
An RNA-Based Vaccine Platform for Use against Mycobacterium tuberculosis
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
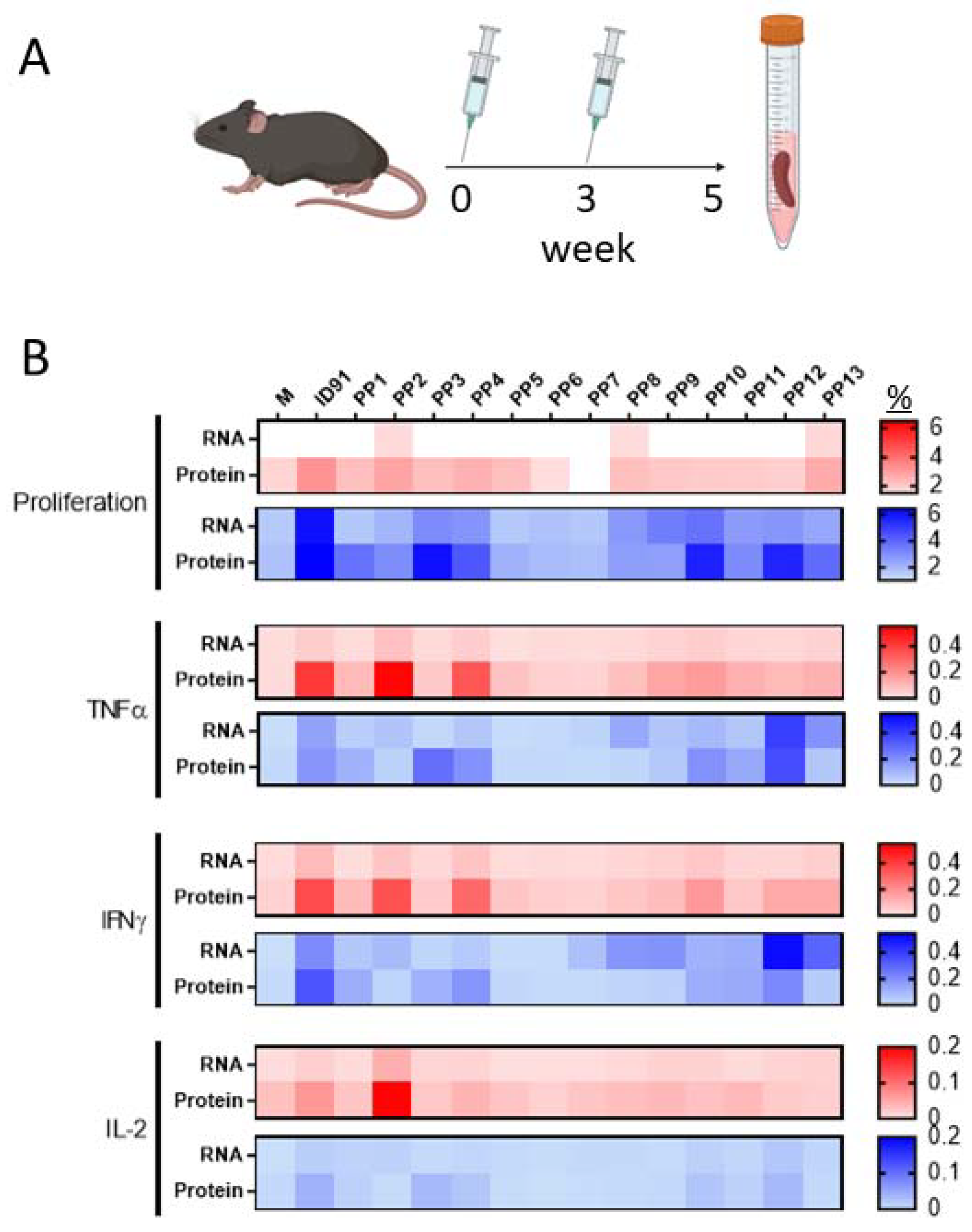
3.1. M.tb RNA Candidate Vaccine
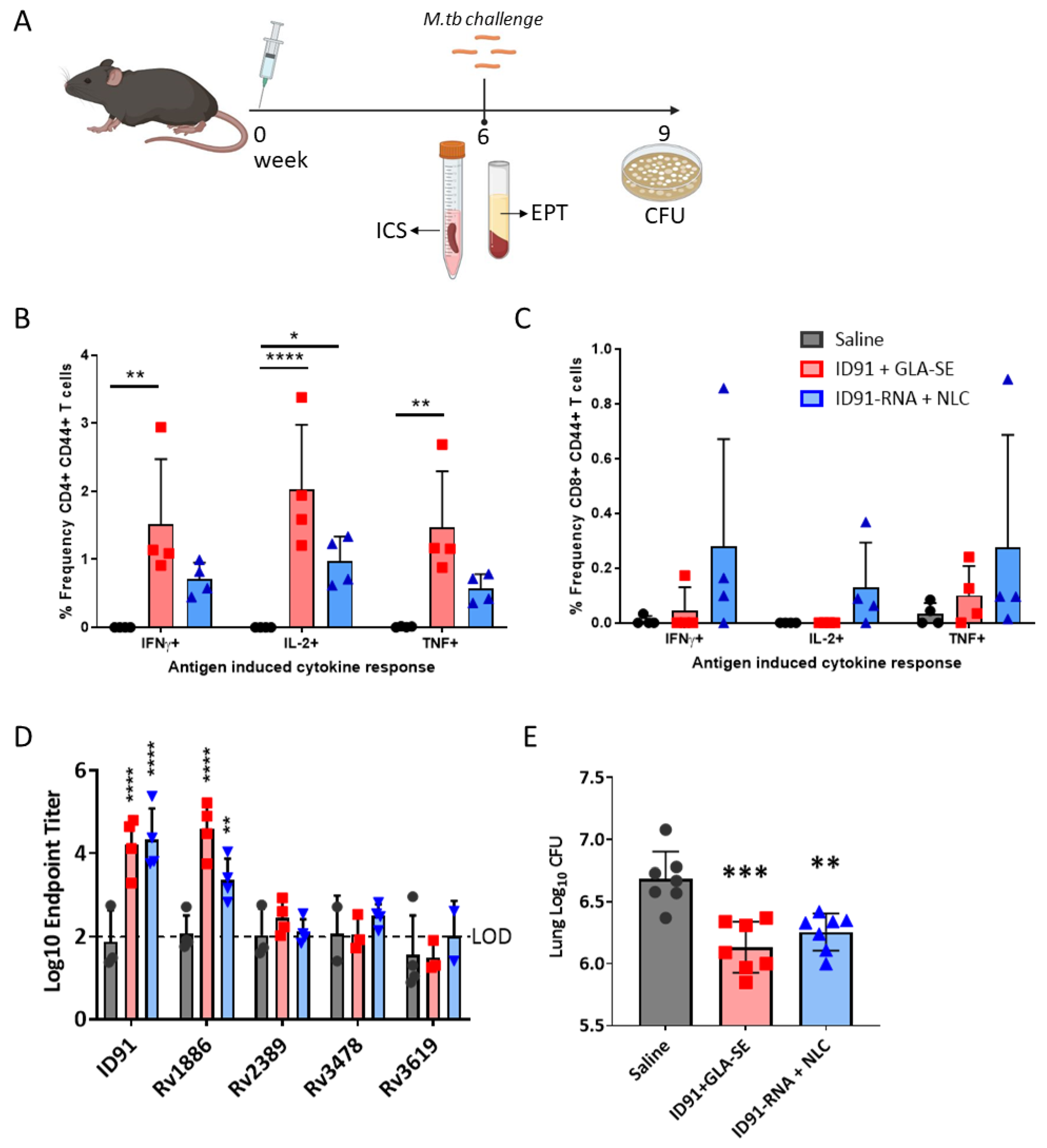
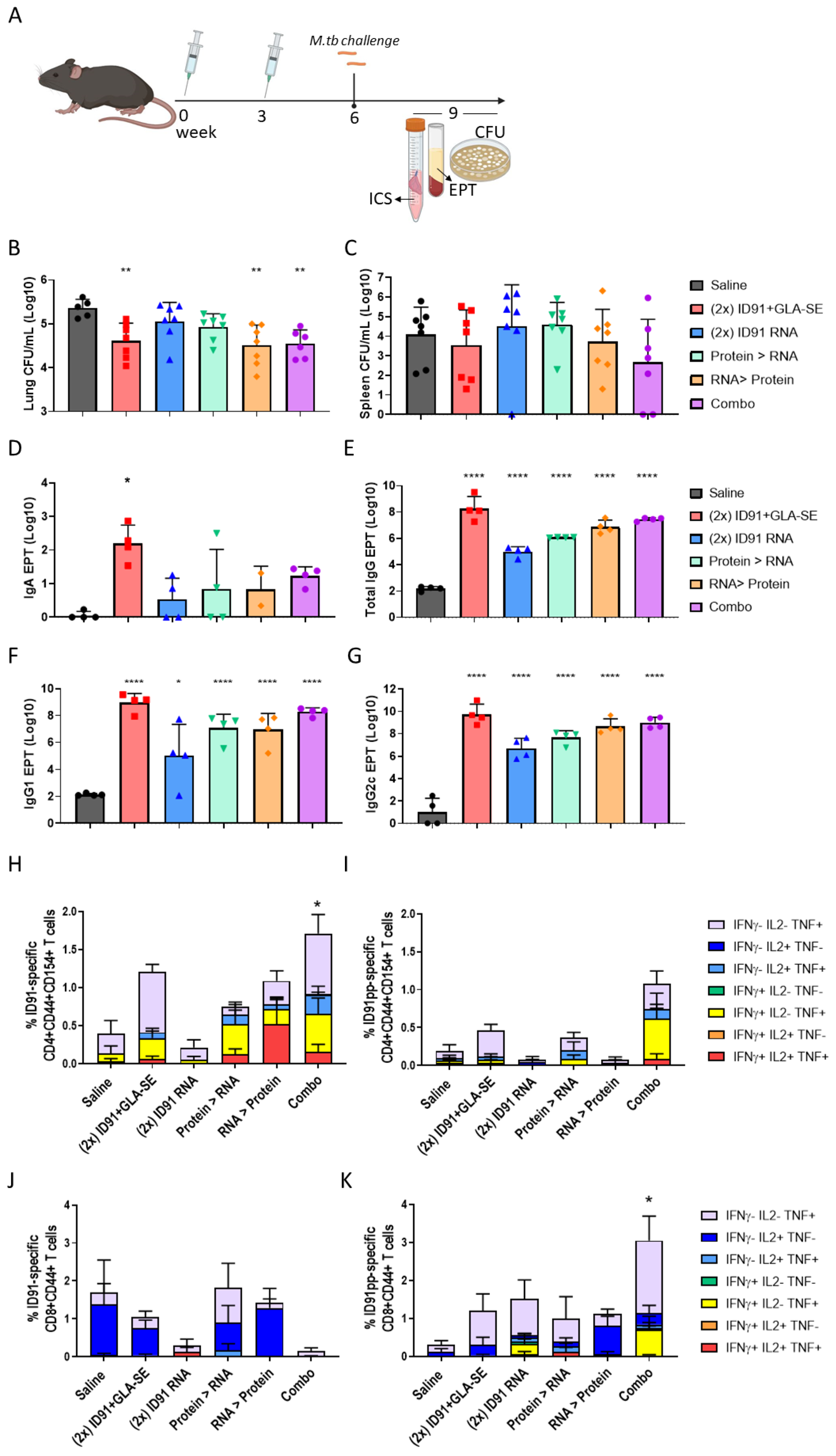
3.2. Immunogenicity and Prophylactic Protective Efficacy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2020: Executive Summary; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- World Health Organization. Global TB Report; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization Global TB Programme. Impact of the COVID-19 Pandemic on TB Detection and Mortality in 2020; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Zaman, K. Tuberculosis: A global health problem. J. Health Popul. Nutr. 2010, 28, 111–113. [Google Scholar] [CrossRef] [Green Version]
