
Targeting Innate Immunity in Cancer Therapy

Section of Hematology/Oncology, Department of Medicine, University of Chicago, Chicago, IL 60637, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Vaccines 2021, 9(2), 138; https://doi.org/10.3390/vaccines9020138
Submission received: 19 December 2020
/
Revised: 30 January 2021
/
Accepted: 2 February 2021
/
Published: 9 February 2021
(This article belongs to the Special Issue Cancer Immunotherapy: Advances and Future Prospects)
Abstract
:The majority of current cancer immunotherapy strategies target and potentiate antitumor adaptive immune responses. Unfortunately, the efficacy of these treatments has been limited to a fraction of patients within a subset of tumor types, with an aggregate response rate of approximately 20% to date across all malignancies. The success of therapeutic inhibition of programmed death protein 1 (PD-1), protein death ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) with immune checkpoint inhibitors (ICI) has been limited to “hot” tumors characterized by preexisting T cell infiltration, whereas “cold” tumors, which lack T cell infiltration, have not achieved durable benefit. There are several mechanisms by which “cold” tumors fail to generate spontaneous immune infiltration, which converge upon the generation of an immunosuppressive tumor microenvironment (TME). The role of the innate immune system in tumor immunosurveillance and generation of antitumor immune responses has been long recognized. In recent years, novel strategies to target innate immunity in cancer therapy have emerged, including therapeutic stimulation of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs); the DNA sensing cGAS/STING pathway; nucleotide-binding oligomerization domain-like receptors (NLRs), such as NLRP3; and the retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs). In addition, therapeutic modulation of key innate immune cell types, such as macrophages and natural killer cells, has been investigated. Herein, we review therapeutic approaches to activate innate immunity within the TME to enhance antitumor immune responses, with the goal of disease eradication in “cold” tumors. In addition, we discuss rational immune-oncology combination strategies that activate both innate and adaptive immunity, with the potential to enhance the efficacy of current immunotherapeutic approaches.
1. Introduction
There is growing evidence that successful immune-mediated elimination of cancer requires coordination between the innate and adaptive arms of the immune system (Figure 1). Innate immune cells, such as dendritic cells (DCs), detect early cancers by a variety of mechanisms, including presentation of tumor-associated neoantigens or through sensing tumor-derived pathogen or damage-associated molecular patterns (PAMP/DAMPs) by pattern recognition receptors (PRRs) [1,2,3,4]. These mechanisms trigger proinflammatory programs and the release of proinflammatory cytokines, chemokines and type I interferons with accompanying DC maturation and trafficking to lymph nodes, where they engage the adaptive immune system and prime and activate antigen-specific T cells. T cells traffic and infiltrate the tumor bed through chemokine and cytokine gradients and mediate tumor elimination following interaction with their cognate antigen [5]. In addition, innate immune cells, such as macrophages and natural killer cells, contribute to tumor elimination through direct tumor cell killing by phagocytosis and cytotoxic mechanisms, respectively.
In recent years, evasion of antitumor immune response has been recognized as a hallmark of cancer and can be mediated by multiple mechanisms [6]. In the context of solid tumors, the absence of T cell infiltration on histopathologic analysis indicates one mechanism for evasion of antitumor immunity [7]. Gene expression analyses of the TME in solid tumors has identified genomic signatures which correlate with the presence or absence of T cell infiltration, referred to as “hot” and “cold” tumors, respectively [8,9,10]. More comprehensive classification has suggested four categories of TMEs: hot, altered-excluded, altered-immunosuppressed and cold [11]. These immune phenotypes have been observed to exist in a distribution, with certain cancer types having a higher proportion of “hot” immune phenotype, such as lung adenocarcinoma and clear cell renal cell carcinoma [12]. In addition, these immune phenotypes deploy distinct mechanisms to avoid immune-mediated elimination. For example, in response to T cell infiltration, “hot” tumors upregulate immune checkpoints, such as PD-1/PD-L1, within the TME, which directly suppress T cell effector mechanisms [13]. In contrast, “cold” tumors fail to generate spontaneous immune infiltration altogether through either “lack of antigenicity”, which results from defects in antigen processing or presentation, or “lack of immunogenicity” due to absence of tumor antigens capable of stimulating the immune system [14]. Of increasingly recognized significance, “cold” tumors prohibit T cell infiltration through orchestration of an immunosuppressive TME characterized by cell types, such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) [14,15,16].
“Hot” and “cold” tumor immune phenotypes carry prognostic significance, as solid tumors with T cell infiltration have been observed to have improved outcomes in multiple treatment settings [7,9,17,18,19]. To date, the success of therapeutic inhibition of PD-1/PD-L1 and CTLA-4 with immune checkpoint inhibitors (ICI) has been limited to “hot” tumors, while “cold” tumors have typically failed to benefit from ICI [9,19,20,21,22]. A large percentage of common malignancies, including prostate, breast and pancreatic cancers, are characterized as having “cold” TMEs and have historically not benefited from ICI. As such, there remains a critical unmet need to develop therapeutic approaches which drive innate immune activation and promote T cell infiltration in immunologically “cold” tumors. Herein, we review the roles of key innate immune cell types and discuss emerging therapeutic strategies to target these cell types to enhance antitumor immune responses, with the goal of disease eradication in “cold” tumors.
2. Key Cellular Components of Innate Immunity
Innate immunity comprises a diverse cadre of cell types which function as the body’s “first defense” against microbes. Each distinct cell type has a unique role; however, there exists overlap in function and cellular machinery, including expression of PRRs and proinflammatory response to detection of PAMPs/DAMPs. The expression of PRR and other innate immune pathways by innate immune cell type is summarized in (Table 1).
2.1. Dendritic Cells (DCs)
DCs are a family of antigen-presenting cells (APC) that perform critical functions in the initiation of antigen-specific immunity and tolerance. DCs express a diverse array of PRRs which, upon detecting PAMPs and DAMPs, lead to the upregulation of Major Histocompatibility Complex (MHC), costimulatory molecules required for T cell activation and CCR7 expression, the latter a key chemokine receptor that directs migration into tumor draining lymph nodes (TDLN) [23,24]. In TDLNs, DCs present tumor-associated antigens and generate antigen-specific CD8+ T cells as part of the cancer immunity cycle.
Dendritic cells derive from common myeloid progenitor cells and can differentiate into four primary DC phenotypes, which include conventional type I DCs (cDC1s), conventional type 2 DCs (cDC2s), plasmacytoid DCs (pDCs) and monocyte-derived DCs (MoDCs) [25]. cDC1s, characterized by BATF3 and IRF8 expression, are the most effective inducers of cell-mediated immunity due to their adept antigen processing and cross presentation. Multiple studies have demonstrated that BATF3-deficient mice are unable to eliminate immunogenic tumors or respond to immune-mediated therapies [4,26]. cDC2s have been found to enrich intratumoral CD4+ T cell density, which supports CD8+ T cell activity [27]. pDCs are a rare subtype of DCs that were initially termed interferon-producing cells (IPCs) due to their capacity to produce interferons and stimulate innate immunity when exposed to viral stimuli [28]. pDCs also function to directly regulate T cell activity; however, they have a comparatively blunted capacity to prime naïve T cells compared to conventional DC subtypes. More recent research suggests that pDCs may play a protumor role, as their infiltration into tumors is a poor prognostic factor in multiple cancers. The role of MoDCs in antitumor immunity is less clear; however, they have been shown to contribute to sustaining immunity following chemotherapy- or radiotherapy-driven cell death [24].
2.2. Macrophages
Macrophages are a type of myeloid cell that reside in healthy tissues throughout the body and perform critical functions to maintain tissue homeostasis and orchestrate innate immune responses [29]. TAMs exist in a continuum of polarization states between protumorigenic M2 macrophages and antitumorigenic M1 macrophages which correspond to dynamic gene expression programs [30,31,32,33]. In many cancer types, macrophages are driven to a M2 functional program which supports tumor growth, analogous to their role in tissue remodeling/wound healing, rather than anticancer immune activation. M2 TAMs mediate local immunosuppression via the production of IL-10 and TGF-β, suppression of T cell proliferation via extracellular arginine-depletion and enrichment of regulatory T cells via secretion of CCL2 [34,35,36]. Differentiation of a monocyte precursor to an M2 macrophage phenotype is promoted by hypoxia and immunosuppressive cytokines, such as IL-4, IL-10 and IL-13 [37,38]. In contrast, antitumorigenic M1 macrophages facilitate tumor control via multiple mechanisms, including phagocytosis and secretion of proinflammatory cytokines, such as IFN-γ, IFN-β and IFN-α [39]. Recent efforts in single-cell RNA sequencing of solid tumors have identified multiple subclusters of TAMs, which suggests that macrophage functionality exists across a continuum of states, and therefore, binary classification inadequately represents complex TAM phenotypes. For example, one study in pancreatic adenocarcinoma identified five subsets of TAMs each with distinct gene expression profiles which correlate with the diverse immunosuppressive or immune-stimulatory functions listed above [40,41]. Histologic analysis of clinical specimens demonstrated that tumor-associated macrophages (TAMs) are associated with worse overall survival [42]. In preclinical models, TAM depletion enhanced efficacy of radiation, chemotherapy and ICI [43,44,45].
2.3. Neutrophils
Neutrophils are circulating myeloid cells which function in the innate immune system’s response to bacterial infection. Neutrophils have been observed in high proportions within the immune infiltrate of many solid tumors, and elevations in both tumor infiltrating neutrophils (or tumor-associated neutrophils, TANs) and peripheral blood neutrophils associate with unfavorable outcomes [46,47,48]. Protumorigenic mechanisms of TANs include promotion of neoangiogenesis, tumor migration and invasion and local immunosuppression [49,50,51]. In contrast, TANs have been observed to exert antitumorigenic effects via direct tumor cell cytotoxicity, production of reactive oxygen species and secretion of proinflammatory cytokines [52,53,54]. The functional plasticity of TANs has led to a bipolar classification similar to that of TAMs with protumorigenic N2 TANs and antitumorigenic N1 TANs. Research has asserted that N1 TANs predominate in early tumor development. However, TGF-β, IL-10 and IL-6 signaling supports protumorigenic N2 TANs differentiation over time [55,56,57]. Despite recent advances, the tumor immunobiology of TANs remains largely under active investigation.
2.4. Myeloid Derived Suppressor Cells (MDSCs)
MDSCs are a heterogenous group of myeloid cells distinguished from other myeloid cell types by their predominantly immunosuppressive properties. Differentiation to MDSC phenotype is associated with chronic inflammation and low-level exposure to growth factors and inflammatory mediators responsible for normal maturation of myeloid cells [58]. Morphologically, MDSCs exist as mononuclear or polymorphonuclear subsets referred to as Mo-MDSC and PMN-MDSC, respectively. Compared to classical neutrophils and monocytes, MDSCs have an increased expression of immunosuppressive molecules, such as nitric oxide (NO) and IL-10, weaker phagocytic abilities and a higher expression of the immunosuppressive enzyme arginase-1 [59]. In many solid tumor types, MDSCs have been observed to suppress both innate and adaptive arms of the immune system and contribute to a “cold” TME [60,61]. MDSC infiltration into the tumor site correlates with increased cancer stage and tumor burden as well as worse prognosis [59,62]. These properties highlight the relevance of MDSCs to therapeutic strategies aimed at overcoming an immunosuppressive TME.
