocompatible materials approved by the Food and Drug Administration (FDA) for clinical use and are, therefore, suitable for nanoparticle-based vaccines. Recently, another candidate platform for innovative vaccines based on extracellular vesicles (EVs) has been shown to efficiently co-deliver antigens and adjuvants. This review will discuss the potential use of PLGA-NPs, liposomes, and EVs as carriers of peptides, adjuvants, mRNA, and DNA for the development of next-generation vaccines against endemic and emerging viruses in light of the recent COVID-19 pandemic.
1. Introduction
Vertebrates have developed defense mechanisms consisting of innate and adaptive immunity [1] that collaborate to build an effective immune response against microbial invaders. Innate immunity is an ancient, fast, unspecific, local, and antigen-independent process, which is stimulated when membrane-associated or cytosolic pattern recognition receptors (PRRs) expressed on immune cells [i.e., dendritic cells (DCs), monocytes, macrophages, neutrophils, natural killer (NK) cells] recognize pathogen-associated molecular patterns (PAMPs) widely expressed in microbes [2]. Upon PAMP recognition, the host defense mechanism is activated, resulting in acute inflammation, crucial to recruiting immune cells to the site of infection [3], and activate adaptive immunity.
During viral infection, cellular immune responses mediated by CD4+ T helper (TH) cells and CD8+ cytotoxic T lymphocytes (CTLs) are crucial for host defense. TH cells include TH1 cells, which potentiate phagocyte and NK cell cytotoxicity by secreting IL-2 and IFN-γ, and TH2, which secrete IL-4, IL-5, and IL-6, thereby enhancing antibody production by B lymphocytes [4,5]. CTLs recognize virus-infected cells and induce their apoptosis in order to clear the invading pathogens [6]. Besides eliciting an effector response that eradicates the infection, these processes contribute to the development of an immunological memory that can trigger an effective response when the same pathogen is encountered a second time [7]. Thus, by exposing the body to harmless forms of the pathogen, vaccines can build up an immunological memory in the absence of a true pathological infection [8]. This review will initially focus on first-, second-, and third-generation vaccines and will then address next-generation vaccines (graphical abstract).
2. Classical Vaccines
Classical vaccines were invented by Louis Pasteur in the late 1800s, following from the initial observation made by Edward Jenner about one century earlier that cowpox infection induced an immune condition that protected from smallpox infection, devising a new procedure nowadays known as vaccination—a term derived from the Latin word for cow “vacca” [9]. Later on, Pasteur succeeded in developing several other vaccines by isolating and inactivating disease-specific pathogens through different methods, de facto revolutionizing the biomedical field [10]. His work inspired classical vaccination techniques based on the use of whole microbes, which were either live attenuated (LA) or inactivated/killed [11].
LA vaccines (LAVs) are indeed developed from weakened pathogens that are able to proliferate in the host, causing mild disease or none at all. The immune response elicited by LAVs is similar to that induced by virulent pathogens and often confers long-term immunity in a single dose, without the need of adjuvants [12]. The main risk associated with LAVs is the reversion to virulence of the pathogen, which may lead to severe infection in immunocompromised individuals and may harm the fetus during pregnancy. In addition, LAVs generally need a temperature-controlled supply chain to preserve the living vaccine [13]. Despite these limitations, current LAVs for measles, rotavirus, and yellow fever, as well as the oral polio vaccine, are considered safe and suited for commercialization [11,13].
Inactivated vaccines consist of pathogens killed by chemical treatments. They are safer and more stable than LAVs and cannot revert to virulence or induce infection even in immunocompromised individuals. However, being less effective than LAVs in inducing immunity, they usually require several doses to generate a humoral response, often not permanent. Current examples are vaccines against poliomyelitis (i.e., inactivated polio vaccine—IPV), hepatitis A, rabies, and influenza [11,13].
Other types of vaccines are derived from antigenic subunits of the pathogen, generally polysaccharides or proteins [11]. Even though they are safe and stable, they can mainly induce a humoral response and thus require a careful choice of the antigen, which must be immunogenic enough to induce protective immunity against the target pathogen. [13,14]. Among this category, we find toxoid vaccines, which can inactivate tetanus or diphtheria exotoxins, and sub-viral particle-based vaccines, which can inhibit hepatitis B viral entry [11,13,14]. A limitation of these protein-based vaccines is that the denaturation and renaturation steps required for their production may alter the exposed epitope, thereby affecting its immunogenicity [8,13,14]. Moreover, the proteins may be degraded by proteases before antigen recognition [14].
The creation of protein-based vaccines has been greatly improved by the use of recombinant DNA technology combined with nano/microparticle delivery systems. Examples include vaccines for human papillomavirus (HPV), containing two or four copies of the capsid protein L1, and hepatitis B, containing the hepatitis B virus surface antigen (HBsAg). These vaccines are widely used and confer protection with rare side effects [8,13]. Viral proteins can also multimerize into virus-like particles (VLPs) that can be more effectively recognized by the immune system upon vaccination [15,16].
To correct the poor immunogenicity of inactivated and subunit-based vaccines, they are usually co-administered with adjuvants. These compounds significantly increase the vaccine immunogenicity by acting as both immunopotentiators and delivery systems [14,17]—described later in this review. The most common adjuvant is aluminum, in the form of several salts—e.g., aluminum phosphate, aluminum hydroxide, and aluminum potassium sulfate (Alum)—which has been administered with diphtheria vaccines since the 1930s [18,19,20]. Alum promotes the recruitment of antigen presenting cells (APCs), thereby increasing their antigen uptake and presentation. Furthermore, it induces APC maturation and migration to the draining lymph node, and it stimulates a TH2 response, supporting antibody production [19]. Aluminum compounds combined with monophosphoryl lipid A (MPLA) have also been used to create a vaccine against human papillomavirus (HPV). Other types of adjuvants include emulsions, such as MF59, an adjuvant composed by squalene in citric acid buffer found in influenza vaccines [21], and DNA sequences, such as the CpG 1018 oligonucleotide, a Toll-like receptor 9 (TLR) agonist adjuvant that, when co-administered with HBsAg, boosts immunity against hepatitis B [22].
3. Nucleic Acid Vaccines
Upon injection in a tissue, exogenous nucleic acids (DNA or RNA) can be captured by cells and eventually translated into protein(s), which are then released or presented to immune cells [23]. DNA vaccines are usually developed by incorporating eukaryotic DNA constructs into bacteria-derived or semi/fully synthetic plasmids [23]. Importantly, plasmids never replicate in mammalian hosts, nor do they integrate in genomic DNA [24]. DNA vaccines are easy and fast to produce, are highly stable, and present no risk of infection since no live pathogen is injected [23,25]. Additionally, they induce both cell-mediated and humoral immune responses since the plasmid can enter not only structural tissue cells, such as myocytes, but also APCs, capable of presenting the vaccine antigens through MHC class I and II molecules. Therefore, these vaccines can induce a complete immune response involving CD4+ T helper cells—TH1 and TH2—CD8+ cytotoxic T cells, and B cells [23,26]. Even though studies performed in different species demonstrated a safe and non-integrative profile of DNA vaccines [25,27,28,29], these concerns have cast doubt on their safety so that, to date, they have yet to be approved for use in humans, whereas four of them have already been licensed for veterinary use [25]. Recently, several clinical trials of DNA vaccines for Ebola, Zika, and influenza H5N1 viruses have reported promising results regarding safety and efficacy, opening new avenues for their future use against viral diseases [23].
RNA vaccines can be made with non-replicating mRNA or self-replicating mRNA [23,30,31]. Non-replicating mRNA contains the sequence of the antigen of interest flanked by two untranslated regions (UTRs) at both the 5′ and 3′ ends of the sequence. It results from a linearized DNA plasmid transcribed in an in vitro system (e.g., E. coli) by a DNA-dependent RNA polymerase, usually derived from the T3, T7, or Sp6 phage. After purification, this process leads to a fully mature mRNA similar to that of typical eukaryotic transcripts, including a 5′ cap and a poly(A) tail for stability and translation enhancement [23,30]. Remarkably, this technique has allowed for the fast development of vaccines against SARS-CoV-2 that have been emergency-approved for mass immunization during the COVID-19 pandemic—further discussed in Section 5 [31].
Self-replicating or self-amplifying mRNAs (saRNAs) are constructs based on the alphavirus genome. These constructs retain the replication machinery of alphavirus thanks to the genes encoding the nsP1-4 complex, which assembles into an RNA-dependent RNA polymerase, whereas the genes encoding the alphavirus structural proteins are replaced by the mRNA encoding the target antigen. After entering the host cells, this mRNA construct can amplify itself, leading to high levels of antigen production and to a potentially robust immune response, thereby reducing the need of vaccine recalls [23,30,32]. Preclinical and clinical studies using saRNA vaccines for influenza, AIDS, rabies, and SARS-CoV-2 are underway—reviewed in [32].
In cases of large transcripts, it is possible to use a trans-amplifying approach based on the combination of two mRNA constructs: one encoding the alphavirus replication genes, and the other encoding the gene of interest. In this case, the nsP1-4 complex can also replicate the non-self-amplifying mRNA transcript with the same advantages as those afforded by self-amplifying mRNAs [32,33]. Tests performed in mice have shown that this approach can confer immunity against influenza upon the administration of two doses, 21 days apart [33].
4. Next-Generation Vaccines Based on Nano/Microparticle Delivery Systems
A further step of vaccine development is represented by engineered nanoparticles (NPs) used as vaccine delivery platforms that are able to protect the antigenic component of the vaccine while delivering innovative adjuvants that can finely tune the immune response. In addition to their delivery function, NPs display an intrinsic adjuvant activity, which makes them particularly suitable as vaccine platforms. Once internalized by APCs, NPs can in fact trigger inflammasome complex formation, which promotes the inflammation and recruitment of immune cells (Figure 1) [36,37,38]. Thus, NPs are promising antigen carriers and immune cell activators for the preparation of more effective vaccines. Of note is the fact that NPs can also be engineered to function as negative modulators of immune activation makes them attractive candidate for inverse vaccination—discussed in Section 4.2.5.
Several NPs are being tested to deliver protein- and nucleic acid-based vaccines. Among the most promising are biodegrading polymers in the form of poly (lactic-co-glycolic acid) NPs (PLGA-NPs), liposomes, and extracellular vesicles (EVs), which will be described in this review (Table 1).