2.5. Mast Cells
Mast cells are granulated innate immune cells that reside in peripheral tissues and secrete a wide array of signaling molecules that facilitate tissue repair and local immune responses. The presence of mast cells in the tumor stroma of several solid tumors has been associated with poor prognosis. However, in breast cancer, the presence of mast cells was found to be a positive prognostic factor [63,64,65]. Precise localization of mast cells within the TME may also impact tumor development [66]. Molecules secreted by mast cells, such as vascular endothelial growth factor (VEGF) and matrix metalloproteases, have been observed to support tumor growth and metastatic potential through the promotion of angiogenesis and lymphangiogenesis and modification of the extracellular matrix [67,68]. Tryptase secreted by mast cells acts as an agonist of proteinase-activated receptor-2 (PAR-2) to further stimulate endothelial cell proliferation and has been associated with tumor cell migration [69]. Anticancer activities of tumor-associated mast cells include mediating tumor cell apoptosis through inflammatory mediators and the peroxidase system [70,71]. The comprehensive understanding of the function of mast cells in the TME remains an area of active investigation.
2.6. Natural Killer (NK) Cells
NK cells belong to a heterogenous family of innate lymphoid cells which lack genetically rearranged antigen receptors and do not require APC-dependent antigen presentation and selection for their cytotoxic activity. NK cells detect stress-induced molecules and altered or downregulated MHC class-I and exert perforin and granzyme-dependent antitumor cytotoxicity similar to CD8+ T cells [72]. In addition, NK cells are activated upon simultaneous binding of multiple receptors, including NKp46, NKG2D, 2B4, CD2 and DNAM, all of which are upregulated in the presence of cellular stress [72]. In addition to cytolytic functions, NK cells can also mediate Fas-ligand-induced target cell apoptosis [73]. It has been long known that NK cells exert early control of transformed cells, thereby serving an important role in tumor immunosurveillance [74]. Early work exploring the antitumor activity of NK cells revealed that NK cell depletion in MCA-induced fibrosarcoma led to significant tumor growth [75]. More recent studies have shown that the release of IFN-γ and chemokines CCL5 and CXCL1/2 by NK cells can potentiate adaptive immune responses through DC activation and induction of M1 macrophages [76,77,78]. Studies have also revealed that NKG2D activation stimulates macrophages and CD8+ T cells [79]. Histologic analysis of human pulmonary squamous cell carcinoma and adenocarcinoma specimens revealed an inverse correlation between NK cells and metastatic disease burden and mortality [80,81,82]. Taken together, the biological rationale and preclinical findings described above suggest a significant potential of NK cell-directed therapies in generating meaningful anticancer responses.
3. Activating the Innate Immune System in Cancer Therapy
Given the complex interplay of innate immune cells within the TME, targeting its key components could provide therapeutic benefits in cancer. There are several approaches to target innate immunity in cancer (Table 2). These include therapeutic stimulation of PRRs, such as Toll-like receptors (TLRs); the DNA sensing cGAS/STING pathway; nucleotide-binding oligomerization domain-like receptors (NLRs), such as NLRP3; and the retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs) with synthetic agonists which resemble cognate PAMP/DAMPs (Figure 2). Direct therapeutic targeting of TAMs, NK cells and DCs are also being evaluated (Figure 3). Within each section, we review rational immune-oncology combination strategies that activate innate immunity, thereby enhancing the efficacy of current immunotherapies.
3.1. Toll-like Receptors (TLRs)
TLRs are highly conserved transmembrane and intracellular PRRs found in a variety of cell types and play a critical role in the detection of microbial pathogens by innate immune cells [83]. In humans, ten TLRs have been identified, and they are expressed by T cells, B-cells, APCs as well as many non-immune cells, including epithelial and endothelial cells. TLRs localize to two different regions of the cell: the plasma membrane and the endosome. TLR 1, 2, 5 and 6 are found specifically in the plasma membrane. TLR 3, 4, 7, 8 and 9 are found specifically in the endosomes, while TLR4 can signal at both locations [84]. TLRs located on the cell membrane recognize lipids and proteins, and TLRs located on the membrane of intracellular endosomes recognize nucleic acids. TLR signaling occurs through activation of adaptor proteins that enter the nucleus and regulate the expression of proinflammatory mediators. These adaptor proteins include MyD88, interferon regulatory factor 3 (IRF-3), NF-kB and activator protein-1 (AP-1) [85].
TLRs are expressed by both cancer cells and immune cells within tumors, and therefore, have pleiotropic effects on immunomodulation of the TME. When activated on immune cells, TLRs mediate a broad range of immunostimulatory effects that promote antitumor T cell responses [86]. Studies demonstrated that chemotherapy-induced DAMP release activated TLR4 and resulted in DC maturation and other immune activating effects [87]. TLR9 signaling has been shown to activate pDCs and result in the secretion of high levels of type I interferon [28]. TLR7 and TLR8 activation led to the reprogramming of tumor promoting M2 TAM phenotype to antitumor M1 phenotype [88,89]. On T cell populations, TLRs have diverse effects, including reduced suppressive function of T-regulatory (Treg) cells and enhancement of the survival, proliferation and cytokine production of CD8+ T cells [90,91,92]. Within cancer cells, TLR activation has been shown to trigger both apoptosis and cell survival [93]. In addition, constitutive activation of TLRs can lead to chronic inflammatory states which are associated with the recruitment of immunosuppressive cell types, such as myeloid derived suppressor cells (MDSCs), leading to tumor progression [94,95,96].
The diverse immunostimulatory functions of TLR activation provide ample rationale for therapeutic targeting for the treatment of tumors with a “cold” TME. Therapeutic administration of synthetic TLR agonists by intratumoral injection have revealed multiple anticancer effects in preclinical models. Activation of TLR9 by synthetic CpG-oligodeoxynucleotides was shown to revert resistance to PD-1 blockade by expanding multifunctional CD8+ T cells [97,98]. TLR9 agonist candidates SD-101 and CMP-001 were well tolerated in early phase clinical trials and demonstrated clinical activity in combination with anti-PD-1 treatment in melanoma and head and neck squamous cell carcinoma (HNSCC) [99,100]. These candidates are currently undergoing phase 2 trials for the treatment of solid tumors in combination with other forms of immunotherapy [NCT01042379, NCT04050085, NCT03007732, NCT03084640, NCT02554812, NCT03438318]. TLR7/8 agonist NKTR-262, in combination with the systemic CD122-biased IL-2 pathway agonist, bempegaldesleukin, promoted antigen presentation and CD8+ T cell infiltration in preclinical models [101]. This combination was well tolerated in early phase clinical trials and is currently being investigated in combination with anti-PD-1 therapy [NCT03435640]. Another TLR7/8 agonist, MEDI9197, was shown to activate pDCs and macrophages leading to interferon-α (IFN-α), IL-12 and IFN-γ release and subsequent antitumor T cell response and tumor regression in syngeneic murine models [102]. This molecule is currently being evaluated in early phase clinical trials [102]. Motilomod, a small molecule TLR8 agonist, is undergoing investigation in combination with nivolumab for HNSCC [NCT03906526].
Three TLR agonists are currently FDA-approved and in clinical use. Bacillus Calmette-Guerin (BCG), an attenuated strain of Mycobacterium bovis is a TLR2/4 ligand that is approved for treatment of superficial, non-muscle invasive bladder cancer [103]. In addition, the TLR4 agonist monophosphoryl A is approved as a vaccine adjuvant and TLR7 agonist imiquimod is approved for the treatment of genital warts and basal cell carcinoma [104,105,106].
3.2. cGAS/STING Pathway
The cGAS-STING pathway detects cytosolic DNA associated with viral infection and tumorigenesis. The cellular mechanisms by which the pathway mediates immune activation have been previously well described [107,108,109]. In brief, cGAS senses cytosolic DNA and activates Stimulator of IFN genes (STING) through synthesis of the cyclic dinucleotide, cyclic GMP-AMP (cGAMP) [110,111]. Upon activation at the endoplasmic reticulum and subsequent translocation to the golgi, STING activates IRF-3 and NF-kB transcriptional programs, resulting in the expression and release of type I IFN [112,113]. STING is expressed by multiple immune and non-immune cells, and its ability to sense tumor-derived DNA can be harnessed for cancer therapeutic purposes. In murine tumor models, STING-dependent cytosolic DNA sensing by tumor-resident DCs was found to induce type I IFN production and was required for CD8+ T cell infiltration and rejection of immunogenic tumors [108]. Intratumoral injection of STING agonists in preclinical models recapitulated these proinflammatory effects and induced profound tumor regression [114].
Therapeutic STING activation has been most successful with synthetic cyclic dinucleotides (CDNs) due to their structural versatility and ability to bind prevalent allelic variants in human STING [114]. Currently, there are multiple ongoing clinical trials with synthetic CDN STING agonists, which are comprehensively reviewed elsewhere [115]. MK1454 is undergoing clinical evaluation as monotherapy or in combination with pembrolizumab for the treatment of advanced solid tumors [NCT03010176, NCT04220866] [116]. ADU-S100/MIW815 is being investigated in phase II clinical trials in combination with ICI as a first line treatment for HNSCC [NCT03937141] [117]. Exploration of the dose-dependent effects of ADU-S100 revealed that while higher doses were more effective at clearing injected tumors, lower doses elicited IFN-γ-driven CD8+ T cell expansion and demonstrated synergy with ICI [118]. While most STING agonists have been administered via intratumoral injection, BMS-986301 is being evaluated for systemic intramuscular administration. Additionally, GSK3745417 is an intravenous STING agonist being evaluated alone and in combination with ICI in advanced solid tumors [NCT03956680, NCT03843359] [119]. A newly developed non-CDN amidobenzimidazole-based small molecule given intravenously displayed potent STING agonism and durable preclinical antitumor activity, thus representing an important step in adapting STING modulators for systemic administration [120]. Clinical evaluation of tolerability and safety is ongoing. Other emerging approaches include engineered liposomal nanoparticle packaging, ex vivo loading of STING into exosomes and bacterial modification to optimize treatment delivery [121].
The mechanistic underpinnings of the cGAS-STING pathway make STING agonists an attractive adjuvant to cancer vaccines. PancVAX is a vaccine composed of synthetic peptides and a STING agonist-based adjuvant, ADU-V19, which when used in combination with an OX40 agonist and anti-PD-1 therapy in preclinical models leads to significant tumor regression and improved survival [122]. The STINGVAX vaccine consists of a CDN ligand formulated with GM-CSF, which has potent antitumor activity as monotherapy across multiple murine models. It was shown to upregulate PD-L1 on the TME and resulted in combinatorial regression of tumors resistant to anti-PD-1 monotherapy [123]. STING agonists used in combination with chemotherapy and radiation therapy have been shown to amplify antitumor immune responses in both preclinical and clinical settings [124,125]. Interestingly, preclinical evaluation of poly ADP-ribose polymerase (PARP) inhibitors demonstrated a STING-dependent immune response which enhanced the efficacy of ICI [126].
Despite the encouraging framework for STING agonism in the treatment of cancer, some nuances with respect to context and dose-dependent effects are emerging. Recent research has demonstrated that cancers with high chromosomal instability (CIN) have increased levels of cytosolic DNA which contributes to endogenous cGAS/STING activation which, if sustained over time, can promote tumorigenesis, immune evasion and metastasis [111,127,128]. The existence of this phenotype represents an important consideration in developing rational strategies that incorporate STING agonists and pave the way towards mechanistic evaluation of intermittent vs. continuous STING pathway activation in generating durable antitumor responses.
3.3. Retinoic Acid Inducible Gene-I-like Receptors (RLRs) and RIG-I
RLRs are PRRs that detect cytosolic RNA physiologically in the context of viral infections [129]. The best studied RLRs are RIG-I, MDA5 and LGP2. RIG-I primarily binds shorter dsRNA, whereas MDA-5 interacts with longer fragments [130]. Upon binding of RNA, RLRs undergo a conformational change that exposes a CARD domain, which activates downstream effectors that promote the transcription factors IRF-1, IRF-3, IRF-7, NF-kb and IFN response elements, similar to the cGAS/STING pathway [127,128]. In addition, the CARD domain plays a role in inflammasome activation [131]. RIG-I mediates a wide range of immunostimulatory functions, including DC maturation, priming of T cells and enhancement of NK cell degranulation and cytolytic activity [132,133]. RIG-I also initiates programmed cell death (PCD) through both the intrinsic and extrinsic apoptotic pathways as well as pyroptosis, an inflammatory variant of PCD. The factors determining which form of cell death is driven by RIG-I is very context dependent and not clearly defined [132]. Furthermore, recent work has revealed that RIG-I is required for adequate response to anti-CTLA-4 treatment by inducing caspase-3-mediated tumor cell death, following which the tumor-associated antigens are processed and presented, leading to a robust CD8+ T cell-mediated adaptive immune response [134].
These proinflammatory mechanisms provide rationale for RIG-I as an innate immunotherapeutic target. Synthetic agonists specific to RIG-I or to RLRs more broadly, classified as RLR mimetics, are being investigated in multiple cancer types [135,136]. Intralesional RGT100 (MK-4621), a synthetic RIG-I agonist, demonstrated tolerability in a Phase I clinical trial and is now being studied as monotherapy or in combination with pembrolizumab for the treatment of advanced solid tumors [NCT03739138] [137]. In addition, synthetic stem-loop RNA (SLR) sequences that are highly specific for the RIG-I RNA binding pocket have been developed. SLRs represent a potent strategy for stimulating RIG-I due to precise structural optimizations and resistance to nucleases [138]. In vivo studies have shown that intratumoral delivery of SLR14 activated both NK cell and CD8+ T cell populations, resulting in significant antitumor effects [139]. A novel oral RIG-I agonist, SB9200, has demonstrated strong antiviral activity against resistant hepatitis C infection through induction of type I IFNs, and as such may have a role in future immunotherapeutic strategies [132,140].
Methods to stimulate RLRs more broadly include the use of poly-ICLC, a synthetic double-stranded RNA that activates TLR-3; MDA5; and, to a lesser extent, RIG-I [141,142]. In a pilot trial conducted with patients with refractory HNSCC and melanoma, an “autovaccination” strategy utilizing intralesional and intramuscular administration of poly-ICLC was well tolerated and led to the generation of antitumor T cell activation. This novel approach is now being further investigated in a phase II clinical trial [NCT02423863] [143].
3.4. CD40
Activation of an antigen-specific T cell response requires T cell receptor-mediated antigen presentation and interaction between costimulatory molecules, such as CD28 and the B7 family. CD40 is a well-characterized costimulatory molecule and member of the tumor necrosis factor (TNF) receptor superfamily and is highly expressed on APCs, including DCs, macrophages, B cells and many non-immune cells [144]. CD40 ligand (CD40L) is expressed by T cells and other non-immune cells. Upon cross-linking, CD40 triggers cell proliferation, upregulation of MHC and other costimulatory molecules and secretion of cytokines, including IFN-γ and IL-12a [145,146,147,148]. CD40-mediated CD8+ T cell activation has been shown to be independent of CD4+ T cells and innate immune sensors [149,150]. In addition, CD40 activates tumor-associated macrophages to the activated M1 phenotype. In one study, this resulted in the secretion of matrix metalloproteinases that modified tumor stroma and enhanced the effects of chemotherapy in a murine model of pancreatic adenocarcinoma [151,152].
Preclinical and early clinical experience has highlighted the efficacy of combining CD40 agonist with chemotherapy, radiation and peptide vaccines [153,154,155]. In an immunologically cold pancreatic ductal adenocarcinoma (PDA) murine model, treatment with a CD40 agonist alone yielded minimal response. However, the addition of chemotherapy resulted in a vaccine-like effect in which potent antigen-specific T cells were generated as a consequence of chemotherapy-induced cell death and antigen release [150]. Of note, CD40 agonism in combination with gemcitabine caused lethal hepatotoxicity in mice, ameliorated by giving CD40 agonist five days or more prior to chemotherapy [156]. This data established a rationale for a clinical trial of neoadjuvant and adjuvant selicrelumab, a CD40 agonist, in combination with nab-paclitaxel in resectable PDA [NCT02588443]. The addition of anti-PD-1 treatment further enhanced tumor regression, and a clinical trial in combination with anti-PD-1 therapy in PDA is underway and has observed encouraging early phase results [NCT02482168, NCT02304393] [157,158].
In recent preclinical studies utilizing murine models of non-immunogenic solid malignancies, triple therapy with a CD40 agonist, an anti-PD-1 antibody and a T cell activating vaccine stimulated macrophages and DCs reduced T cell exhaustion and generated effector memory CD4+ T cells [155]. In murine tumor models with high PD1+ T cells, CD40 agonism reversed T cell exhaustion and enhanced response to anti-PD-1 and anti-CTLA-4 immune checkpoint inhibition, suggesting a re-sensitization benefit for patients who experience resistance to ICI [159]. Furthermore, CD40 agonism and anti-CTLA-4 antibody achieved promising response rates and demonstrated increased T cell infiltration and activation in patients with melanoma [160]. From a clinical translational standpoint, multiple CD40 agonists are in early phase clinical trials for solid tumors, with each agonist exhibiting unique properties [161,162]. In CD40-expressing malignancies, such as chronic lymphocytic leukemia, CD40 monoclonal antibodies (mAbs) mediate direct tumor cell death via antibody-dependent cellular cytotoxicity [163,164].
3.5. NLRP3-Inflammasome
Inflammasomes are large cytosolic multiprotein complexes that mediate critical inflammatory innate immune responses in the host defense against microbial pathogens. The nucleotide-binding oligomerization domain-like receptors, or NOD-like receptors (NLRs) are a diverse family of intracellular PRRs. The nucleotide-binding domain and leucine-rich repeat family pyrin domain (NLRP) is a subfamily of NLRs and an important component of the inflammasome. The most well-characterized NLRP is NLRP3, which is expressed in macrophages, DCs and lymphocytes, in addition to non-immune populations, such as epithelial cells [165]. NLRP3 inflammasome activation is driven by the recognition of PAMPs or DAMPs generated in response to cellular stress. Examples include ATP, extracellular glucose and reactive oxidative species (ROS) [166,167,168,169,170]. When activated, NLRP3 forms the NLRP3 inflammasome by a multistep sequential process of priming and activation [166]. NLRP3 oligomerizes and cleaves procaspase-1 to caspase-1 and culminates in the release and proteolysis of the cytokines IL-18 and IL-1β. Gasdermin-D, a critical component of the inflammasome complex, permeabilizes the cell membrane and facilitates pyroptosis, as opposed to apoptosis [171,172,173]. Inflammasome-induced pyroptosis is canonically a caspase-1-dependent, immunostimulatory form of PCD in which target cell cytoplasmic contents are released to induce inflammation [174].
The impact of the NLRP3 inflammasome activation on tumorigenesis is complex. Preclinical evaluation has demonstrated that NLRP3 inflammasome mediated IL-1β and IL-18 release results in IFN-γ production and CD8+ T cell-dependent tumor regression [175,176]. In a preclinical model of hepatocellular carcinoma (HCC), estrogen receptor signaling increased cancer cell death through NLRP3 inflammasome-initiated, caspase-1-dependent pyroptosis and inhibition of autophagy in cancer cells [177]. In a preclinical model of colorectal cancer liver metastases, mice deficient in NLRP3 inflammasome had increased metastatic growth due to impaired IL-18 signaling and maturation of hepatic NK cells [178]. In the clinical setting, analysis of HCC patient tissue samples revealed an association between low expression of NLRP3 inflammasome components and more advanced HCC [179].
Dysregulated NLRP3 inflammasome activation has also been observed to promote tumorigenesis in multiple solid tumor murine models. 3′methylcholanthrene (MCA)-induced sarcomas in mice deficient in NLRP3 had decreased tumor burden compared to wild-type mice, attributed to NLRP3-mediated suppression of NK cell immune surveillance [180]. In oral squamous cell carcinoma cell lines, NLRP3 inflammasome activation and IL-1β secretion were associated with tumor progression, metastases and infiltration of immune suppressive myeloid cells, such as TAMs and MDSCs into the TME [181,182,183]. In addition, recent studies have shown that NLRP3 inflammasome activation within tumor cells can drive resistance to anti-PD-1 checkpoint inhibitor treatment. In anti-PD-1 treated solid tumor murine models, activated CD8+ T cells induced NLRP3 inflammasome activation within tumor cells, which resulted in downstream Wnt5a-mediated CXCR2 ligand expression and MDSC recruitment into the tumor tissue. This effect was abrogated by genetic and pharmacologic inhibition of NLRP3 [184].
Therapeutic targeting of NLRP3 is an emerging strategy, and a rationale exists for both NLRP3 inflammasome activation and inhibition. The novel NLRP3 agonist BMS-986299 is being studied in a phase I clinical trial as monotherapy and in combination with nivolumab and ipilimumab in advanced solid tumors [NCT03444753]. Saponins, derived from tree bark, contain molecules with strong proinflammatory properties that can activate the NLRP3 inflammasome and are being investigated for use as a vaccine adjuvant [185,186]. Though preclinical evaluations of NLRP3 inhibitors are expanding, they have not yet entered clinical trial testing [187]. In the HNSCC murine model, a novel NLRP3 inhibitor MCC950 delayed tumor growth, reduced MDSCs, Tregs and TAMs, while also increasing T cell infiltration within the TME [188].
Several recent studies have explored the mechanistic relationship between pyroptosis core proteins in cancer. Decreased expression of a primary mediator of pyroptosis, gasdermin D, is linked to enhanced cancer cell proliferation in vitro and tumor growth in vivo, whereas increased gasdermin E expression enhanced drug sensitivity of tumor cells [189,190]. These findings have led to therapeutic interest in activating pyroptosis as an antitumor strategy [191].
3.6. Dendritic Cell Directed Strategies
DCs are a key component of the innate immune system’s antitumor response. Strategies targeting PRRs, such as TLRs and RIG-I agonism, mediate their antitumor effect through activation of DCs [24]. Other DC activating and mobilizing agents, such as FLT3 ligands, have shown preclinical promise and are being evaluated in clinical trials [NCT03789097] [192]. Alternatively, activation of DCs can be achieved by blunting suppressive programs, which has led to the exploration of STAT3 and indoleamine 2,3-dioxygenase (IDO) inhibitors, now in various phases of clinical evaluation [193,194]. An increasingly nuanced understanding of DC biology has led to clinical breakthroughs in many DC-based therapeutic strategies that have earned FDA approval or advanced in clinical trials for the treatment of solid cancers [24].
3.7. Adoptive DC Strategies
Ex vivo vaccination of DCs with tumor-associated antigens (TAAs) and personalized tumor-specific antigens (TSAs) have also been explored. In these methods, autologous DC subsets are isolated from blood, activated ex vivo and loaded with antigen and reintroduced into the patient. Such DC vaccination strategies have long been an area of interest, with upwards of 200 clinical trials exploring their potential. Sipuleucel-T is an FDA-approved autologous DC vaccine in which DCs are incubated ex vivo with a fusion protein of GM-CSF and prostate-specific antigen (PSA) to treat metastatic, castrate-resistant prostate cancer [195]. Novel methods for optimizing the adoptive transfer of autologous antigen-loaded DCs are currently being developed, including personalized antigen selection and ex vivo activation with adjuvants [196]. In addition, combination with ICI is currently being evaluated to help overcome hostile immunosuppressive TMEs.