4.1. PLGA
For several decades, poly(lactic-co-glycolic acid) (PLGA) has been used as a constituent of NPs because of its excellent biocompatibility, biodegradability, and safety profile [39]. Indeed, its use in humans was approved by the Food and Drug Administration (FDA) more than 30 years ago [40,41]. PLGA undergoes hydrolysis in the body to produce lactic acid and glycolic acid, which are efficiently metabolized through the Krebs cycle, thus avoiding toxicity [42].
PLGA-NPs have been validated as effective drug delivery systems by several studies in vivo. In particular, these compounds were shown to function as transporters of orally-administered insulin [43] and to be effective in delivering drugs to various body districts (e.g., cochlea, liver, and kidneys [44]), inflamed sites due to inflammatory diseases (e.g., arthritis [45] and Bowel disease [46]), and neoplastic tissues. They were also employed to produce tolerogenic vaccines for autoimmune diseases, such as experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis [47,48]. In particular, PLGA was shown to be an excellent biocompatible polymer for NPs because PLGA-NPs could be loaded with a wide variety of molecules and their surface functionalized in order to improve their delivery to target tissues. In this regard, it is important to point out that vaccine delivery is highly influenced by the size of the NP. Indeed, small NPs elicit stronger humoral and cellular immune responses because they can more easily reach the lymph nodes and are more efficiently captured by APCs [49].
4.1.1. Protein—Based PLGA Viral Vaccines
Influenza A virus, characterized by a high mutation rate, is known to cause seasonal infection waves worldwide [50]. Vaccines for influenza A induce the production of antibodies against the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) and must, therefore, be reformulated every year in order to be adapted to the antigenic drift of the virus. In this regard, a PLGA-NP-based vaccine for the H1N1 strain of influenza was obtained by loading NPs with the HA protein, together with MLPA and muramyl dipeptide (MDP), used as adjuvants that are capable of triggering a PRR-mediated response. This vaccine was shown to induce IFN-γ-producing CD4+ T cells and a strong antibody response. Interestingly, mice immunized with HA-PLGA-NPs plus the adjuvants were significantly more resistant to the lethal challenge with H1N1 virus compared to mice immunized without adjuvants [51].
Dengue virus (DENV) causes hemorrhagic fever and shock syndrome and represents a public health threat in Southeast Asia and Central and South America. A Dengue vaccine consisting of PLGA/PEG NPs loaded with viral nonstructural protein 1 (NS1) in the absence of adjuvants was shown to be effective in mice [52]. Subsequently, Metz et al. tested the efficacy of immunization with a tetravalent recombinant envelope (rE) protein subunit vaccine adsorbed into the PLGA-NP surface. This strategy led to a uniform antibody response against all four DENV serotypes, compared to the sole use of soluble antigens, demonstrating the promising potential of this approach for vaccine development [53].
A different strategy came from studies by Zhu et al. on immunization against hepatitis B virus. In order to promote a continuous release of HBsAg, the protein was loaded into PLGA-NPs. In addition, to increase the antigen uptake by APCs, the NP surface was functionalized by mannosylation so as to target mannose receptors. Mannose-grafted PLGA-NPs loaded with HBsAg induced successful antigen presentation, CD8+ T cell response, and secretion of IFN-γ and IL-2 [54]. In another study, HBsAg entrapped in PLGA-NPs positively charged with cationic particles (i.e., stearyl amine and polyethyleneimine) was given as aerosol to female Sprague Dawley rats to reach the lungs. This vaccination induced the production of antigen-specific IgG in the serum and IgA in the oral, vaginal, and bronchoalveolar lavages. The respiratory route of administration also induced a cell-mediated immune response, triggering the production of IFN-γ and IL-2 [55]. In another study, PLGA-NPs were loaded with hepatitis B core antigen (HBcAg), with or without MPLA. The results showed that the vaccine containing MPLA was highly effective at inducing a strong HBcAg-specific TH1 immune response [56].
PLGA-NPs can also be used for the delivery of poor soluble proteins. Roopngam et al. encapsulated the insoluble form of E2 envelope glycoprotein subtype 1b of hepatitis C virus (HCV1b-E2) in PLGA microspheres, showing that its continuous release from these microspheres induced a strong CD8+ T-cell immune response, as well as IFN-γ secretion in vaccinated mice [57]. Lastly, PLGA NPs have been used to deliver a multi-epitope vaccine against human T-cell leukemia/lymphoma virus type 1 (HTLV-1), an oncogenic RNA virus responsible for T-cell leukemia. Specifically, a multi-epitope chimera, consisting of the Tax, env, and gag immunodominant HTLV-1 epitopes, was encapsulated in PLGA-NPs together with the CpG-oligonucleotide adjuvant. The results showed that the vaccine induced a strong humoral response, and that NP encapsulation was crucial to improve antigen presentation and induce a strong cellular and mucosal immune response [58].
4.1.2. PLGA in DNA Vaccines
Despite the advantages of DNA vaccines, only a few studies have shown that naked-DNA vaccines can induce a robust immune response in humans. One study showed that a HBsAg DNA vaccine was safe and well tolerated in a cohort of 12 healthy hepatitis-naïve human volunteers, where it induced an adequate immune response leading to virus clearance [59]. Because DNA plasmids are susceptible to fast degradation by nucleases, various particle formulations have been employed to precisely deliver DNA vaccines to tissues and overcome this degradation. In this regard, PLGA-NPs represent an interesting approach since they seem to provide a continuous DNA release while inducing a strong T-cell response. Indeed, oral administration of a single dose of PLGA-NPs loaded with HBsAg-DNA induced a long-lasting antigen-specific antibody response in BALB/c mice. Moreover, an effective antigen specific CTL response was detected in the spleen and gut-associated lymphoid tissue upon in vitro re-stimulation with HBsAg [60].
Another potential application of this approach is against Ebola virus, which causes hemorrhagic fever and multiorgan failure. To date, no human vaccine for Ebola has been approved. A feasible method of vaccination proposed by Yang et al. consists in Ebola DNA vaccine coated on PLGA-poly- l-lysine/poly-γ-glutamic acid (PLGA-PLL/γPGA) NPs, which is capable of inducing a strong immune response in mice [61].
Finally, the development of PLGA microspheres loaded with complexes of DNA and polyethylenimine (PEI) holds great promise for the design of vaccines against human immunodeficiency virus type 1 (HIV-1), which has so far remained elusive to vaccination thanks to its ability to impair and evade the host immune system. This approach may lead to more effective vaccines because PEI protects DNA from degradation during encapsulation and, upon intramuscular injection, the microspheres can release intact and penetrative PEI/DNA complexes for several days. Indeed, this vaccine induced strong antibody and CTL responses to HIV in mice [62].
Altogether, these results show that PLGA-NPs can effectively transfer the DNA vaccine to DCs and stimulate efficient CD4+ and CD8+ T-cell immune responses.
4.1.3. PLGA in mRNA Vaccines
PLGA-NPs have recently gained increasing attention as potential platforms to deliver mRNA vaccines because of their ability to escape from endosomes alongside their excellent biodegradability and biocompatibility profile [63]. However, the negative charge of PLGA severely impairs the mRNA incorporation efficacy, which might account for the poor success in developing these vaccines thus far [64]. However, promising results have been obtained with PLGA/PEI NPs, which were shown to deliver mRNA encoding for green fluorescent protein (GFP) to human monocyte-derived DCs in order to elicit the host immune response and eliminate any hypothetical pathogen [65].
4.1.4. PLGA as Adjuvant in Vaccine Formulations
NPs have the potential to boost the immune response even without the encapsulated antigen. For instance, Seth et al., immediately before injection in BALB/c mice, mixed PLGA-NPs with a modular capsomere comprising the antigenic M2e peptide (CapM2e) of influenza A virus, obtaining higher levels of anti-M2e IgG1 compared to the control [66]. Similarly, Zhang et al. used PLGA as an adjuvant for influenza A immunization. These authors administered PLGA-NPs, loaded or not, with the TLR-7 agonist imiquimod (IMQ), to mice along with HA derived from an H5N1 influenza vaccine (A/Anhui/1/2005). The results showed the upregulation of the anti-HA antibody response in mice injected with HA adjuvanted with either empty PLGA-NPs or PLGA-NPs loaded with IMQ compared with control mice injected with HA alone. Furthermore, this formulation also increased IFN-γ production detected in splenocytes compared to that induced by control immunization with HA alone or HA plus Alum as adjuvant, in an ex-vivo setting [67].
4.1.5. PLGA NP in Inverse Vaccination
Inverse vaccination is aimed at specifically inhibiting pathologic immune responses, such as those responsible for autoimmune and allergic diseases, by inducing peripheral tolerance. Tolerogenic vaccines aim to preserve the host immune defense while avoiding severe opportunistic infections [68] which may occur using immunosuppressive drugs. While conventional vaccines induce humoral and cellular effector immunity, tolerogenic vaccines inhibit existing pathogenic effector/memory T cells by inducing either their anergy/deletion or suppression through regulatory T cells (Tregs) capable of maintaining long-lasting immune tolerance. Tregs may derive from pre-existing Tregs or differentiated CD4+ naive T cells [69].
A promising approach of tolerogenic vaccination takes advantage of NPs to deliver both antigens and “tolerogenic adjuvants” to trigger suppressive responses [70]. In particular, our group investigated the tolerogenic effect of PLGA-NPs loaded with the myelin oligodendrocyte glycoprotein (MOG)35–55 autoantigen and recombinant interleukin-10 (r IL-10), used as a tolerogenic adjuvant, in experimental autoimmune encephalomyelitis (EAE). Results showed that this combination was effective in ameliorating EAE and reducing both demyelination and TH1 and TH17 responses [47]. Subsequent studies confirmed the efficacy of this approach in other autoimmune diseases, as reviewed in [48]. Interestingly, a very recent study has suggested the effectiveness of subcutaneous injection of MOG35–55 PEGylated-containing PLGA-NPs without tolerogenic adjuvants in ameliorating the EAE course [71]. Moreover, two other studies performed in mice have shown that oral vaccination with collagen II [72] or nasal vaccination with HSP70 [73]—in both cases, the antigens were incapsulated in PLGA-NPs—conferred a high level of protection against rheumatoid arthritis-like disease in the absence of inverse adjuvants.
4.2. Liposomes
Liposomes are spherical artificial vesicles derived from natural phospholipids and cholesterol [74] that, due to their excellent versatility and plasticity, are emerging as promising tools for vaccine development. Liposomes are safe and have already been successfully translated into clinical use [75]. In the context of vaccination, liposomes passively target their contents to APCs, diffuse into the lymph nodes, and ultimately enhance immune responses [76,77].