3.8. TAM Directed Strategies
Multiple therapeutic strategies have been found to drive polarization of protumorigenic M2 TAMs towards antitumor M1 TAMs, which include activation of CD40, TLR3, TLR4, TLR7/8 and TLR9 [89,90,151,155,197], covered in prior sections. Here, we discuss additional strategies being pursued in clinical trials.
3.8.1. Colony Stimulating Factor 1 (CSF-1)
Blockade of macrophage CSF-1 binding to its cognate receptor (CSF-1R) resulted in the depletion of M2 TAMs, enrichment in M1 TAMs and tumor control in murine preclinical models [198,199,200]. Synergy in combination with anti-PD-1 and anti-CTLA-4 therapy has also been demonstrated [201]. Clinical trials of antibodies and small molecule inhibitors targeting CSF-1R are ongoing as monotherapy, and in combination with chemotherapy and ICI [197,202,203].
3.8.2. PI3K-γ Inhibition
PI3K activation via the p110γ isoform within macrophages drives polarization to a protumorigenic M2 phenotype [204]. Consistent with this observation, inhibition of PI3K-γ isoform has been demonstrated to promote M1 polarization of TAMs and increase proinflammatory cytokines, resulting in antitumor immune activation and suppression of tumor growth in multiple solid tumor cancer models [204,205,206]. PI3K-γ inhibitor IPI-549 is currently undergoing clinical development for the treatment of solid tumors [207].
3.8.3. CD47- Signal-Recognition Protein Alpha (SIRPα)
Therapeutic targeting of CD47, a surface glycoprotein and “don’t eat me” signal expressed on immune and tumor cells, which interacts with SIRPα on macrophages to suppress phagocytosis, is also being investigated. CD47-SIRPα blockade using CD47 antibodies was shown to restore phagocytosis of tumor cells by TAMs in vitro, stimulate antigen-specific T cells and limit tumor growth in murine models [208,209]. Multiple CD47 antibodies are undergoing clinical development for the treatment of solid tumors [210,211].
3.8.4. Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Non-Integrin (DC-SIGN)
DC-SIGN, also known as CD209, is a marker of immunosuppressive TAMs. DC-SIGN+ TAM infiltration in clinical specimens is associated with an increased proportion of immunosuppressive regulatory T cells (Treg) and exhausted CD8+ T cells (TIGIT+ LAG3+). Treatment of human muscle-invasive bladder cancer single cell suspensions with monoclonal antibody targeting DC-SIGN in combination with PD-1 blockade demonstrated heightened antitumor activity as compared to monotherapy, establishing a rationale for clinical development [212].
3.8.5. Other Macrophage-Directed Strategies
Inhibition of Arginase-1 (Arg1) has resulted in reduced tumor growth in preclinical models, and inhibitors of this enzyme have entered clinical development for the treatment of solid tumors [213,214]. Therapeutic blockade of CCR5, the receptor of protumoral chemokine CCL5, resulted in M2 to M1 macrophage polarization and a reduction in T-reg trafficking in a patient-derived functional in vitro organotypic culture model of hepatic colorectal metastases. This strategy demonstrated some activity in a Phase I clinical trial in patients with hepatic metastases of refractory colorectal carcinoma [215]. Class IIa histone deacetylase inhibitor, TMP195, demonstrated tumor reduction in breast cancer murine models by increasing the abundance of M1 macrophages [216]. Finally, cancer stem cell-derived WNT paracrine signaling in an ovarian cancer model upregulated M2 macrophages, which was reversed by WNT knockdown, suggesting this pathway as a potential target for TAM polarization [217]. It is noteworthy that inhibition of CCL2, an important chemokine that recruits monocytes, demonstrated promising preclinical activity; however, minimal clinical benefit was observed in clinical trials [218,219,220].
3.9. NK Cell Directed Strategies
While therapeutic targeting of activating receptors on NK cells has received much interest, most approaches have yet to reach clinical trials.
3.9.1. Natural-Killer Group 2, Member D (NKG2D) Ligands
The most extensively studied activating receptor is NKG2D and its ligands, major histocompatibility complex (MHC) class I chain-related protein A (MICA) and MHC class I chain-related protein B (MICB). MICA and MICB are polymorphic proteins that are induced upon cell stress, damage or transformation and act as a “kill me” signal through the NKG2D receptor expressed on cytotoxic lymphocytes [221]. Unfortunately, many cancers shed MICA and MICB through proteolytic cleavage via ADAM10, ADAM17 and MMP14 matrix metalloproteases [222]. To combat MICA and MICB proteolytic shedding, small molecular inhibitors of matrix metalloproteases have been recently developed [223]. Additional strategies to reduce MICA and MICB shedding include antibody targeting of the α3 domains of MICA/B. These antibodies were shown to result in a significant NK cell-mediated antitumor response in an immunocompetent mouse model [224]. Similarly, proteosome inhibitors, such as bortezomib, may prevent MICA/B degradation and augment NK cell targeting of tumor cells [225]. Cancer cells with NKG2D-resistant variants can emerge through immune editing and epigenetic mechanisms which alter NKG2D ligand expression [226,227]. Thus, histone deacetylase inhibitors or chemotherapies have been investigated as treatments to upregulate the membrane expression of NKG2D ligands [228,229].
3.9.2. NK Cell Engagers (NKCEs)
Bi- and tri-specific antibody constructs can bind multiple antigens and redirect NK cells to proximity of tumor cells and trigger an immune response and tumor cell death [230]. Multifunctional NKCEs with a tri-specific engager targeting two activating receptors, NKp46 and CD16, on NK cells and tumor antigen on cancer cells were observed to mediate significant tumor control in solid tumor murine models without notable toxicity [231]. These studies establish preclinical rationale for the clinical development of these molecules.
3.9.3. NKG2A Inhibition
NKG2A, an inhibitory immune checkpoint molecule expressed on NK cells, and a subset of α/β T cells, has recently emerged as a potential therapeutic target for cancer therapy [232,233,234]. Monalizumab, a first in class ICI that targets the NKG2A receptor and simultaneously activates both NK cells and CD8+ T cells, has recently advanced to phase III clinical trials for head and neck squamous carcinoma, signifying an important advancement in NK cell therapy [235].
3.9.4. Adoptive NK Cell Strategies
Due to the potent physiologic role of NK cells in tumor immunosurveillance, adoptive NK therapy (ACT) strategies have been an area of active investigation. Most current trials use allogeneic NK cells which are isolated from peripheral blood, propagated ex vivo and reinfused after lymphodepleting chemotherapy [236]. Genetic engineering of activating receptors on NK cells, such as the NKG2D receptor, has been used to optimize antitumor activity. NK cell ACT has demonstrated robust control of early metastasis, tumor specificity and more favorable toxicity profiles, when compared to adoptive T cell strategies. However, concerns over efficacy in solid tumors for NK cell ACT exist, including limited proliferative capacity, ability to infiltrate the tumor and presence of local immunosuppressive mechanisms within the TME [237,238]. Methods to increase the persistence of NK cells after infusion are being investigated, such as HLA knockdown to prevent rejection by the recipient’s immune system [239]. Tumors can escape NK cell-driven cytotoxicity by secreting immunosuppressive factors, such as TGF-β and adenosine, increasing immunosuppressive tryptophan metabolites via upregulating IDO, shedding MICA and MICB proteins and recruiting suppressive populations, such as Tregs, MDSCs and M2 TAMs [240,241,242,243]. Therapies targeting these mechanisms may also serve to reinvigorate a suppressed NK cell compartment.
4. Conclusions and Future Directions
There have been significant advances made in our understanding of the innate immune system’s contribution and response to tumorigenesis. Rational immuno-oncology combination strategies to modulate these pathways and activate both innate and adaptive immunity carry the potential to improve immunotherapy outcomes in all patients, particularly those with immunologically “cold” tumors.
Enthusiasm over novel innate immune strategies must be met with appropriate consideration regarding the risk of immune-related adverse events (irAEs). By virtue of their desired antigen-independent on-target effects to stimulate highly potent and conserved proinflammatory mechanisms, the risk of off-target toxicity is not insignificant. As clinical use expands, it will be important to distinguish irAEs specific to innate immune investigational agents from potentiation of irAEs caused by ICI, as many investigational strategies involve combination with ICI. There is an unmet clinical need to develop novel evidence-based protocols for toxicity management, acknowledging the expanding role of combinatorial approaches.
Despite considerable scientific and clinical progress, there remain gaps in our understanding of the dynamic interplay between cancer and immune cells within the TME following therapeutic intervention. Of critical importance will be to further define context-dependent roles of innate immune pathways in different tumor and genomic subtypes. For example, activation of innate immunity can be a “double-edged sword” and have pro and antitumorigenic roles in tumor development and progression, depending on tumor type, genetic/epigenetic, metabolic and microenvironmental context. However, the selective context-specific targeting of the innate immune system has the potential to become a cornerstone of immunotherapy strategies for the treatment of solid tumors.