The research efforts on liposome-based vaccines have expanded enormously in the course of the last year in the attempt to develop effective vaccines against SARS-CoV-2. Thanks to the possibility to modulate liposome features (i.e., lipid composition, charge, and size), both hydrophilic and lipophilic molecules, such as proteins, peptides, nucleic acid, and adjuvants, can be entrapped within the liposome lipid layer or exposed on the liposome surface through chemical linking.
4.2.1. Protein-Based Liposome Vaccines
Synthetic peptides are safe and can be prepared as pure immunogens in large quantities, which makes them ideal tools for vaccination. However, these peptides are weakly immunogenic and need the help of adjuvants to overcome this limitation. In this regard, liposomes are ideal to provide adjuvant activity as they induce innate immune responses and improve antigen delivery, which mounts robust adaptive immune responses. Moreover, peptide-vaccines can be equally effective with the peptides being either encapsulated into the liposomes or chemically coupled with the liposome surface. Liposome-coupled peptides are taken up by APCs through either direct fusion with the plasma membrane or pinocytosis [78]. Senchi et al. synthesized an oligomannose-coated liposome vaccine against human parainfluenza virus type 3 (HPIV3), which causes acute respiratory infections and asthma in children, by combining the HPIV3 hemagglutinin-neuraminidase antigen with the adjuvant poly(I:C). Intranasal administration of low doses of this vaccine protected mice from HPIV3 infection through induction of antigen-specific IgG and IgA [79].
Intradermal injection of a liposomal cationic adjuvant formulation (CAF09) and a mixture of peptides (pepmix) spanning the entire sequence of the HCV nonstructural protein 3 (NS3) induced a vigorous CD4+ T-cell response. Importantly, it induced immunity against subdominant T-cell epitopes that were not efficiently targeted by either vaccination with full-length recombinant rNS3 or even infection with HCV [80]. Likewise, subcutaneous immunization with HCV-derived antigenic peptides coupled with the liposome surface (Lip-603) promoted a robust CD8+ T cell-mediated anti-viral immunity. This response was more effective than that obtained using the same peptides emulsified in incomplete Freund’s adjuvant [81]. Moreover, Ohno et al. selected two peptides from SARS-CoV, the virus causing severe acute respiratory syndrome (SARS), as HLA-A*0201-restricted CTL epitopes and went on showing that, upon linkage to the liposomal surface (Lip-N223 and Lip-N227), they were highly effective in inducing peptide-specific CTLs in HLA-A*0201 transgenic mice [82].
4.2.2. Liposomes in DNA Vaccines
Cationic liposomes can protect DNA from degradation and, by interacting with negatively charged cell membranes, can be taken up by APCs where they eventually disassemble, thereby favoring DNA plasmid entry into the nucleus.
Qiao et al. showed that, upon intramuscular immunization with mannosylated zwitterionic-based cationic liposomes (man-ZCL) decorated with an HIV DNA plasmid Env, mice developed TH1/TH2 mixed immune responses [83]. Rodriguez et al. used liposomes to deliver DNA plasmids encoding bovine herpesvirus type 1, demonstrating that immunized mice were able to develop specific IgG responses [84]. In another study, upon oral administration of a cationic liposome/DNA vaccine encoding the M1 gene of influenza A virus, immunized mice developed not only an M1-specific IgG antibody response but also antigen-specific CTLs [85]. Another group generated cationic liposomes loaded with a DNA vaccine encoding middle (pre-S2 plus S) envelope proteins of HBV together with the CpG-oligodeoxynucleotide adjuvant. This vaccine was successively transcutaneously injected into the mouse skin through microneedle to achieve a sustained release and long-lasting gene expression. Results showed that this transcutaneous immunization led to a balanced Th1/Th2 cell response [86]. Finally, cationic liposome-DNA complexes (CLDCs) were used as adjuvants for a vaccine containing an influenza H5N1 subunit and intramuscularly injected in mice. This immunization not only induced robust serum antibody and TH1/IFN-γ responses compared those observed in mice immunized with an unadjuvanted vaccine, but it also protected mice from influenza virus infection after just one jab [87].
4.2.3. Liposomes in mRNA-Based Vaccines
mRNA-based vaccines loaded in liposomes represent a promising alternative to conventional vaccines to fight viral infection [30] and are known to induce anti-cancer immunity [88]. One of the advantages of using mRNA-liposome vaccines lies in their ability to elicit both humoral and cellular immunity, which are both required to eradicate intracellular pathogens, whereas subunit vaccines and killed/inactivated vaccines mainly elicit humoral immunity. This distinctive feature of mRNA-liposome vaccines is due to their ability to deliver the mRNA directly to the cytoplasm of DCs, where the exogenous mRNA is rapidly translated into the antigenic proteins that will then be processed by the proteasome to generate peptide epitopes to be presented to CD8+ T cells through MHC class I molecules [89]. Liposome mRNA-vaccines are regarded as safe since the exogenous mRNA, unlike DNA, cannot integrate into the host genome [90].
The pioneering study attempting to load mRNA in liposomes was published in 1978 by Dimitriadis et al. [91]. The authors entrapped rabbit globin mRNA sequences into liposomes and then successfully transfected them into mouse lymphocytes, thus providing the proof-of-concept of their approach. About fifteen years later, Martinon et al. demonstrated in vivo that injection of liposomes containing mRNA encoding the influenza virus nucleoprotein induced strong CTL responses in mice [92]. Another study showed that intranasal injection of liposome loaded with double-stranded RNA (LE-PolyICLC) was effective in eradicating influenza virus (H5N1-HPIV) by inhibiting virus replication, reducing viral titers, increasing survival of infected mice, and attenuating pulmonary fibrosis. Moreover, this compound displayed adjuvant activity when combined with an inactivated H5N1 vaccine, leading to enhanced humoral and cellular responses [93]. Others showed that liposome could be used also for passive immunization as intramuscular injection of an mRNA encoding ZIKV-117—a human anti-Zika neutralizing antibody—encapsulated in liposomes, conferred protection against Zika in mice [94]. Liposomes were also employed by Pardi et al. to develop an mRNA vaccine encoding influenza virus HA. Specifically, the authors demonstrated that immunization with HA mRNA-liposomes induced antibody responses against the HA stalk domain of influenza virus in mice, rabbits, and ferrets [95].
An additional advantage of mRNA vaccines is that they have been proven to be much more effective than LAVs. This was initially shown by Monslow et al. reporting that mRNA encoding the gE antigen of varicella-zoster virus (VZV) encapsulated in lipid NPs conferred a stronger immune response than that elicited by live attenuated VZV [96]. Liposomes have also been used to immunize guinea pigs with Ebola envelope (env) mRNA, leading to a strong response in terms of specific neutralizing IgG and 100% survival following Ebola virus infection [97]. Importantly, liposomes encapsulating the spike protein mRNA of SARS-CoV-2 are effective in inducing immunity against SARS-CoV-2. These vaccines are being currently administered worldwide and are under evaluation for the assessment of long protective responses [98]. A specific paragraph is dedicated to this topic in Section 5.
4.2.4. Liposomes as Adjuvants in Vaccine Formulations
Liposomes are highly effective in overcoming the weak immunogenicity of subunit vaccines as they can carry adjuvants to further enhance the immune response. For instance, liposomes can be loaded with pathogen-derived molecules capable of triggering PPRs, such as TLRs or C-type lectin receptors (CLRs) [99]. In particular, Wui et al. developed a lyophilized vaccine, mixing cationic liposomes with the TLR4 agonist de-O-acylated lipooligosaccharide (dLOS), Quillaja saponin fraction QS-21, and the recombinant varicella zoster virus (VZV) glycoprotein E. This formulation was shown to induce a strong TH1 response in immunized mice [100]. Another study showed that the administration of trivalent influenza vaccine with the cationic liposome adjuvant system CAF01 enhanced both humoral and cellular immune responses in mice, followed by increased IL-1β, IL-2, IL-12, IFN-γ, and TNF-α [101]. Wørzne et al. reported that a single immunization of mice with the SARS-CoV-2 spike protein together with the adjuvant CAF01 significantly enhanced spike-specific CD4+ TH responses, producing IFN-γ and IL-17, compared to other adjuvants, such as squalene emulsion (SE) and aluminum hydroxide. By contrast, the antibody responses against the spike receptor binding domain (RBD) was similar for all adjuvants [102]. Vaccines against hepatitis virus E mainly target the structural capsid protein open-reading-frame-2 (ORF-2) of the virus. Joshi et al. demonstrated that recombinant neutralizing epitope protein (rNEp), which is part of ORF-2, adjuvanted with liposomes, elicited a balanced TH1/TH2 response driven by DCs, while other adjuvants (i.e., Alum) induced a TH2 response driven by macrophages [103].
4.2.5. Liposomes in Inverse Vaccination
Liposomes are not immunogenic per se, which makes them particularly suitable to inhibit autoimmune and allergic diseases through inverse vaccination, a process where vaccines are used to induce antigen-specific inhibition of autoimmune responses. In this regard, Kenison and colleagues found that a nanoliposome-based platform encapsulating the ligand of aryl hydrocarbon receptor and MOG35–55 suppressed EAE, in both prophylactic and therapeutic settings, by inducing several Treg cell types [104].
The rationale behind the use of liposomes for inverse vaccination is that DCs may acquire tolerogenic activity upon endocytosis of apoptotic cell material [105]. Thus, the fact that phosphatidyl-serine (PS) is exposed only on the apoptotic cell membrane [106] makes liposomes enriched in PS and loaded with autoantigens the ideal vectors to generate tolerogenic DCs with which to inhibit immune responses in autoimmune disease. For example, the intraperitoneal injection of PS-rich liposomes loaded with MOG40–55, before EAE onset, was shown to suppress EAE development and induce splenic forkhead box P3+ (foxp3) Tregs in mice [107]. The same strategy was also effective in non-obese diabetic mice, a model of type 1 diabetes, in which PS-liposomes loaded with insulin peptides induced tolerogenic DCs, impaired autoreactive T-cell proliferation, and inhibited the development of diabetes [108]. Furthermore, PS-enriched liposomes loaded with ovalbumin peptide 323 (OVA323) were shown to induce Tregs in wild-type mice that had received an adoptive transfer of splenocytes from OT-II mice, transgenic for an anti-OVA323 TCR, one day before immunization. Moreover, liposomes containing the anionic phospholipid 1,2-distearoyl-sn-glycero-3-phosphoglycerol induced Treg proliferation and reduced atherosclerotic plaque formation in apolipoprotein E (ApoE−/−) mice, a model of atherosclerosis [109]. Finally, in the murine OVA-induced model of allergic diarrhea, the treatment of sensitized mice with OVA loaded in oligomannose-coated liposomes induced regulatory CD8+ T cells, triggered IL-10 production, and ameliorated allergic diarrhea [110].