Author Contributions
S.R., B.W.L., A.A., A.P. contributed to conceptualization, original draft preparation, review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef] [Green Version]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Spranger, S.; Luke, J.J.; Bao, R.; Zha, Y.; Hernandez, K.M.; Li, Y.; Gajewski, A.P.; Andrade, J.; Gajewski, T.F. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7759–E7768. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, T.A.; Amir, E. HYPE or HOPE: The prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 2018, 118, e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Louahed, J.; Brichard, V.G. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J. 2010, 16, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Paulete, A.R.; Cueto, F.J.; Martinez-Lopez, M.; Labiano, S.; Morales-Kastresana, A.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 2019, 177, 556–571.e516. [Google Scholar] [CrossRef]
- Liu, Y.J. IPC: Professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Allavena, P.; Garlanda, C.; Locati, M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum. Immunol. 2009, 70, 325–330. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dalen, F.J.; van Stevendaal, M.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brune, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19. [Google Scholar] [CrossRef]
- Zaidi, N.E.; Shazali, N.A.H.; Chor, A.L.T.; Osman, M.A.; Ibrahim, K.; Jaoi-Edward, M.; Afizan Nik Abd Rahman, N.M. Time-Lapse 2D Imaging of Phagocytic Activity in M1 Macrophage-4T1 Mouse Mammary Carcinoma Cells in Co-cultures. J. Vis. Exp. 2019. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiao, S.L.; Ruffell, B.; DeNardo, D.G.; Faddegon, B.A.; Park, C.C.; Coussens, L.M. TH2-Polarized CD4(+) T Cells and Macrophages Limit Efficacy of Radiotherapy. Cancer Immunol. Res. 2015, 3, 518–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarpia, M.; Karachaliou, N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol. Med. 2015, 12, 74–78. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Rao, H.L.; Chen, J.W.; Li, M.; Xiao, Y.B.; Fu, J.; Zeng, Y.X.; Cai, M.Y.; Xie, D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS ONE 2012, 7, e30806. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-associated neutrophils as a new prognostic factor in cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.; Bastholt, L.; Geertsen, P.; Christensen, I.J.; Larsen, S.; Gehl, J.; von der Maase, H. Elevated neutrophil and monocyte counts in peripheral blood are associated with poor survival in patients with metastatic melanoma: A prognostic model. Br. J. Cancer 2005, 93, 273–278. [Google Scholar] [CrossRef]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benevides, L.; da Fonseca, D.M.; Donate, P.B.; Tiezzi, D.G.; De Carvalho, D.D.; de Andrade, J.M.; Martins, G.A.; Silva, J.S. IL17 Promotes Mammary Tumor Progression by Changing the Behavior of Tumor Cells and Eliciting Tumorigenic Neutrophils Recruitment. Cancer Res. 2015, 75, 3788–3799. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Gerrard, T.L.; Cohen, D.J.; Kaplan, A.M. Human neutrophil-mediated cytotoxicity to tumor cells. J. Natl. Cancer Inst. 1981, 66, 483–488. [Google Scholar]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Mishalian, I.; Singhal, S.; Cheng, G.; Kapoor, V.; Horng, W.; Fridlender, G.; Bayuh, R.; Worthen, G.S.; et al. Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS ONE 2012, 7, e31524. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Bird, L. Controlling neutrophil plasticity. Nat. Rev. Immunol. 2010, 10, 752. [Google Scholar] [CrossRef] [PubMed]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wang, K.; Huang, X.J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef] [Green Version]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikstrom, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Welsh, T.J.; Green, R.H.; Richardson, D.; Waller, D.A.; O’Byrne, K.J.; Bradding, P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. J. Clin. Oncol. 2005, 23, 8959–8967. [Google Scholar] [CrossRef]
- Rajput, A.B.; Turbin, D.A.; Cheang, M.C.; Voduc, D.K.; Leung, S.; Gelmon, K.A.; Gilks, C.B.; Huntsman, D.G. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: A study of 4444 cases. Breast Cancer Res. Treat. 2008, 107, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth-Jakatics, R.; Jimi, S.; Takebayashi, S.; Kawamoto, N. Cutaneous malignant melanoma: Correlation between neovascularization and peritumor accumulation of mast cells overexpressing vascular endothelial growth factor. Hum. Pathol. 2000, 31, 955–960. [Google Scholar] [CrossRef]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, D.R.; Ding, Y.; Ricks, T.K.; Gullapalli, A.; Wolfe, B.L.; Trejo, J. Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006, 66, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Gooch, J.L.; Lee, A.V.; Yee, D. Interleukin 4 inhibits growth and induces apoptosis in human breast cancer cells. Cancer Res. 1998, 58, 4199–4205. [Google Scholar]
- Henderson, W.R.; Chi, E.Y.; Jong, E.C.; Klebanoff, S.J. Mast cell-mediated tumor-cell cytotoxicity. Role of the peroxidase system. J. Exp. Med. 1981, 153, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Hashimoto, W.; Osaki, T.; Okamura, H.; Robbins, P.D.; Kurimoto, M.; Nagata, S.; Lotze, M.T.; Tahara, H. Differential antitumor effects of administration of recombinant IL-18 or recombinant IL-12 are mediated primarily by Fas-Fas ligand- and perforin-induced tumor apoptosis, respectively. J. Immunol. 1999, 163, 583–589. [Google Scholar] [PubMed]
- Trinchieri, G. Biology of natural killer cells. Adv. Immunol. 1989, 47, 187–376. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Crowe, N.Y.; Godfrey, D.I. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int. Immunol. 2001, 13, 459–463. [Google Scholar] [CrossRef] [Green Version]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, T.; Saddawi-Konefka, R.; Vermi, W.; Koebel, C.M.; Arthur, C.; White, J.M.; Uppaluri, R.; Andrews, D.M.; Ngiow, S.F.; Teng, M.W.; et al. Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 2012, 209, 1869–1882. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Kastelein, R.A.; Hunter, C.A. IL-10 enhances NK cell proliferation, cytotoxicity and production of IFN-γ when combined with IL-18. Eur. J. Immunol. 1999, 29, 2658–2665. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The Role of the NKG2D Immunoreceptor in Immune Cell Activation and Natural Killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Takanami, I.; Takeuchi, K.; Giga, M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J. Thorac. Cardiovasc. Surg. 2001, 121, 1058–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villegas, F.R.; Coca, S.; Villarrubia, V.G.; Jiménez, R.; Chillón, M.a.J.; Jareño, J.; Zuil, M.; Callol, L. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer 2002, 35, 23–28. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangloff, M. Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem. Sci. 2012, 37, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Adams, S. Toll-like receptor agonists in cancer therapy. Immunotherapy 2009, 1, 949–964. [Google Scholar] [CrossRef] [Green Version]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, C.T.; Huang, X.; Pardoll, D.M. Persistent Toll-like receptor signals are required for reversal of regulatory T cell-mediated CD8 tolerance. Nat. Immunol. 2004, 5, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, F.; Cai, Y.; Liu, N.; Wang, L.; Xu, D.; Chu, Y. TLR1/TLR2 agonist induces tumor regression by reciprocal modulation of effector and regulatory T cells. J. Immunol. 2011, 186, 1963–1969. [Google Scholar] [CrossRef] [Green Version]
- Jin, B.; Sun, T.; Yu, X.H.; Yang, Y.X.; Yeo, A.E. The effects of TLR activation on T-cell development and differentiation. Clin. Dev. Immunol. 2012, 2012, 836485. [Google Scholar] [CrossRef] [PubMed]
- Salaun, B.; Coste, I.; Rissoan, M.C.; Lebecque, S.J.; Renno, T. TLR3 can directly trigger apoptosis in human cancer cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajon, M.; Iribarren, K.; Petitprez, F.; Marmier, S.; Lupo, A.; Gillard, M.; Ouakrim, H.; Victor, N.; Vincenzo, D.B.; Joubert, P.E.; et al. Toll like receptor 7 expressed by malignant cells promotes tumor progression and metastasis through the recruitment of myeloid derived suppressor cells. Oncoimmunology 2019, 8, e1505174. [Google Scholar] [CrossRef]
- Hao, B.; Chen, Z.; Bi, B.; Yu, M.; Yao, S.; Feng, Y.; Yu, Y.; Pan, L.; Di, D.; Luo, G.; et al. Role of TLR4 as a prognostic factor for survival in various cancers: A meta-analysis. Oncotarget 2018, 9, 13088–13099. [Google Scholar] [CrossRef] [Green Version]