4.3. EVs as Delivery Systems in Vaccines
EVs are small lipid-based bilayer particles that are naturally secreted by almost all cell types [111]. Due to their size, origin, and content, they can be easily distinguished from exosomes, microvesicles, and apoptotic bodies. Both exosomes and microvesicles are physiologically involved in cell-to-cell communication and play a role in cancer cell-mediated modulation of the tumor microenvironment [112,113]. In addition, viral infection of mammalian cells can affect cellular EV content and secretion. For example, cytomegalovirus (CMV)-infected endothelial cells release viral antigen-containing EVs, which trigger CD4+ T cell activation [114]. Moreover, HIV-1 infection stimulates the release of T cell-derived EVs containing HIV Gag [115], an essential structural viral protein that contributes to the assembly, secretion, and maturation of HIV-1 [116].
EVs are natural carriers of several types of molecules, including nucleic acids (DNA and RNA), proteins and lipids, and have been shown to be safe, efficient, non-toxic, and weakly immunogenic carriers [112,117]. Furthermore, EVs can be engineered to express different surface markers, which can turn them into “antigen-presenting EVs”. Intriguingly, Tregs are able to release EVs with tolerogenic properties, which contributes to modulating the immune response without the need of direct cell-to-cell interaction. This activity could be either ascribed to the transfer of miRNAs or proteins to the target cells or attributed to the activity of surface proteins expressed on the vesicles [118]. Moreover, several studies performed in mice have shown that EVs derived from foxp3+ Tregs display suppressive functions mediated by molecules, such as CD73, which impair cytokine release from T cells by converting AMP into adenosine [119], or let-7d miRNA, which suppresses TH1 proliferation and IFN-γ release by inhibiting cyclooxygenase-2 (Cox-2) [120].
Altogether, the aforementioned features make EVs attractive candidates as delivery platforms in vaccine settings. Albeit pioneering studies have been focused on anti-cancer therapies, the attention has now shifted to viral disease. Of note, EVs are also used as biomarkers of several human diseases, including viral infections [121,122].
4.3.1. Protein-Based EV Vaccines
Several types of engineered EVs containing viral proteins have been recently generated for immunization against viral infections. In particular, Admyre et al. treated monocyte-derived DCs with EVs loaded with 23 different peptide sequences (i.e., CEF peptide mix), originated from CMV, influenza, and Epstein-Barr virus, to test the immune response in vitro. The authors found that these EVs induced high levels of IFN-γ production from CD8+ T cells, which directly correlated with the number of EVs and expression levels of MHC class-I molecules [123].
More recently, Martins et al. fused bacterial EVs—also referred to as outer membrane vesicles (OMVs)—derived from Neisseria meningitidis with the envelope proteins of Zika virus in an attempt to create a vaccine against Zika infection. The immunization of mice with these particles induced an immune response, producing antibodies, IL-2 and IL-4. The authors proposed that the use of this innocuous and rapidly generated vehicle might be a promising approach for vaccine formulation [124].
4.3.2. EVs in DNA-Based vaccines
Several efforts have been made to use EVs to produce valuable alternatives to conventional DNA vaccines. Di Bonito et al. showed that a DNA plasmid encoding a mutated HIV Nef protein (Nefmut), unable to downregulate CD4 and MHC class-I, fused with HPV E7 protein, induced the production of EVs expressing the protein chimera due to the membrane-anchoring properties of Nef. Intramuscular injection of this DNA plasmid induced a powerful CTL response against both Nef and E7, which was not achieved using wild-type Nef or E7 alone [125]. Subsequently, the authors replicated these results by fusing the mutant Nef protein with other viral proteins, including hepatitis C virus (HCV)-NS3, Ebola virus (EboV)-VP24, EboV-VP40, and EboV-NP, West Nile virus (WNV)-NS3, influenza (Flu)-NP, and Crimean–Congo hemorrhagic fever (CCHFV)-NP [126]. More recently, Polak et al. generated a prototype of EV-based anti-SARS-CoV-2 vaccine that combines both DNA and peptide-based techniques. Immunization with this vaccine required a primary immunization with a DNA vector inducing in vivo production of SARS-CoV-2 spike protein-expressing EVs and subsequent boost immunizations using EVs expressing the spike proteins produced in mammalian cells in vitro. This vaccine induced potent humoral and cellular responses in mice, without the need of adjuvants [127].
4.3.3. EVs as Adjuvants in Vaccine Formulation
EVs can also be exploited as adjuvants regardless of their antigen expression. Their role as immunopotentiators has been explored by Jesus et al. using EVs isolated from LPS-activated THP-1 monocytes. Immunization of mice with these EVs mixed with either a solution of HBsAg or a suspension of HBsAg-loaded poly(ɛ-caprolactone)/chitosan NPs induced a stronger cell-mediated immune response, marked by IFN-γ production, even though the humoral response was comparable to that induced by vaccination in the absence of EVs [111].
4.4. Limitations of Nano/Microparticle-Based Vaccines
The knowledge of the biological behavior of NPs in terms of distribution in vivo, at both the organ and cellular level, is still lacking. We do not know whether exposure to NPs over long periods of time may affect the human body. Another concern is related to the use of NPs as adjuvants, which may result in chronic inflammatory reactions. Moreover, we have yet to fully explore the physicochemical properties of NPs. If on the one hand NPs might increase the uptake by APCs, on the other hand they may reach other organs/tissues potentially leading to adverse biological effects (i.e., apoptosis or necrosis). Finally, another important issue concerns the ability of NPs to aggregate, which may block the blood vessels in the host leading to thrombosis.
5. COVID-19 Vaccines Based on Nano/Microparticle Platforms
Since late 2019, the novel β-coronavirus-SARS-CoV-2 has spread across the globe causing the coronavirus disease 2019 (COVID-19) pandemic [128]. Coronaviruses are single-stranded enveloped RNA viruses with a tropism for the lower respiratory tract. Upon binding of the virus spike (S) glycoproteins expressed on the viral surface to angiotensin converting enzyme 2 (ACE2) receptor expressed on type II pneumocytes, SARS-CoV-2 infects the host cells and induces a severe inflammatory reaction, producing high levels of pro-inflammatory cytokines (“cytokine storm”), such as IL-1β, IL-2, IL-6, IFN-γ, and TNF-α which are responsible for severe tissue damage and thrombosis [129]. Clinical symptoms of COVID-19 range from asymptomatic infection to respiratory failure—requiring mechanical ventilation and intensive care unit (ICU) admission—acute respiratory distress syndrome (ARDS), sepsis, and multi-organ dysfunction syndrome (MODS) [130].
Immediately after the declaration of pandemic state by the World Health Organization (WHO) on 11 March 2020, and as soon as the SARS-CoV-2 sequence became available in March 2020, many research centers around the world started developing safe and effective vaccines against SARS-CoV-2 [131]. To date, a large number of different approaches have been proposed. Among the 198 vaccine candidates at the time of writing this review, 44 are in clinical trials, and 10 are in late-stage clinical development [132]. The first two vaccines approved by the European Medicines Agency (EMA) were the BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna), two mRNA vaccines encapsulated in liposomes targeting the viral spike protein [131,133].
Both BNT162b2 and mRNA-1273 were able to induce the synthesis of the viral spike proteins in the host and induce an effective and protective immune response against SARS-CoV-2. They are administered in two-doses, 21 days apart for BNT162b2 and 28 days apart for mRNA-1273 [133]. Notably, Polack et al. has shown that the efficacy of BNT162b2 ranges from 89% to 100% after the second dose, eliciting a strong induction of TH1 and CD8+ T cells and neutralizing antibody response [131]. Similarly, recent data have reported a 94.1% efficacy of mRNA-1273 in preventing COVID-19 disease [133]. These vaccines together with more traditional ones based on recombinant adenoviruses carrying the SARS-CoV-2 spike genes [134,135] are playing a key role in battling the pandemic.
6. Conclusions
There is a great need for harmonization and simplification of the roadmap for the design and development of novel effective vaccines. The use of nanotechnological platforms in vaccine development holds great promise and will likely allow the generation of safe and affordable formulations for preventing multiple infections, possibly in a single shot. It is, however, mandatory to understand the mechanisms of NP entry into the cells and activation of the adaptive immune responses, including any related toxicity effects, especially inflammation.
In the future, we need to devise NPs that can specifically bind to target cells to ensure that only these carriers adhere to those cells. In addition, the increasing proportion of the human population requiring vaccination and the emerging threat of new viruses and drift variants have highlighted the need to develop affordable broad-spectrum vaccines. The data reviewed here indicate that nano/microparticles platforms will play a central role in achieving this goal. Finally, the effectiveness of these platforms in loading large amounts of adjuvants and modulating the immune response opens new avenues for their use in tolerogenic vaccination, which is expected to revolutionize the management of autoimmune diseases.