- Jouhi, L.; Renkonen, S.; Atula, T.; Makitie, A.; Haglund, C.; Hagstrom, J. Different Toll-Like Receptor Expression Patterns in Progression toward Cancer. Front. Immunol. 2014, 5, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Campos, J.; Gallotta, M.; Gong, M.; Crain, C.; Naik, E.; Coffman, R.L.; Guiducci, C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [Google Scholar] [CrossRef] [Green Version]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Ribas, A.; Medina, T.; Kummar, S.; Amin, A.; Kalbasi, A.; Drabick, J.J.; Barve, M.; Daniels, G.A.; Wong, D.J.; Schmidt, E.V.; et al. SD-101 in Combination with Pembrolizumab in Advanced Melanoma: Results of a Phase Ib, Multicenter Study. Cancer Discov. 2018, 8, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.E.W.; Nabell, L.; Wong, D.J.L.; Day, T.A.; Daniels, G.A.; Milhem, M.M.; Deva, S.; Jameson, M.B.; Guntinas-Lichius, O.; Almubarak, M.; et al. Phase 1b/2, open label, multicenter study of intratumoral SD-101 in combination with pembrolizumab in anti-PD-1 treatment naïve patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2019, 37, 6039. [Google Scholar] [CrossRef]
- Kivimae, S.; Hennessy, M.; Pena, R.; Kirksey, Y.; Nieves, W.; Quatch, P.; Cetz, J.; Ren, Z.; Cai, H.; Deng, B.L.; et al. Comprehensive antitumor immune activation by a novel TLR7/8 targeting agent NKTR-262 combined with CD122-biased immunostimulatory cytokine NKTR-214. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2018 2018, 78, 3755. [Google Scholar]
- Mullins, S.R.; Vasilakos, J.P.; Deschler, K.; Grigsby, I.; Gillis, P.; John, J.; Elder, M.J.; Swales, J.; Timosenko, E.; Cooper, Z.; et al. Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J. Immunother. Cancer 2019, 7, 244. [Google Scholar] [CrossRef] [PubMed]
- Morales, A. Long-term results and complications of intracavitary bacillus Calmette-Guerin therapy for bladder cancer. J. Urol. 1984, 132, 457–459. [Google Scholar] [CrossRef]
- Beutner, K.R.; Spruance, S.L.; Hougham, A.J.; Fox, T.L.; Owens, M.L.; Douglas, J.M., Jr. Treatment of genital warts with an immune-response modifier (imiquimod). J. Am. Acad. Dermatol. 1998, 38, 230–239. [Google Scholar] [CrossRef]
- Casella, C.R.; Mitchell, T.C. Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol. Life Sci. 2008, 65, 3231–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, V.; Keating, G.M.; Perry, C.M. Imiquimod: In superficial basal cell carcinoma. Am. J. Clin. Dermatol. 2005, 6, 195–200; discussion 192–201. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.J.; Zhao, L.; Hu, H.G.; Li, W.H.; Li, Y.M. Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev. 2020, 40, 1117–1141. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Song, Z.; Shen, A.; Chen, T.; Zhang, A. Small molecules targeting the innate immune cGAS–STING–TBK1 signaling pathway. Acta Pharm. Sin. B 2020, in press, pre-proof. [Google Scholar] [CrossRef]
- Harrington, K.J.; Brody, J.; Ingham, M.; Strauss, J.; Cemerski, S.; Wang, M.; Tse, A.; Khilnani, A.; Marabelle, A.; Golan, T. Preliminary results of the first-in-human (FIH) study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with pembrolizumab (pembro) in patients with advanced solid tumors or lymphomas. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Sandhu, S.K.; Hamid, O.; Spreafico, A.; Kasper, S.; Dummer, R.; Shimizu, T.; Steeghs, N.; Lewis, N.; Talluto, C.C.; et al. Phase Ib study of MIW815 (ADU-S100) in combination with spartalizumab (PDR001) in patients (pts) with advanced/metastatic solid tumors or lymphomas. J. Clin. Oncol. 2019, 37, 2507. [Google Scholar] [CrossRef]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2019, 29, 785–789. [Google Scholar] [CrossRef]
- Schieven, G.; Brown, J.; Swanson, J.; Stromko, B.S.C.; Ho, C.-P.; Zhang, R.; Li, W.B.; Qiu, H.; Sun, H.; Fink, B.; et al. Preclinical characterization of BMS-986301, a differentiated STING agonist with robust antitumor activity as monotherapy or in combination with anti-PD-1. In Proceedings of the 33rd Annual Meeting & Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2018), Washington, DC, USA, 7–11 November 2018. [Google Scholar]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.Y.; Tran, J.L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Flood, B.A.; Higgs, E.F.; Li, S.; Luke, J.J.; Gajewski, T.F. STING pathway agonism as a cancer therapeutic. Immunol. Rev. 2019, 290, 24–38. [Google Scholar] [CrossRef]
- Kinkead, H.L.; Hopkins, A.; Lutz, E.; Wu, A.A.; Yarchoan, M.; Cruz, K.; Woolman, S.; Vithayathil, T.; Glickman, L.H.; Ndubaku, C.O.; et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaffari, A.; Peterson, N.; Khalaj, K.; Vitkin, N.; Robinson, A.; Francis, J.A.; Koti, M. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Poeck, H.; Bscheider, M.; Gross, O.; Finger, K.; Roth, S.; Rebsamen, M.; Hannesschlager, N.; Schlee, M.; Rothenfusser, S.; Barchet, W.; et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 2010, 11, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Elion, D.L.; Cook, R.S. Harnessing RIG-I and intrinsic immunity in the tumor microenvironment for therapeutic cancer treatment. Oncotarget 2018, 9, 29007–29017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Heidegger, S.; Wintges, A.; Stritzke, F.; Bek, S.; Steiger, K.; Koenig, P.A.; Gottert, S.; Engleitner, T.; Ollinger, R.; Nedelko, T.; et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Terheyden, P.; Weishaupt, C.; Heinzerling, L.; Klinkhardt, U.; Krauss, J.; Mohr, P.; Kiecker, F.; Becker, J.C.; Dähling, A.; Döner, F.; et al. Phase I dose-escalation and expansion study of intratumoral CV8102, a RNA-based TLR- and RIG-1 agonist in patients with advanced solid tumors. Ann. Oncol. 2018, 29, viii466. [Google Scholar] [CrossRef]
- Barsoum, J.; Renn, M.; Schuberth, C.; Jakobs, C.; Schwickart, A.; Schlee, M.; van den Boorn, J.; Hartmann, G. Abstract B44: Selective stimulation of RIG-I with a novel synthetic RNA induces strong anti-tumor immunity in mouse tumor models. Cancer Immunol. Res. 2017. [Google Scholar] [CrossRef]
- Middleton, M.R.; Wermke, M.; Calvo, E.; Chartash, E.; Zhou, H.; Zhao, X.; Niewel, M.; Dobrenkov, K.; Moreno, V. Phase I/II, multicenter, open-label study of intratumoral/intralesional administration of the retinoic acid–inducible gene I (RIG-I) activator MK-4621 in patients with advanced or recurrent tumors. Ann. Oncol. 2018, 29, viii712. [Google Scholar] [CrossRef]
- Linehan, M.M.; Dickey, T.H.; Molinari, E.S.; Fitzgerald, M.E.; Potapova, O.; Iwasaki, A.; Pyle, A.M. A minimal RNA ligand for potent RIG-I activation in living mice. Sci. Adv. 2018, 4, e1701854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Muthusamy, V.; Fedorova, O.; Kong, Y.; Kim, D.J.; Bosenberg, M.; Pyle, A.M.; Iwasaki, A. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 2019, 216, 2854–2868. [Google Scholar] [CrossRef]
- Jones, M.; Cunningham, M.E.; Wing, P.; DeSilva, S.; Challa, R.; Sheri, A.; Padmanabhan, S.; Iyer, R.P.; Korba, B.E.; Afdhal, N.; et al. SB 9200, a novel agonist of innate immunity, shows potent antiviral activity against resistant HCV variants. J. Med. Virol. 2017, 89, 1620–1628. [Google Scholar] [CrossRef]
- Kyi, C.; Roudko, V.; Sabado, R.; Saenger, Y.; Loging, W.; Mandeli, J.; Thin, T.H.; Lehrer, D.; Donovan, M.; Posner, M.; et al. Therapeutic Immune Modulation against Solid Cancers with Intratumoral Poly-ICLC: A Pilot Trial. Clin. Cancer Res. 2018, 24, 4937–4948. [Google Scholar] [CrossRef] [Green Version]
- Dauletbaev, N.; Cammisano, M.; Herscovitch, K.; Lands, L.C. Stimulation of the RIG-I/MAVS Pathway by Polyinosinic:Polycytidylic Acid Upregulates IFN-beta in Airway Epithelial Cells with Minimal Costimulation of IL-8. J. Immunol. 2015, 195, 2829–2841. [Google Scholar] [CrossRef] [Green Version]
- Salazar, A.M.; Erlich, R.B.; Mark, A.; Bhardwaj, N.; Herberman, R.B. Therapeutic in situ autovaccination against solid cancers with intratumoral poly-ICLC: Case report, hypothesis, and clinical trial. Cancer Immunol. Res. 2014, 2, 720–724. [Google Scholar] [CrossRef] [Green Version]
- van Kooten, C.; Banchereau, J. CD40-CD40 ligand. J. Leukoc. Biol. 2000, 67, 2–17. [Google Scholar] [CrossRef]
- Todryk, S.M.; Tutt, A.L.; Green, M.H.; Smallwood, J.A.; Halanek, N.; Dalgleish, A.G.; Glennie, M.J. CD40 ligation for immunotherapy of solid tumours. J. Immunol. Methods 2001, 248, 139–147. [Google Scholar] [CrossRef]
- van Mierlo, G.J.; den Boer, A.T.; Medema, J.P.; van der Voort, E.I.; Fransen, M.F.; Offringa, R.; Melief, C.J.; Toes, R.E. CD40 stimulation leads to effective therapy of CD40(-) tumors through induction of strong systemic cytotoxic T lymphocyte immunity. Proc. Natl. Acad. Sci. USA 2002, 99, 5561–5566. [Google Scholar] [CrossRef] [Green Version]