Funding
This research was funded by Fondazione Italiana Sclerosi Multipla (2020/PR-Single/021 (to GC); Italian Ministry of Education, University and Research (MIUR) program “Departments of Excellence 2018–2022”, FOHN (to AC); Fondazione Cariplo 2017–0535 (to UD) and 2019-3277 (to AC); European Union’s Horizon 2020 Research and Innovation Program under Grant Agreement No.953121—project FLAMIN-GO; Associazione Italiana Ricerca sul Cancro—IG 20714, Milano (to UD).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to the Associazione per la ricerca che cura-ARCA onlus (to CC).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Owen, J.A.; Punt, J.; Stranford, S.A.; Jones, P.P.; Kuby, J. Kuby Immunology, 7th ed.; W.H. Freeman: New York, NY, USA, 2013. [Google Scholar]
- Peng, J.; Thakur, A.; Zhang, S.; Dong, Y.; Wang, X.; Yuan, R.; Zhang, K.; Guo, X. Expressions of MiR-181a and MiR-20a in RPMI8226 Cell Line and Their Potential as Biomarkers for Multiple Myeloma. Tumor Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kogut, M.H.; Lee, A.; Santin, E. Microbiome and Pathogen Interaction with the Immune System. Poult. Sci. 2020, 99. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive Immunity. J. Allergy Clin. Immunol. 2010, 125. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Toll-like Receptor Control of the Adaptive Immune Responses. Nat. Immunol. 2004, 5. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.M.; Metkar, S.S.; Froelich, C.J. Cytotoxic Granule-Mediated Apoptosis: Unraveling the Complex Mechanism. Curr. Opin. Immunol. 2003, 15. [Google Scholar] [CrossRef]
- Pradeu, T.; Du Pasquier, L. Immunological Memory: What’s in a Name? Immunol. Rev. 2018, 283. [Google Scholar] [CrossRef]
- Loomis, R.J.; Johnson, P.R. Emerging Vaccine Technologies. Vaccines 2015, 3, 429–447. [Google Scholar] [CrossRef] [Green Version]
- Willis, N.J. Edward Jenner and the Eradication of Smallpox. Scott. Med. J. 1997. [Google Scholar] [CrossRef]
- Berche, P. Louis Pasteur, from Crystals of Life to Vaccination. Clin. Microbiol. Infect. 2012. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention. Principles of Vaccination. In Pink Book Webinar Series; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2020; Chapter 1; pp. 1–7. [Google Scholar]
- Mok, D.Z.L.; Chan, K.R. The Effects of Pre-Existing Antibodies on Live-Attenuated Viral Vaccines. Viruses 2020, 12, 520. [Google Scholar] [CrossRef]
- World Health Organization. Module 2: Types of vaccines and adverse reactions. In Vaccine Safety Basics Learning Manual; World Health Organization: Geneva, Switzerland, 2013; pp. 38–60. [Google Scholar]
- Moyle, P.M.; Toth, I. Modern Subunit Vaccines: Development, Components, and Research Opportunities. ChemMedChem 2013, 8, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major Findings and Recent Advances in Virus–like Particle (VLP)-Based Vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Syomin, B.V.; Ilyin, Y.V. Virus-Like Particles as an Instrument of Vaccine Production. Mol. Biol. 2019, 53. [Google Scholar] [CrossRef]
- Foged, C.; Hansen, J.; Agger, E.M. License to Kill: Formulation Requirements for Optimal Priming of CD8 + CTL Responses with Particulate Vaccine Delivery Systems. Eur. J. Pharm. Sci. 2012, 45. [Google Scholar] [CrossRef]
- Park, W.H.; Schroder, M.C. Diphtheria Toxin-Antitoxin and Toxoid. Am. J. Public Health Nations Health 1932, 22, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, T.R. The Mechanisms of Action of Vaccines Containing Aluminum Adjuvants: An in Vitro vs in Vivo Paradigm. Springerplus 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Glenny, A.T.; Buttle, G.A.H.; Stevens, M.F. Rate of Disappearance of Diphtheria Toxoid Injected into Rabbits and Guinea—Pigs: Toxoid Precipitated with Alum. J. Pathol. Bacteriol. 1931, 34, 267–275. [Google Scholar] [CrossRef]
- Ko, E.J.; Kang, S.M. Immunology and Efficacy of MF59-Adjuvanted Vaccines. Hum. Vaccines Immunother. 2018, 14. [Google Scholar] [CrossRef]
- Campbell, J.D. Development of the CpG Adjuvant 1018: A Case Study. Methods Mol. Biol. 2017, 1494. [Google Scholar] [CrossRef]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, J.J.; Ulmer, J.B.; Liu, M.A. DNA Vaccines. Dev. Biol. Stand. 1997, 15, 617–648. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A. A Comparison of Plasmid DNA and MRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Robinson, H.L.; Pertmer, T.M. DNA Vaccines for Viral Infections: Basic Studies and Applications. Adv. Virus Res. 2000, 55. [Google Scholar] [CrossRef]
- Sheets, R.L.; Stein, J.; Manetz, T.S.; Duffy, C.; Nason, M.; Andrews, C.; Kong, W.P.; Nabel, G.J.; Gomez, P.L. Biodistribution of DNA Plasmid Vaccines against HIV-1, Ebola, Severe Acute Respiratory Syndrome, or West Nile Virus Is Similar, without Integration, despite Differing Plasmid Backbones or Gene Inserts. Toxicol. Sci. 2006, 91, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Ledwith, B.J.; Manam, S.; Troilo, P.J.; Barnum, A.B.; Pauley, C.J.; Griffiths, T.G.; Harper, L.B.; Beare, C.M.; Bagdon, W.J.; Nichols, W.W. Plasmid DNA Vaccines: Investigation of Integration into Host Cellular DNA Following Intramuscular Injection in Mice. Intervirology 2000, 43. [Google Scholar] [CrossRef]
- Houston, R.; Moxon, S.; Nogué, F.; Papadopoulou, N.; Ramon, M.; Waigmann, E. Assessment of the Potential Integration of the DNA Plasmid Vaccine CLYNAV into the Salmon Genome. EFSA J. 2017, 15, 1–15. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines-a New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Forni, G.; Mantovani, A.; Forni, G.; Mantovani, A.; Moretta, L.; Rappuoli, R.; Rezza, G.; Bagnasco, A.; Barsacchi, G.; Bussolati, G.; et al. COVID-19 Vaccines: Where We Stand and Challenges Ahead. Cell Death Differ. 2021, 28. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-Amplifying RNA Vaccines for Infectious Diseases. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-Amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Iavarone, C.; O’hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of Action of MRNA-Based Vaccines. Expert Rev. Vaccines 2017, 16. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. In Current Topics in Microbiology and Immunology; Springer: Berlin, Germany, 2020; pp. 1–40. [Google Scholar]
- Sun, B.; Wang, X.; Ji, Z.; Li, R.; Xia, T. NLRP3 Inflammasome Activation Induced by Engineered Nanomaterials. Small 2013, 9. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 Inflammasome in Dendritic Cells Induces IL-1Β-Dependent Adaptive Immunity against Tumors. Nat. Med. 2009, 15. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, J.M.; Wang, X.; Marin-Muller, C.; Wang, H.; Lin, P.H.; Yao, Q.; Chen, C. Current Advances in Research and Clinical Applications of PLGA-Based Nanotechnology. Expert Rev. Mol. Diagn. 2009, 9. [Google Scholar] [CrossRef] [Green Version]
- Allahyari, M.; Mohit, E. Peptide/Protein Vaccine Delivery System Based on PLGA Particles. Hum. Vaccines Immunother. 2016, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.; Skidmore, S.; Hadar, J.; Garner, J.; Park, H.; Otte, A.; Soh, B.K.; Yoon, G.; Yu, D.; Yun, Y.; et al. Injectable, Long-Acting PLGA Formulations: Analyzing PLGA and Understanding Microparticle Formation. J. Control. Release 2019, 304. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-Based Nanoparticles: An Overview of Biomedical Applications. J. Control. Release 2012, 161. [Google Scholar] [CrossRef]
- Cui, F.-d.; Tao, A.-j.; Cun, D.-m.; Zhang, L.-q.; Shi, K. Preparation of Insulin Loaded PLGA-Hp55 Nanoparticles for Oral Delivery. J. Pharm. Sci. 2007, 96. [Google Scholar] [CrossRef]
- Tamura, T.; Kita, T.; Nakagawa, T.; Endo, T.; Kim, T.S.; Ishihara, T.; Mizushima, Y.; Higaki, M.; Ito, J. Drug Delivery to the Cochlea Using PLGA Nanoparticles. Laryngoscope 2005, 115. [Google Scholar] [CrossRef]
- Higaki, M.; Ishihara, T.; Izumo, N.; Takatsu, M.; Mizushima, Y. Treatment of Experimental Arthritis with Poly(D, L-Lactic/Glycolic Acid) Nanoparticles Encapsulating Betamethasone Sodium Phosphate. Ann. Rheum. Dis. 2005, 64. [Google Scholar] [CrossRef] [Green Version]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Maincent, P.; Kawashima, Y.; Lehr, C.M. Biodegradable Nanoparticles for Targeted Drug Delivery in Treatment of Inflammatory Bowel Disease. J. Pharmacol. Exp. Ther. 2001, 299, 775–781. [Google Scholar]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous Inverse Vaccination with PLGA Particles Loaded with a MOG Peptide and IL-10 Decreases the Severity of Experimental Autoimmune Encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles Target Distinct Dendritic Cell Populations According to Their Size. Eur. J. Immunol. 2008, 38. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Mutation and Epistasis in Influenza Virus Evolution. Viruses 2018, 10, 407. [Google Scholar] [CrossRef] [Green Version]
- Tukhvatulin, A.; Dzharullaeva, A.; Erokhova, A.; Zemskaya, A.; Balyasin, M.; Ozharovskaia, T.; Zubkova, O.; Shevlyagina, N.; Zhukhovitsky, V.; Fedyakina, I.; et al. Adjuvantation of an Influenza Hemagglutinin Antigen with Tlr4 and Nod2 Agonists Encapsulated in Poly(D,l-Lactide-Co-Glycolide) Nanoparticles Enhances Immunogenicity and Protection against Lethal Influenza Virus Infection in Mice. Vaccines 2020, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-s.; Li, I.-H.; Hong, P.-d.; Yeh, M.-k. Evaluation of Protective Efficacy Using a Nonstructural Protein NS1 in DNA Vaccine- Loaded Microspheres against Dengue 2 Virus. Int. J. Nanomed. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Metz, S.W.; Thomas, A.; Brackbill, A.; Xianwen, Y.; Stone, M.; Horvath, K.; Miley, M.J.; Luft, C.; DeSimone, J.M.; Tian, S.; et al. Nanoparticle Delivery of a Tetravalent E Protein Subunit Vaccine Induces Balanced, Type-Specific Neutralizing Antibodies to Each Dengue Virus Serotype. PLoS Negl. Trop. Dis. 2018, 12. [Google Scholar] [CrossRef]