- Buhtoiarov, I.N.; Lum, H.; Berke, G.; Paulnock, D.M.; Sondel, P.M.; Rakhmilevich, A.L. CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. J. Immunol. 2005, 174, 6013–6022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.T.; Vonderheide, R.H. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, K.B.; Gladney, W.L.; Tooker, G.M.; Graham, K.; Fraietta, J.A.; Beatty, G.L. IFNgamma and CCL2 Cooperate to Redirect Tumor-Infiltrating Monocytes to Degrade Fibrosis and Enhance Chemotherapy Efficacy in Pancreatic Carcinoma. Cancer Discov. 2016, 6, 400–413. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.K.; Cook, A.M.; McDonnell, A.M.; Millward, M.J.; Creaney, J.; Francis, R.J.; Hasani, A.; Segal, A.; Musk, A.W.; Turlach, B.A.; et al. A phase 1b clinical trial of the CD40-activating antibody CP-870,893 in combination with cisplatin and pemetrexed in malignant pleural mesothelioma. Ann. Oncol. 2015, 26, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.S.; Poudel, B.; Torres, E.R.; Sidhom, J.W.; Robinson, T.M.; Christmas, B.; Scott, B.; Cruz, K.; Woolman, S.; Wall, V.Z.; et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell-Mediated Anticancer Activity. Cancer Immunol. Res. 2019, 7, 428–442. [Google Scholar] [CrossRef]
- Byrne, K.T.; Leisenring, N.H.; Bajor, D.L.; Vonderheide, R.H. CSF-1R-Dependent Lethal Hepatotoxicity When Agonistic CD40 Antibody Is Given before but Not after Chemotherapy. J. Immunol. 2016, 197, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [Green Version]
- O’Hara, M.H.; O’Reilly, E.M.; Rosemarie, M.; Varadhachary, G.; Wainberg, Z.A.; Ko, A.; Fisher, G.A.; Rahma, O.; Lyman, J.P.; Cabanski, C.R.; et al. A phase 1b study of CD40 agonistic monoclonal antibody APX005M together with gemcitabine and nab-paclitaxel with or without nivolumab in untreated metastatic pancreatic ductal adenocarcinoma (PDAC) patients. In Proceedings of the American Association for Cancer Research Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Ngiow, S.F.; Young, A.; Blake, S.J.; Hill, G.R.; Yagita, H.; Teng, M.W.; Korman, A.J.; Smyth, M.J. Agonistic CD40 mAb-Driven IL12 Reverses Resistance to Anti-PD1 in a T-cell-Rich Tumor. Cancer Res. 2016, 76, 6266–6277. [Google Scholar] [CrossRef] [Green Version]
- Bajor, D.L.; Mick, R.; Riese, M.J.; Huang, A.C.; Sullivan, B.; Richman, L.P.; Torigian, D.A.; George, S.M.; Stelekati, E.; Chen, F.; et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 2018, 7, e1468956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Chan, H.T.C.; Orr, C.M.; Dadas, O.; Booth, S.G.; Dahal, L.N.; Penfold, C.A.; O’Brien, L.; Mockridge, C.I.; French, R.R.; et al. Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 2018, 33, 664–675.e664. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Li, Y.; Long, K.B. Cancer immunotherapy: Activating innate and adaptive immunity through CD40 agonists. Expert Rev. Anticancer. Ther. 2017, 17, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luqman, M.; Klabunde, S.; Lin, K.; Georgakis, G.V.; Cherukuri, A.; Holash, J.; Goldbeck, C.; Xu, X.; Kadel, E.E., 3rd; Lee, S.H.; et al. The antileukemia activity of a human anti-CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood 2008, 112, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Khubchandani, S.; Czuczman, M.S.; Hernandez-Ilizaliturri, F.J. Dacetuzumab, a humanized mAb against CD40 for the treatment of hematological malignancies. Curr. Opin. Investig. Drugs 2009, 10, 579–587. [Google Scholar]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Garcia, J.J.; Martinez-Banaclocha, H.; Angosto-Bazarra, D.; de Torre-Minguela, C.; Baroja-Mazo, A.; Alarcon-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martin-Sanchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, R.; Man, S.M.; Kanneganti, T.D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.M.; Wang, E.; Ma, W.; Haines, D.; O’HUigin, C.; et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Zhu, R.; Zhu, J.; Zhao, R.; Li, M. E2-Induced Activation of the NLRP3 Inflammasome Triggers Pyroptosis and Inhibits Autophagy in HCC Cells. Oncol. Res. 2019, 27, 827–834. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Rodrigue-Gervais, I.G.; Breton, V.; Colpitts, S.L.; Beauchemin, N.; et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Mu, K.; Li, T.; Zhang, Y.; Yang, Z.; Jia, X.; Zhao, W.; Huai, W.; Guo, P.; Han, L. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab. Investig. 2014, 94, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Moller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.D.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty-Roix, R.; Vladimer, G.I.; Pouliot, K.; Weng, D.; Buglione-Corbett, R.; West, K.; MacMicking, J.D.; Chee, J.D.; Wang, S.; Lu, S.; et al. Identification of QS-21 as an Inflammasome-activating Molecular Component of Saponin Adjuvants. J. Biol. Chem. 2016, 291, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Kopalli, S.R.; Kang, T.-B.; Lee, K.-H.; Koppula, S. NLRP3 Inflammasome Activation Inhibitors in Inflammation-Associated Cancer Immunotherapy: An Update on the Recent Patents. Recent. Pat Anticancer Drug Discov. 2018, 13, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, C.-F.; Li, Y.-C.; Deng, W.-W.; Mao, L.; Wu, L.; Zhang, W.-F.; Zhang, L.; Sun, Z.-J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-”host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Qiu, X.; Xi, G.; Liu, H.; Zhang, F.; Lv, T.; Song, Y. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol. Rep. 2018, 40, 1971–1984. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, K.; Soundarapandian, K.; Thorne, R.F.; Li, D.; Li, D. Activation of Pyroptotic Cell Death Pathways in Cancer: An Alternative Therapeutic Approach. Transl. Oncol. 2019, 12, 925–931. [Google Scholar] [CrossRef]
- Saito, T.; Takayama, T.; Osaki, T.; Nagai, S.; Suzuki, T.; Sato, M.; Kuwano, H.; Tahara, H. Combined mobilization and stimulation of tumor-infiltrating dendritic cells and natural killer cells with Flt3 ligand and IL-18 in vivo induces systemic antitumor immunity. Cancer Sci. 2008, 99, 2028–2036. [Google Scholar] [CrossRef]
- Nefedova, Y.; Cheng, P.; Gilkes, D.; Blaskovich, M.; Beg, A.A.; Sebti, S.M.; Gabrilovich, D.I. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J. Immunol. 2005, 175, 4338–4346. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Autio, K.A.; Klebanoff, C.A.; Schaer, D.; Kauh, J.S.; Slovin, S.F.; Blinder, V.S.; Comen, E.A.; Danila, D.C.; Hoffman, D.M.J.; Kang, S.; et al. Phase 1 study of LY3022855, a colony-stimulating factor-1 receptor (CSF-1R) inhibitor, in patients with metastatic breast cancer (MBC) or metastatic castration-resistant prostate cancer (MCRPC). J. Clin. Oncol. 2019, 37, 2548. [Google Scholar] [CrossRef]
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang-Gillam, A.; O’Reilly, E.M.; Bendell, J.C.; Wainberg, Z.A.; Borazanci, E.H.; Bahary, N.; O’Hara, M.H.; Beatty, G.L.; Pant, S.; Cohen, D.J.; et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2019, 37, TPS465. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kgamma is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kgamma Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varner, J.A. Taming the beast: Strategies to target the immune-suppressive macrophage to enhance cancer immune therapy. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2019, 2019, 79. [Google Scholar]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [Green Version]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [Green Version]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPalphaFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Hu, B.; Wang, Z.; Zeng, H.; Qi, Y.; Chen, Y.; Wang, T.; Wang, J.; Chang, Y.; Bai, Q.; Xia, Y.; et al. Blockade of DC-SIGN+ tumor-associated macrophages reactivates anti-tumor immunity and improves immunotherapy in muscle-invasive bladder cancer. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.B.; Yao, M.; Brummer, G.; Acevedo, D.; Alhakamy, N.; Berkland, C.; Cheng, N. Targeted gene silencing of CCL2 inhibits triple negative breast cancer progression by blocking cancer stem cell renewal and M2 macrophage recruitment. Oncotarget 2016, 7, 49349–49367. [Google Scholar] [CrossRef]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Furst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int. J. Cancer 2013, 133, 1557–1566. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Vales-Gomez, M.; Chisholm, S.E.; Cassady-Cain, R.L.; Roda-Navarro, P.; Reyburn, H.T. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. 2008, 68, 1546–1554. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Soto, A.; Folgueras, A.R.; Seto, E.; Gonzalez, S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: Potential implications for the immunosurveillance of cancer. Oncogene 2009, 28, 2370–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, T.; Dunn, G.P.; Lacoursiere, D.Y.; Schreiber, R.D.; Bui, J.D. Cancer immunoediting of the NK group 2D ligand H60a. J. Immunol. 2011, 187, 3538–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armeanu, S.; Bitzer, M.; Lauer, U.M.; Venturelli, S.; Pathil, A.; Krusch, M.; Kaiser, S.; Jobst, J.; Smirnow, I.; Wagner, A.; et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005, 65, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Hervieu, A.; Rebe, C.; Vegran, F.; Chalmin, F.; Bruchard, M.; Vabres, P.; Apetoh, L.; Ghiringhelli, F.; Mignot, G. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J. Investig. Dermatol. 2013, 133, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, E.; Kosmopoulou, M.N.; Chrysina, E.D.; Leonidas, D.D.; Hadjiloi, T.; Tiraidis, C.; Zographos, S.E.; Gyorgydeak, Z.; Somsak, L.; Docsa, T.; et al. Crystallographic studies on two bioisosteric analogues, N-acetyl-beta-D-glucopyranosylamine and N-trifluoroacetyl-beta-D-glucopyranosylamine, potent inhibitors of muscle glycogen phosphorylase. Bioorg. Med. Chem. 2006, 14, 181–189. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef] [Green Version]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; Andre, P.; et al. NKG2A Blockade Potentiates CD8 T Cell Immunity Induced by Cancer Vaccines. Cell 2018, 175, 1744–1755. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Antonia, S.J. The NKG2A immune checkpoint - a new direction in cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.S.; Lanier, L.L. Natural Killer Cells in Cancer Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell-cancer cycle: Advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Baginska, J.; Viry, E.; Paggetti, J.; Medves, S.; Berchem, G.; Moussay, E.; Janji, B. The critical role of the tumor microenvironment in shaping natural killer cell-mediated anti-tumor immunity. Front. Immunol. 2013, 4, 490. [Google Scholar] [CrossRef] [Green Version]