- Zhu, J.; Qin, F.; Ji, Z.; Fei, W.; Tan, Z.; Hu, Y.; Zheng, C. Mannose-Modified PLGA Nanoparticles for Sustained and Targeted Delivery in Hepatitis B Virus Immunoprophylaxis. AAPS PharmSciTech 2020, 21. [Google Scholar] [CrossRef]
- Thomas, C.; Gupta, V.; Ahsan, F. Influence of Surface Charge of PLGA Particles of Recombinant Hepatitis B Surface Antigen in Enhancing Systemic and Mucosal Immune Responses. Int. J. Pharm. 2009, 379. [Google Scholar] [CrossRef]
- Chong, C.S.W.; Cao, M.; Wong, W.W.; Fischer, K.P.; Addison, W.R.; Kwon, G.S.; Tyrrell, D.L.; Samuel, J. Enhancement of T Helper Type 1 Immune Responses against Hepatitis B Virus Core Antigen by PLGA Nanoparticle Vaccine Delivery. J. Control. Release 2005, 102. [Google Scholar] [CrossRef]
- Roopngam, P.; Liu, K.; Mei, L.; Zheng, Y.; Zhu, X.; Tsai, H.I.; Huang, L. Hepatitis C Virus E2 Protein Encapsulation into Poly D, L-Lactic-Co-Glycolide Microspheres Could Induce Mice Cytotoxic T-Cell Response. Int. J. Nanomed. 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabiri, M.; Sankian, M.; Sadri, K.; Tafaghodi, M. Robust Mucosal and Systemic Responses against HTLV-1 by Delivery of Multi-Epitope Vaccine in PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2018, 133. [Google Scholar] [CrossRef]
- Roy, M.J.; Wu, M.S.; Barr, L.J.; Fuller, J.T.; Tussey, L.G.; Speller, S.; Culp, J.; Burkholder, J.K.; Swain, W.F.; Dixon, R.M.; et al. Induction of Antigen-Specific CD8+ T Cells, T Helper Cells, and Protective Levels of Antibody in Humans by Particle-Mediated Administration of a Hepatitis B Virus DNA Vaccine. Vaccine 2000, 19. [Google Scholar] [CrossRef]
- He, X.W.; Wang, F.; Jiang, L.; Li, J.; Liu, S.K.; Xiao, Z.Y.; Jin, X.Q.; Zhang, Y.N.; He, Y.; Li, K.; et al. Induction of Mucosal and Systemic Immune Response by Single-Dose Oral Immunization with Biodegradable Microparticles Containing DNA Encoding HBsAg. J. Gen. Virol. 2005, 86. [Google Scholar] [CrossRef]
- Yang, H.W.; Ye, L.; Guo, X.D.; Yang, C.; Compans, R.W.; Prausnitz, M.R. Ebola Vaccination Using a DNA Vaccine Coated on PLGA-PLL/ΓPGA Nanoparticles Administered Using a Microneedle Patch. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, B.; Yu, X.; Zha, X.; Zhang, X.; Wang, X.; Jin, Y.; Wu, Y.; Chen, Y.; Shan, Y.; et al. Controlled Release of PEI/DNA Complexes from PLGA Microspheres as a Potent Delivery System to Enhance Immune Response to HIV Vaccine DNA Prime/MVA Boost Regime. Eur. J. Pharm. Biopharm. 2008, 68. [Google Scholar] [CrossRef] [PubMed]
- Elmowafy, E.M.; Tiboni, M.; Soliman, M.E. Biocompatibility, Biodegradation and Biomedical Applications of Poly(Lactic Acid)/Poly(Lactic-Co-Glycolic Acid) Micro and Nanoparticles. J. Pharm. Investig. 2019, 49. [Google Scholar] [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of Mrna-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Sharifnia, Z.; Bandehpour, M.; Hamishehkar, H.; Mosaffa, N.; Kazemi, B.; Zarghami, N. In-Vitro Transcribed Mrna Delivery Using Plga/Pei Nanoparticles into Human Monocyte-Derived Dendritic Cells. Iran. J. Pharm. Res. 2019, 18. [Google Scholar] [CrossRef]
- Seth, A.; Ritchie, F.K.; Wibowo, N.; Lua, L.H.L.; Middelberg, A.P.J. Non-Carrier Nanoparticles Adjuvant Modular Protein Vaccine in a Particle-Dependent Manner. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, L.; Yang, T.; Liu, Y.; Chen, X.; Liu, Q.; Jia, J.; Ma, G. Immunopotentiator-Loaded Polymeric Microparticles as Robust Adjuvant to Improve Vaccine Efficacy. Pharm. Res. 2015, 32. [Google Scholar] [CrossRef]
- Hasegawa, H.; Matsumoto, T. Mechanisms of Tolerance Induction by Dendritic Cells in Vivo. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Waldmann, H.; Adams, E.; Fairchild, P.J.; Cobbold, S. Infectious Tolerance and the Long-Term Acceptance of Transplanted Tissue. Immunol. Rev. 2006, 212. [Google Scholar] [CrossRef]
- Capurso, N.A.; Look, M.; Jeanbart, L.; Nowyhed, H.; Abraham, C.; Craft, J.; Fahmy, T.M. Development of a Nanoparticulate Formulation of Retinoic Acid That Suppresses Th17 Cells and Upregulates Regulatory T Cells. Self Nonself Immune Recognit. Signal. 2010, 1. [Google Scholar] [CrossRef] [Green Version]
- Li, P.Y.; Bearoff, F.; Zhu, P.; Fan, Z.; Zhu, Y.; Fan, M.; Cort, L.; Kambayashi, T.; Blankenhorn, E.P.; Cheng, H. PEGylation Enables Subcutaneously Administered Nanoparticles to Induce Antigen-Specific Immune Tolerance. J. Control. Release 2021, 331. [Google Scholar] [CrossRef]
- Zhu, P.; Li, X.Y.; Wang, H.K.; Jia, J.F.; Zheng, Z.H.; Ding, J.; Fan, C.M. Oral Administration of Type-II Collagen Peptide 250-270 Suppresses Specific Cellular and Humoral Immune Response in Collagen-Induced Arthritis. Clin. Immunol. 2007, 122. [Google Scholar] [CrossRef]
- Keijzer, C.; Slütter, B.; van der Zee, R.; Jiskoot, W.; van Eden, W.; Broere, F. PLGA, PLGA-TMC and TMC-TPP Nanoparticles Differentially Modulate the Outcome of Nasal Vaccination by Inducing Tolerance or Enhancing Humoral Immunity. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, Preparation, and Applications. Nanoscale Res. Lett. 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Gregoriadis, G. Immunological Adjuvants: A Role for Liposomes. Immunol. Today 1990, 11. [Google Scholar] [CrossRef]
- Frézard, F. Liposomes: From Biophysics to the Design of Peptide Vaccines. Brazilian J. Med. Biol. Res. 1999, 32. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Taneichi, M.; Kasai, M.; Kakiuchi, T.; Uchida, T. Liposome-Coupled Antigens Are Internalized by Antigen-Presenting Cells via Pinocytosis and Cross- Presented to CD8+ T Cells. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Senchi, K.; Matsunaga, S.; Hasegawa, H.; Kimura, H.; Ryo, A. Development of Oligomannose-Coated Liposome-Based Nasal Vaccine against Human Parainfluenza Virus Type 3. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Filskov, J.; Mikkelsen, M.; Hansen, P.R.; Christensen, J.P.; Thomsen, A.R.; Andersen, P.; Bukh, J.; Agger, E.M. Broadening CD4+ and CD8+ T Cell Responses against Hepatitis C Virus by Vaccination with NS3 Overlapping Peptide Panels in Cross-Priming Liposomes. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, A.; Kobayashi, N.; Taneichi, M.; Uchida, T.; Akatsuka, T. Coupling to the Surface of Liposomes Alters the Immunogenicity of Hepatitis C Virus-Derived Peptides and Confers Sterile Immunity. Biochem. Biophys. Res. Commun. 2013, 430. [Google Scholar] [CrossRef]
- Ohno, S.; Kohyama, S.; Taneichi, M.; Moriya, O.; Hayashi, H.; Oda, H.; Mori, M.; Kobayashi, A.; Akatsuka, T.; Uchida, T.; et al. Synthetic Peptides Coupled to the Surface of Liposomes Effectively Induce SARS Coronavirus-Specific Cytotoxic T Lymphocytes and Viral Clearance in HLA-A*0201 Transgenic Mice. Vaccine 2009, 27. [Google Scholar] [CrossRef]
- Qiao, C.; Liu, J.; Yang, J.; Li, Y.; Weng, J.; Shao, Y.; Zhang, X. Enhanced Non-Inflammasome Mediated Immune Responses by Mannosylated Zwitterionic-Based Cationic Liposomes for HIV DNA Vaccines. Biomaterials 2016, 85. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.E.; Zamorano, P.; Wilkowsky, S.; Torrá, F.; Ferreri, L.; Dominguez, M.; Florin-Christensen, M. Delivery of Recombinant Vaccines against Bovine Herpesvirus Type 1 GD and Babesia Bovis MSA-2c to Mice Using Liposomes Derived from Egg Yolk Lipids. Vet. J. 2013, 196. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, J.; Wang, B.; Zeng, S.; Qi, F.; Lu, C.; Kimura, Y.; Liu, B. Oral Vaccination with a Liposome-Encapsulated Influenza DNA Vaccine Protects Mice against Respiratory Challenge Infection. J. Med. Virol. 2014, 86. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Guo, L.; Zhang, S.; Xu, B.; Gao, Y.; Hu, Y.; Hou, J.; Bai, B.; Shen, H.; Mao, P. DNA-Based Vaccination against Hepatitis B Virus Using Dissolving Microneedle Arrays Adjuvanted by Cationic Liposomes and CpG ODN. Drug Deliv. 2016, 23. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Liu, F.; Fairman, J.; Hong, D.K.; Lewis, D.B.; Monath, T.; Warner, J.F.; Belser, J.A.; Patel, J.; Hancock, K.; et al. Cationic Liposome-DNA Complexes (CLDC) Adjuvant Enhances the Immunogenicity and Cross-Protective Efficacy of a Pre-Pandemic Influenza A H5N1 Vaccine in Mice. Vaccine 2012, 30. [Google Scholar] [CrossRef] [PubMed]