- Trzonkowski, P.; Szmit, E.; Mysliwska, J.; Dobyszuk, A.; Mysliwski, A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin. Immunol. 2004, 112, 258–267. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Cancer Immunity Cycle. Innate immune cells facilitate the immune system’s response against a recognized pathogen. This process is initiated by detection of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and other unique tumor antigens by innate immune cells which result in antigen presentation and activation of antigen-specific T cells in tumor draining lymph nodes. These T cells traffic to the tumor and mediate tumor elimination. Abbreviations: PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; PRRs, pattern-recognizing-receptors.
Figure 1.
Cancer Immunity Cycle. Innate immune cells facilitate the immune system’s response against a recognized pathogen. This process is initiated by detection of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and other unique tumor antigens by innate immune cells which result in antigen presentation and activation of antigen-specific T cells in tumor draining lymph nodes. These T cells traffic to the tumor and mediate tumor elimination. Abbreviations: PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; PRRs, pattern-recognizing-receptors.

Figure 2.
Innate Immune sensing pathways. A diverse set of pattern recognition receptors (PRRs) detect damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) or tumor-associated antigens (TAAs) and activate innate immune cells within the TME. These sensors can also be expressed by tumor cells themselves. Multiple PRRs, including cGAS/STING, RIG-I and TLRs, promote transcription of proinflammatory genes via IRF-3 and NF-kb. These processes result in Type I IFN, cytokine and chemokine production, which supports a cytotoxic antitumor response mediated by effector T cells and NK cells. The NLRP3 inflammasome drives a proinflammatory response through IL-1β and IL-18, in addition to mediating pyroptosis. NKG2D ligands activate NK cells.
Figure 2.
Innate Immune sensing pathways. A diverse set of pattern recognition receptors (PRRs) detect damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) or tumor-associated antigens (TAAs) and activate innate immune cells within the TME. These sensors can also be expressed by tumor cells themselves. Multiple PRRs, including cGAS/STING, RIG-I and TLRs, promote transcription of proinflammatory genes via IRF-3 and NF-kb. These processes result in Type I IFN, cytokine and chemokine production, which supports a cytotoxic antitumor response mediated by effector T cells and NK cells. The NLRP3 inflammasome drives a proinflammatory response through IL-1β and IL-18, in addition to mediating pyroptosis. NKG2D ligands activate NK cells.

Figure 3.
Strategies targeting TAMs. Multiple therapeutic strategies exist to target tumor-associated macrophages (TAMs) in cancer immunotherapy. CSF-1R inhibition and CD40 agonism promote TAM polarization to proinflammatory M1 phenotype. CD47-SIRPα blockade restores TAM-mediated phagocytosis. PD-1 blockade reverses TAM-mediated immunosuppression. CCL5 and CCL2 blockade attenuate TAM recruitment.
Figure 3.
Strategies targeting TAMs. Multiple therapeutic strategies exist to target tumor-associated macrophages (TAMs) in cancer immunotherapy. CSF-1R inhibition and CD40 agonism promote TAM polarization to proinflammatory M1 phenotype. CD47-SIRPα blockade restores TAM-mediated phagocytosis. PD-1 blockade reverses TAM-mediated immunosuppression. CCL5 and CCL2 blockade attenuate TAM recruitment.


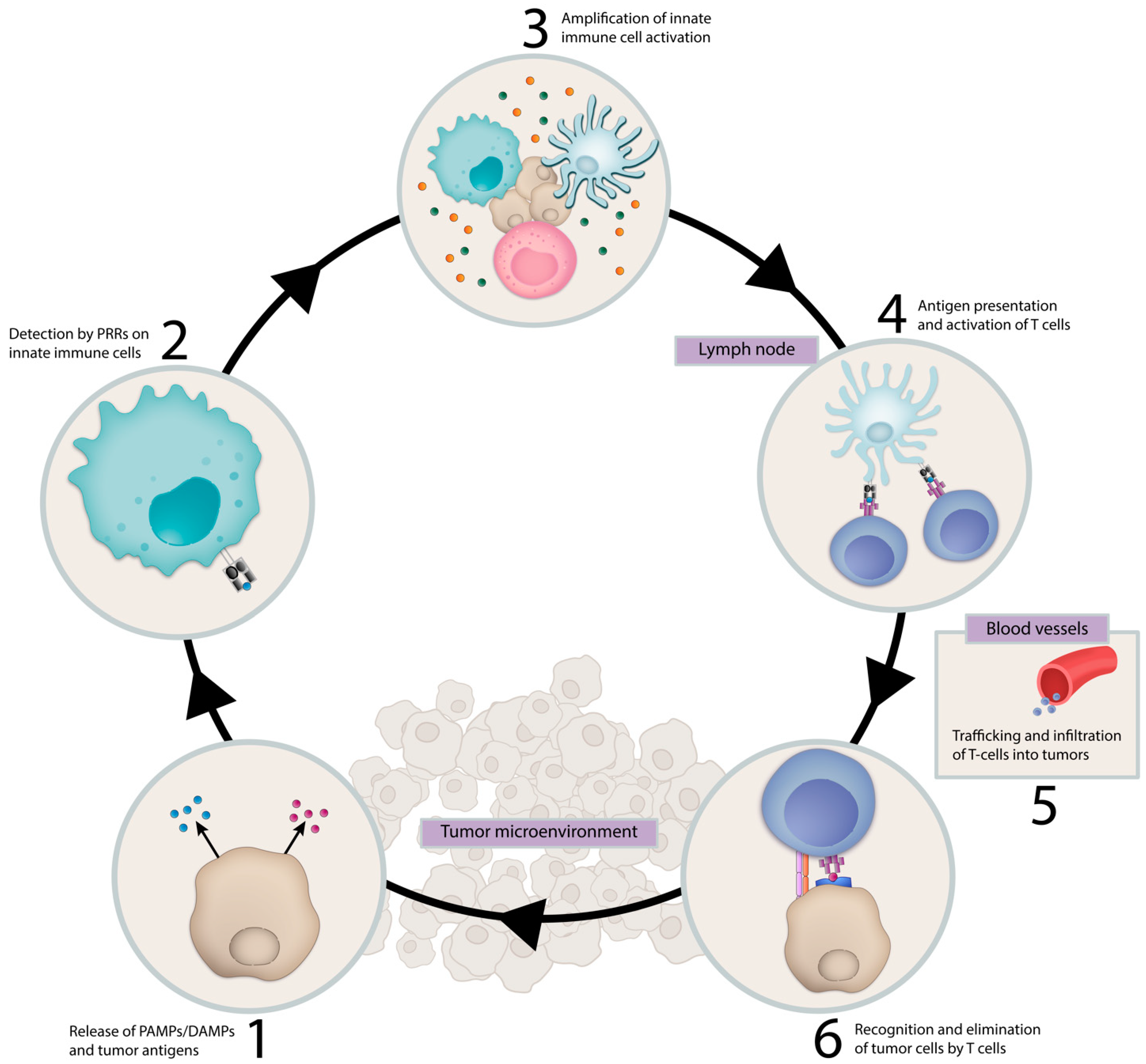{kind=link}
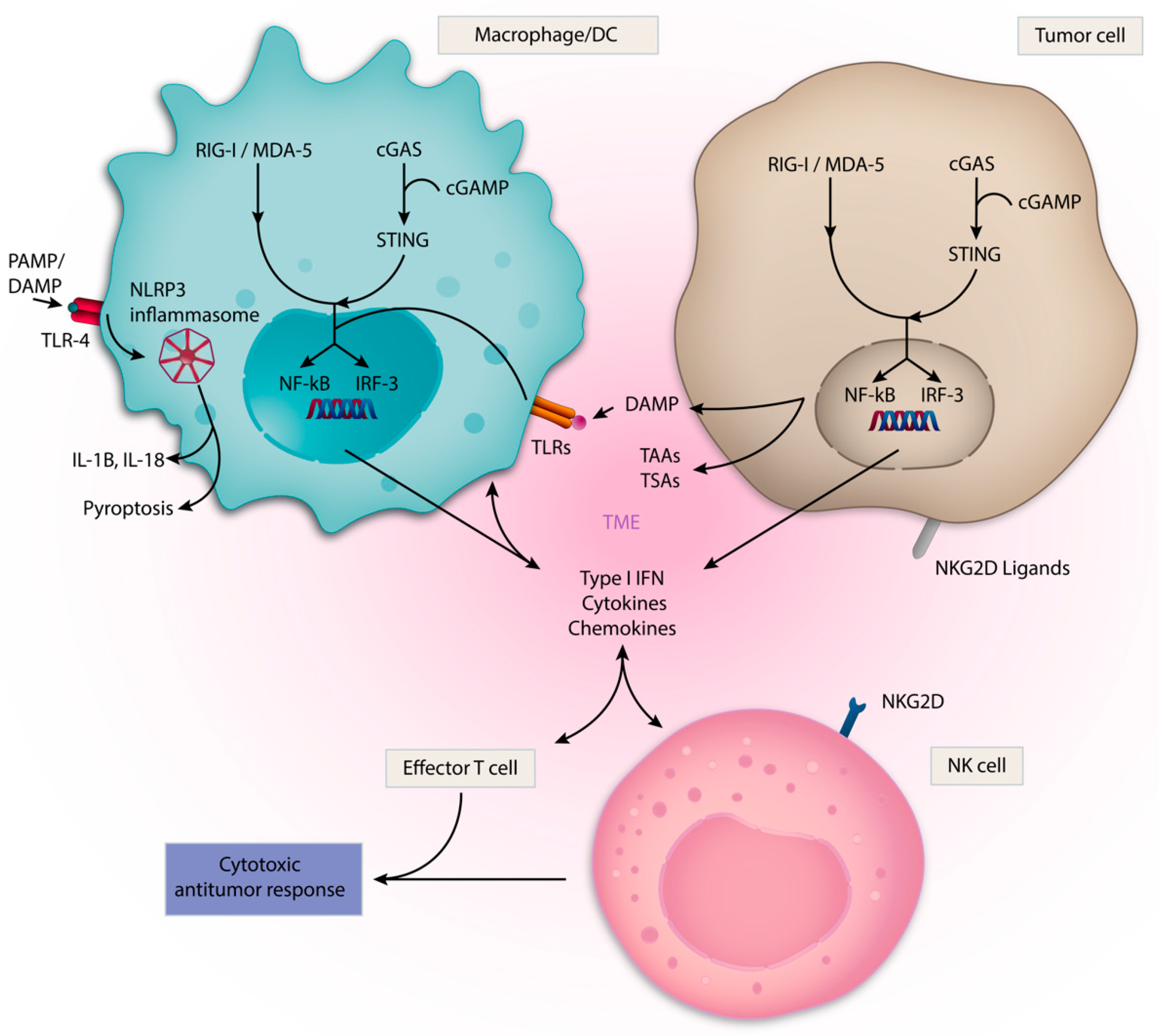{kind=link}
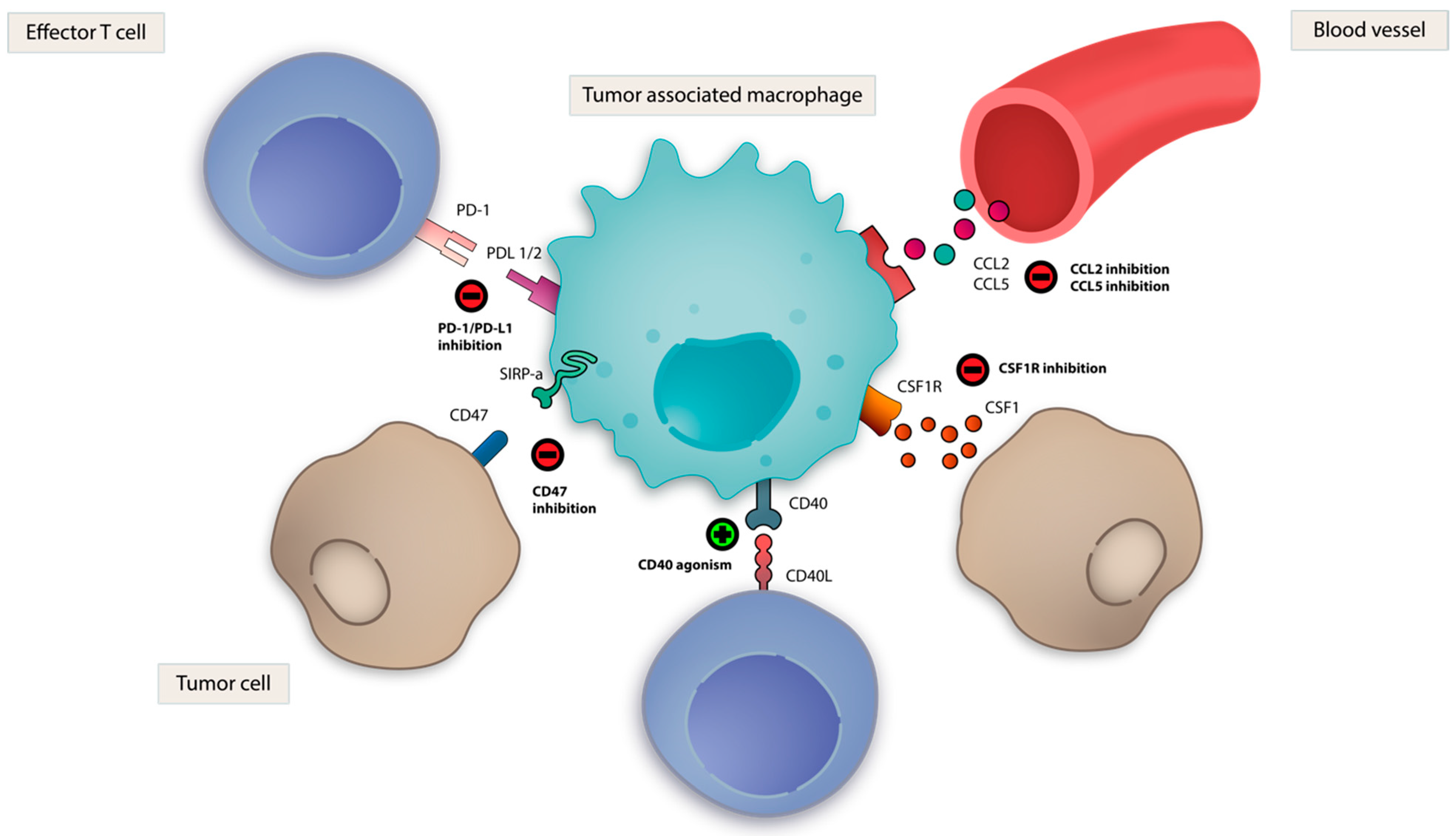{kind=link}
Table 1.
Innate immune cell expression of PRRs and PAMP/DAMP-sensing pathways.
| DC | TAM | Mo-MDSC | PMN-MDSC | Neutrophils | NK Cells | Basophils | Eosinophils | Mast Cells | |
|---|---|---|---|---|---|---|---|---|---|
| TLR7/8 | |||||||||
| TLR9 | |||||||||
| RIG-I | |||||||||
| cGAS-STING | |||||||||
| NLRP3 | |||||||||
| CSF-1R | |||||||||
| CD40 | |||||||||
| NKG2D |
Green = expression of innate immune pathway or receptor is observed in human cell types based on literature cited in main body of text. This is a conceptual and binary illustration which does not reflect contextual and dynamic nuances in expression.
Table 2.
Summary of agents in clinical development.
| PRR | Agent | Molecule Type | Route of Administration | Cancer Type(s) | Clinical Phase of Development | |
|---|---|---|---|---|---|---|
| TLR8 | VTX-2337 (motilomod) | Small molecule | Intratumoral | ovarian, HNSCC | I, II | |
| dTLR7/8 | NKTR-262 | Small molecule | Intratumoral | Solid tumors | I, II | |
| TLR9 | SD-101 | CpG-C class ODN | Intratumoral | Solid tumors | I, II | |
| EMD 1201081 | Synthetic ODN | Subcutaneous Injection | HNSCC | I, II | ||
| CPG 7909 | CpG ODN | Subcutaneous Injection | Lymphomas | I, II | ||
| IMO-2125 (Tilsotolimod) | Synthetic ODN | Intratumoral | melanoma, Solid tumors | I, II | ||
| CMP-001 | CpG ODN | Intratumoral | Solid tumors | I, II | ||
| RIG-I | SLR-14 | Synthetic stem loop RNA | Intratumoral | Solid tumors | Pre-clinical | |
| RGT-100 (MK-4621) | Synthetic oligonucleotide | Intratumoral | Solid tumors | |||
| MDA-5 | BO-112 (poly(I:C)) | Synthetic dsRNA | Intratumoral | Solid tumors | I | |
| STING | E7766 | Novel macrocycle-bridged | Intravenous | Solid tumors | I | |
| GSK3745417 | Small molecule | Intravenous | Solid tumors | I | ||
| MIW815 (ADU-S100) | Synthetic CDN | Intratumoral | Solid tumors | I, II | ||
| MK1454 | Small molecule | Intratumoral | Solid tumors | I, II | ||
| BMS-986301 | Small molecule | Intratumoral * | Solid tumors | I | ||
| NLRP3 | BMS-986299 | First in class agonist * | Intratumoral * | I | ||
| Other innate immune targets | ||||||
| CSF-1R | Cabiralizumab | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| JNJ-40346527 | Monoclonal antibody | Intravenous | Advanced prostate cancer | I, II | ||
| PLX3397 | Small molecule | Oral | Solid tumors | I, II | ||
| MCS110 | Solid tumors | I, II | ||||
| IMC-CS4 | Monoclonal antibody | Intravenous | Solid tumors | I | ||
| CD40 | APX005M | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| CP-870,893 | Monoclonal antibody | Intravenous | Solid tumors | I | ||
| Selicrelumab | Monoclonal antibody | Intravenous | Solid tumors | I, II | ||
| PI3K (delta) Inhibitors | IPI-549 | Small molecule | Oral | Solid tumors | I, II | |
| Class IIa histone deacetylase inhibitor | TMP-195 | Small molecule | Oral | Solid Tumors | Preclinical | |
| IDO inhibitors | Indoximod | Small molecule | Oral | Solid tumors | I, II | |
| STAT3 inhibitors | Siltuximab | Monoclonal antibody | Intravenous | Solid tumors | I, II | |
| WP1066 | Small molecule | Oral | Solid tumors | I | ||
| TT-101 | Small molecule | Oral | Solid tumors | I | ||
* BMS-986301 is being evaluated for systemic intramuscular administration. The novel NLRP3 agonist BMS-986299 is being studied in a phase I clinical trial as mon-otherapy and in combination with nivolumab and ipilimumab in advanced solid tumors [NCT03444753].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rameshbabu, S.; Labadie, B.W.; Argulian, A.; Patnaik, A. Targeting Innate Immunity in Cancer Therapy. Vaccines 2021, 9, 138. https://doi.org/10.3390/vaccines9020138
AMA Style
Rameshbabu S, Labadie BW, Argulian A, Patnaik A. Targeting Innate Immunity in Cancer Therapy. Vaccines. 2021; 9(2):138. https://doi.org/10.3390/vaccines9020138
Chicago/Turabian StyleRameshbabu, Srikrishnan, Brian W. Labadie, Anna Argulian, and Akash Patnaik. 2021. "Targeting Innate Immunity in Cancer Therapy" Vaccines 9, no. 2: 138. https://doi.org/10.3390/vaccines9020138
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.