- Guevara, M.L.; Persano, S.; Persano, F. Lipid-Based Vectors for Therapeutic MRNA-Based Anti-Cancer Vaccines. Curr. Pharm. Des. 2019, 25. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jeklenec, A.; Langer, R.; Blankschtein, D. MRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Deliv. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Pascolo, S. Vaccination with Messenger RNA (MRNA). Handb. Exp. Pharmacol. 2008, 183. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Translation of Rabbit Globin MRNA Introduced by Liposomes into Mouse Lymphocytes. Nature 1978, 274. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of Virus-specific Cytotoxic T Lymphocytes in Vivo by Liposome-entrapped MRNA. Eur. J. Immunol. 1993, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hu, Y.; Jin, Y.; Zhang, G.; Wong, J.; Sun, L.Q.; Wang, M. Prophylactic, Therapeutic and Immune Enhancement Effect of Liposome-Encapsulated PolyICLC on Highly Pathogenic H5N1 Influenza Infection. J. Gene Med. 2011, 13. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Archer, J.; Fuerte-Stone, J.; Khandhar, A.P.; Voigt, E.; Granger, B.; Bombardi, R.G.; Govero, J.; Tan, Q.; Durnell, L.A.; et al. Intramuscular Delivery of Replicon RNA Encoding ZIKV-117 Human Monoclonal Antibody Protects against Zika Virus Infection. Mol. Ther. Methods Clin. Dev. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Parkhouse, K.; Kirkpatrick, E.; McMahon, M.; Zost, S.J.; Mui, B.L.; Tam, Y.K.; Karikó, K.; Barbosa, C.J.; Madden, T.D.; et al. Nucleoside-Modified MRNA Immunization Elicits Influenza Virus Hemagglutinin Stalk-Specific Antibodies. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Monslow, M.A.; Elbashir, S.; Sullivan, N.L.; Thiriot, D.S.; Ahl, P.; Smith, J.; Miller, E.; Cook, J.; Cosmi, S.; Thoryk, E.; et al. Immunogenicity Generated by MRNA Vaccine Encoding VZV GE Antigen Is Comparable to Adjuvanted Subunit Vaccine and Better than Live Attenuated Vaccine in Nonhuman Primates. Vaccine 2020, 38. [Google Scholar] [CrossRef]
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified MRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs from Ebola Virus Disease. J. Infect. Dis. 2018, 217. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An MRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Wui, S.R.; Kim, K.S.; Ryu, J.I.; Ko, A.; Do, H.T.T.; Lee, Y.J.; Kim, H.J.; Lim, S.J.; Park, S.A.; Cho, Y.J.; et al. Efficient Induction of Cell-Mediated Immunity to Varicella-Zoster Virus Glycoprotein E Co-Lyophilized with a Cationic Liposome-Based Adjuvant in Mice. Vaccine 2019, 37. [Google Scholar] [CrossRef] [PubMed]
- Rosenkrands, I.; Vingsbo-Lundberg, C.; Bundgaard, T.J.; Lindenstrøm, T.; Enouf, V.; van der Werf, S.; Andersen, P.; Agger, E.M. Enhanced Humoral and Cell-Mediated Immune Responses after Immunization with Trivalent Influenza Vaccine Adjuvanted with Cationic Liposomes. Vaccine 2011, 29. [Google Scholar] [CrossRef] [PubMed]
- Wørzner, K.; Sheward, D.J.; Schmidt, S.T.; Hanke, L.; Zimmermann, J.; McInerney, G.; Hedestam, G.B.K.; Murrell, B.; Christensen, D.; Pedersen, G.K. Adjuvanted SARS-CoV-2 Spike Protein Elicits Neutralizing Antibodies and CD4 T Cell Responses after a Single Immunization in Mice. EBioMedicine 2021, 63. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Arankalle, V.A. Differential Immune Responses in Mice Immunized with Recombinant Neutralizing Epitope Protein of Hepatitis e Virus Formulated with Liposome and Alum Adjuvants. Viral Immunol. 2016, 29. [Google Scholar] [CrossRef] [PubMed]
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Nowakowska, D.; Plasencia, A.; Stanton, V.P.; Sherr, D.H.; et al. Tolerogenic Nanoparticles Suppress Central Nervous System Inflammation. Proc. Natl. Acad. Sci. USA 2020, 117. [Google Scholar] [CrossRef]
- Vives-Pi, M.; Rodríguez-Fernández, S.; Pujol-Autonell, I. How Apoptotic β-Cells Direct Immune Response to Tolerance or to Autoimmune Diabetes: A Review. Apoptosis 2015, 20. [Google Scholar] [CrossRef] [Green Version]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine Is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ. 2016, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol-Autonell, I.; Mansilla, M.J.; Rodriguez-Fernandez, S.; Cano-Sarabia, M.; Navarro-Barriuso, J.; Ampudia, R.M.; Rius, A.; Garcia-Jimeno, S.; Perna-Barrull, D.; Caceres, E.M.; et al. Liposome-Based Immunotherapy against Autoimmune Diseases: Therapeutic Effect on Multiple Sclerosis. Nanomedicine 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Autonell, I.; Serracant-Prat, A.; Cano-Sarabia, M.; Ampudia, R.M.; Rodriguez-Fernandez, S.; Sanchez, A.; Izquierdo, C.; Stratmann, T.; Puig-Domingo, M.; Maspoch, D.; et al. Use of Autoantigen-Loaded Phosphatidylserine-Liposomes to Arrest Autoimmunity in Type 1 Diabetes. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Benne, N.; van Duijn, J.; Lozano Vigario, F.; Leboux, R.J.T.; van Veelen, P.; Kuiper, J.; Jiskoot, W.; Slütter, B. Anionic 1,2-Distearoyl-Sn-Glycero-3-Phosphoglycerol (DSPG) Liposomes Induce Antigen-Specific Regulatory T Cells and Prevent Atherosclerosis in Mice. J. Control. Release 2018, 291. [Google Scholar] [CrossRef]
- Kawakita, A.; Shirasaki, H.; Yasutomi, M.; Tokuriki, S.; Mayumi, M.; Naiki, H.; Ohshima, Y. Immunotherapy with Oligomannose-Coated Liposomes Ameliorates Allergic Symptoms in a Murine Food Allergy Model. Allergy Eur. J. Allergy Clin. Immunol. 2012, 67. [Google Scholar] [CrossRef]
- Jesus, S.; Soares, E.; Cruz, M.T.; Borges, O. Exosomes as Adjuvants for the Recombinant Hepatitis B Antigen: First Report. Eur. J. Pharm. Biopharm. 2018, 133. [Google Scholar] [CrossRef] [PubMed]
- Sabanovic, B.; Piva, F.; Cecati, M.; Giulietti, M. Promising Extracellular Vesicle-Based Vaccines against Viruses, Including SARS-CoV-2. Biology 2021, 10, 94. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current Knowledge on Exosome Biogenesis and Release. Cell. Mol. Life Sci. 2018, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.D.; Maier, C.L.; Pober, J.S. Cytomegalovirus-Infected Human Endothelial Cells Can Stimulate Allogeneic CD4 + Memory T Cells by Releasing Antigenic Exosomes. J. Immunol. 2009, 182. [Google Scholar] [CrossRef] [Green Version]
- Booth, A.M.; Fang, Y.; Fallon, J.K.; Yang, J.M.; Hildreth, J.E.K.; Gould, S.J.; Sandefur, S.; Varthakavi, V. Exosomes and HIV Gag Bud from Endosome-like Domains of the T Cell Plasma Membrane. J. Cell Biol. 2006, 172. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Freed, E.O. HIV Type 1 Gag as a Target for Antiviral Therapy. AIDS Res. Hum. Retrovir. 2012, 28. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wu, D.; Ma, X.; Wang, J.; Hou, W.; Zhang, W. Exosomes as Drug Carriers for Cancer Therapy and Challenges Regarding Exosome Uptake. Biomed. Pharmacother. 2020, 128. [Google Scholar] [CrossRef]
- Rojas, C.; Campos-Mora, M.; Cárcamo, I.; Villalón, N.; Elhusseiny, A.; Contreras-Kallens, P.; Refisch, A.; Gálvez-Jirón, F.; Emparán, I.; Montoya-Riveros, A.; et al. T Regulatory Cells-Derived Extracellular Vesicles and Their Contribution to the Generation of Immune Tolerance. J. Leukoc. Biol. 2020, 108. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.S.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 Expression on Extracellular Vesicles Derived from CD4+CD25+Foxp3+ T Cells Contributes to Their Regulatory Function. Eur. J. Immunol. 2013, 43. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-Containing T-Regulatory-Cell-Derived Exosomes Suppress Pathogenic T Helper 1 Cells. Immunity 2014, 41. [Google Scholar] [CrossRef] [Green Version]
- Cappellano, G.; Raineri, D.; Rolla, R.; Giordano, M.; Puricelli, C.; Vilardo, B.; Manfredi, M.; Cantaluppi, V.; Sainaghi, P.P.; Castello, L.; et al. Circulating Platelet-Derived Extracellular Vesicles Are a Hallmark of Sars-Cov-2 Infection. Cells 2021, 10, 85. [Google Scholar] [CrossRef]
- Barberis, E.; Vanella, V.V.; Falasca, M.; Caneapero, V.; Cappellano, G.; Raineri, D.; Ghirimoldi, M.; De Giorgis, V.; Puricelli, C.; Vaschetto, R.; et al. Circulating Exosomes Are Strongly Involved in SARS-CoV-2 Infection. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef]
- Admyre, C.; Johansson, S.M.; Paulie, S.; Gabrielsson, S. Direct Exosome Stimulation of Peripheral Human T Cells Detected by ELISPOT. Eur. J. Immunol. 2006, 36. [Google Scholar] [CrossRef]
- Martins, P.; Machado, D.; Theizen, T.H.; Guarnieri, J.P.O.; Bernardes, B.G.; Gomide, G.P.; Corat, M.A.F.; Abbehausen, C.; Módena, J.L.P.; Melo, C.F.O.R.; et al. Outer Membrane Vesicles from Neisseria Meningitidis (Proteossome) Used for Nanostructured Zika Virus Vaccine Production. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-Specific CTL Activity Elicited by in Vivo Engineered Exosomes Produced through DNA Inoculation. Int. J. Nanomed. 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anticoli, S.; Manfredi, F.; Chiozzini, C.; Arenaccio, C.; Olivetta, E.; Ferrantelli, F.; Capocefalo, A.; Falcone, E.; Ruggieri, A.; Federico, M. An Exosome-Based Vaccine Platform Imparts Cytotoxic T Lymphocyte Immunity Against Viral Antigens. Biotechnol. J. 2018, 13. [Google Scholar] [CrossRef]
- Polak, K.; Greze, N.; Lachat, M.; Merle, D.; Chiumento, S.; Bertrand-Gaday, C.; Trentin, B.; Mamoun, R.Z. Extracellular Vesicle-Based Vaccine Platform Displaying Native Viral Envelope Proteins Elicits a Robust Anti-SARS-CoV-2 Response in Mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tregoning, J.S.; Brown, E.S.; Cheeseman, H.M.; Flight, K.E.; Higham, S.L.; Lemm, N.M.; Pierce, B.F.; Stirling, D.C.; Wang, Z.; Pollock, K.M. Vaccines for COVID-19. Clin. Exp. Immunol. 2020, 202. [Google Scholar] [CrossRef]
- Bohn, M.K.; Hall, A.; Sepiashvili, L.; Jung, B.; Steele, S.; Adeli, K. Pathophysiology of COVID-19: Mechanisms Underlying Disease Severity and Progression. Physiology 2020, 35. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Battaglini, D.; Pelosi, P.; Rocco, P.R.M. Multiple Organ Dysfunction in SARS-CoV-2: MODS-CoV-2. Expert Rev. Respir. Med. 2020, 14. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383. [Google Scholar] [CrossRef]
- World Health Organization. Draft Landscape of COVID-19 Candidate Vaccines. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 23 April 2021).
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384. [Google Scholar] [CrossRef]
- Nojszewska, M.; Kalinowska, A.; Adamczyk-Sowa, M.; Kułakowska, A.; Bartosik-Psujek, H. COVID-19 MRNA Vaccines (Pfizer-BioNTech and Moderna) in Patients with Multiple Sclerosis: A Statement by a Working Group Convened by the Section of Multiple Sclerosis and Neuroimmunology of the Polish Neurological Society. Neurol. Neurochir. Pol. 2021, 55. [Google Scholar] [CrossRef]
- Knoll, M.D.; Wonodi, C. Oxford–AstraZeneca COVID-19 Vaccine Efficacy. Lancet 2021, 397. [Google Scholar] [CrossRef]
Figure 1.
Overview of liposomes, EVs and PLGA-NPs as carriers for vaccines against viral infections. Proteins, plasmid DNA and mRNA have been successfully formulated in these NPs; empty NPs can also be used as adjuvant in vaccine formulation. Upon injection, they are internalized by APCs and, by reaching the lymph nodes, they present the viral antigen to T cells in order to induce an immune response. Some of these advanced vaccines were successful in eradicating the related viral infections (shown in bold). HBV: Hepatitis B Virus, HCV: Hepatitis C Virus; DV: Dengue Virus; CMV: cytomegalovirus; HIV: Human Immunodeficiency Virus: Sars-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2.
Figure 1.
Overview of liposomes, EVs and PLGA-NPs as carriers for vaccines against viral infections. Proteins, plasmid DNA and mRNA have been successfully formulated in these NPs; empty NPs can also be used as adjuvant in vaccine formulation. Upon injection, they are internalized by APCs and, by reaching the lymph nodes, they present the viral antigen to T cells in order to induce an immune response. Some of these advanced vaccines were successful in eradicating the related viral infections (shown in bold). HBV: Hepatitis B Virus, HCV: Hepatitis C Virus; DV: Dengue Virus; CMV: cytomegalovirus; HIV: Human Immunodeficiency Virus: Sars-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2.


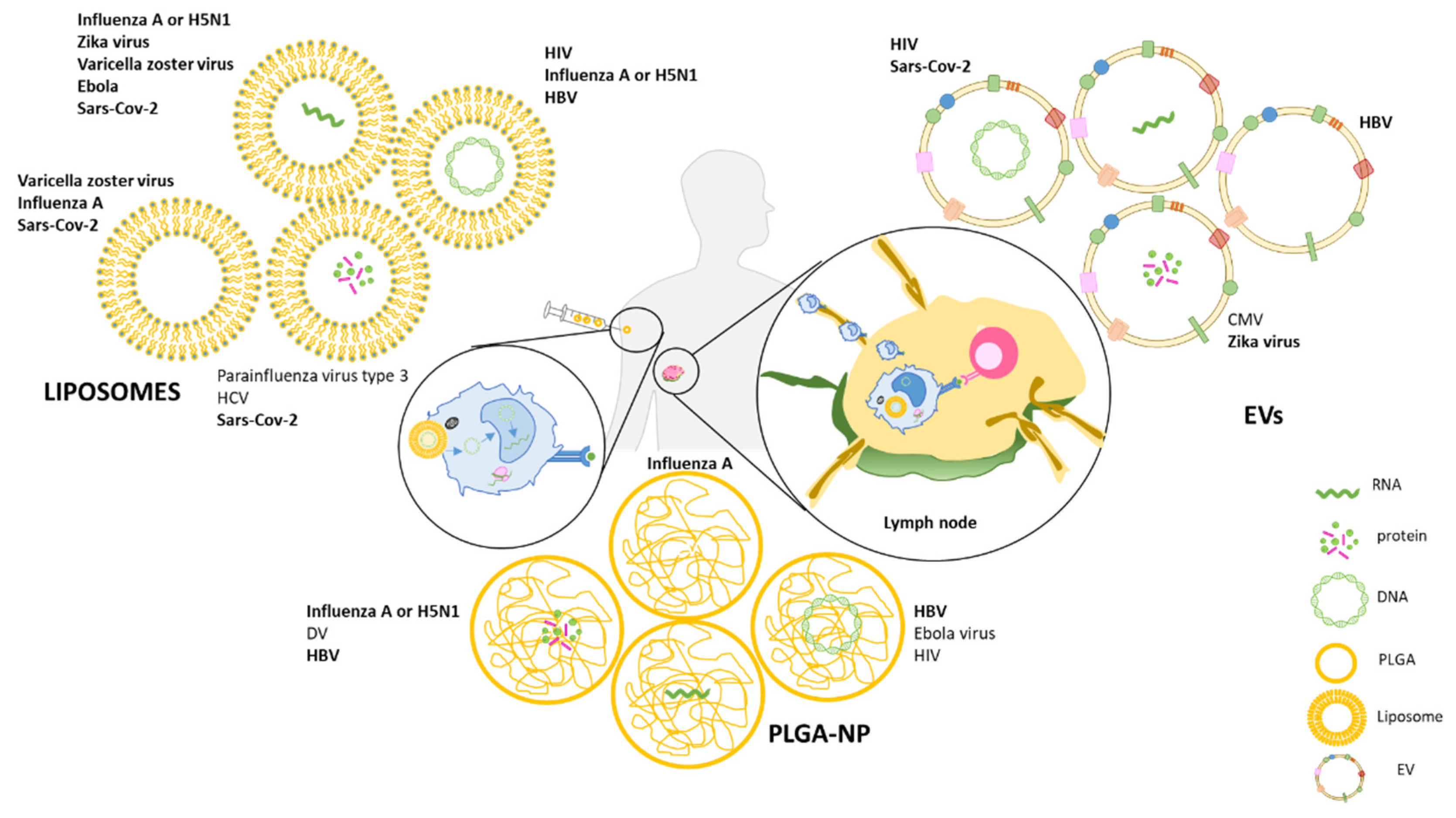{kind=link}
Table 1.
List of advanced vaccines delivered by nano- and microcarriers for the treatment of viral infection.
Table 1.
List of advanced vaccines delivered by nano- and microcarriers for the treatment of viral infection.
| Antigen | Nano/Microparticle Platform | Disease | Animal/Human |
|---|---|---|---|
| Hemagglutinin (HA) | PLGA-NPs adjuvanted with MLPA and muramyl dipeptide (MDP) | Influenza A [51] | BALB/c and C57BL/6 mice |
| Nonstructural protein 1 (NS1) | PLGA/polyethylene glycol (PEG)-NPs | Dengue [52] | BALB/c mice |
| Hepatitis B surface antigen (HBsAg) | mannose-grafted PLGA-NPs | Hepatitis B virus [53] | Balb/c mice |
| Hepatitis B core antigen (HBcAg) | PLGA-NPs with monophospholipid A (MPLA) | chronic hepatitis B infection [53] | C57BL/6J mice |
| Insoluble form of E2 envelope glycoprotein subtype 1b of hepatitis C virus (HCV1b-E2) | PLGA microspheres | Hepatitis C virus (HCV) [57] | Balb/c mice |
| Plasmid DNA encoding HBsAg | PLGA-NPs | hepatitis B virus (HBV) [60] | Balb/c mice |
| M2e peptide (CapM2e) | PLGA-NPs | Influenza A [66] | Balb/c mice |
| Hemagglutinin-neuraminidase (HN) | oligomannose-coated liposome and Poly(I:C) as adjuvant | human parainfluenza virus type 3 (HPIV3) [79] | BALB/c mice |
| Mixture of peptides (pepmix) spanning the entire sequence of nonstructural protein 3 (NS3) | cationic liposomes (CAF09) | chronic hepatitis C virus (HCV) [80] | CB6F1 (C57BL/6 × BALB/c) and C3H mice |
| Four HLA-A*0201-restricted cytotoxic T lymphocytes (CTL) epitopes | Liposomes | Severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV) [82] | HLA-A*0201 transgenic mice |
| Influenza virus nucleoprotein (NP) | Cholesterol/phosphatidylcholine/phosphatidylserine liposomes | Influenza virus [92] | Mice |
| Hemagglutinin (HA) | Lipid nanoparticles (LNPs) | Influenza virus [95] | Mice, rabbits, and ferrets |
| VZV gE antigen | LNPs | Varicella-zoster virus (VZV) [96] | Indian rhesus macaques |
| Ebola envelope glycoprotein | LNPs | Ebola virus [97] | Guinea pigs |
| SARS-CoV-2 spike protein | LNPs | severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [98] | Healthy human adults |
| Recombinant VZV glycoprotein E (gE) | Cationic liposomes with the TLR4 agonist de-O-acylated | Varicella zoster virus (VZV) [100] | BALB/c and C57BL/6 mice |
| Trivalent influenza vaccine (TIV) | Cationic liposome adjuvant system CAF01 | New influenza A (H1N1) [101] | BALB/c mice |
| Spike receptor binding domain (RBD) | Three different adjuvant systems: an aluminum hydroxide (AH), an oil-in-water squalene emulsion (SE) adjuvant resembling MF59™, a cationic liposome-based adjuvant (CAF®01) | Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [102] | C57Bl/6 mice |
| recombinant neutralizing epitope protein (rNEp), a part of the structural capsid protein open-reading-frame-2 (ORF-2) | Liposomes | Hepatitis E virus (HEV) [103] | Mice and rhesus macaques |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cappellano, G.; Abreu, H.; Casale, C.; Dianzani, U.; Chiocchetti, A. Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines 2021, 9, 606. https://doi.org/10.3390/vaccines9060606
AMA Style
Cappellano G, Abreu H, Casale C, Dianzani U, Chiocchetti A. Nano-Microparticle Platforms in Developing Next-Generation Vaccines. Vaccines. 2021; 9(6):606. https://doi.org/10.3390/vaccines9060606
Chicago/Turabian StyleCappellano, Giuseppe, Hugo Abreu, Chiara Casale, Umberto Dianzani, and Annalisa Chiocchetti. 2021. "Nano-Microparticle Platforms in Developing Next-Generation Vaccines" Vaccines 9, no. 6: 606. https://doi.org/10.3390/vaccines9060606
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.