- Mallapaty, S.; Callaway, E.; Kozlov, M.; Ledford, H.; Pickrell, J.; Van Noorden, R. How COVID vaccines shaped 2021 in eight powerful charts. Nature 2021, 600, 580–583. [Google Scholar] [CrossRef]
- Parveen, U.; Sultana, S.; Heba, S.F.; Rafi, R.; Begum, A.; Fatima, N. Pretomanid: A novel therapeutic paradigm for treatment of drug resistant tuberculosis. Indian J. Tuberc. 2021, 68, 106–113. [Google Scholar] [CrossRef]
- Haldar, R.; Narayanan, S.J. A novel ensemble based recommendation approach using network based analysis for identification of effective drugs for Tuberculosis. Math. Biosci. Eng. 2022, 19, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Nandi, S.; Saxena, A.K. An updated patent review on drugs for the treatment of tuberculosis (2018-present). Expert Opin. Ther. Pat. 2022, 32, 401–421. [Google Scholar] [CrossRef] [PubMed]
- Almeida, D.; Converse, P.J.; Li, S.-Y.; Upton, A.M.; Fotouhi, N.; Nuermberger, E.L. Comparative Efficacy of the Novel Diarylquinoline TBAJ-876 and Bedaquiline against a Resistant Rv0678 Mutant in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e0141221. [Google Scholar] [CrossRef]
- Larkins-Ford, J.; Greenstein, T.; Van, N.; Degefu, Y.N.; Olson, M.C.; Sokolov, A.; Aldridge, B.B. Systematic measurement of combination-drug landscapes to predict in vivo treatment outcomes for tuberculosis. Cell Syst. 2021, 12, 1046–1063.e1047. [Google Scholar] [CrossRef]
- Houben, R.M.G.J.; A Menzies, N.; Sumner, T.; Huynh, G.H.; Arinaminpathy, N.; Goldhaber-Fiebert, J.D.; Lin, H.-H.; Wu, C.-Y.; Mandal, S.; Pandey, S.; et al. Feasibility of achieving the 2025 WHO global tuberculosis targets in South Africa, China, and India: A combined analysis of 11 mathematical models. Lancet Glob. Health 2016, 4, e806–e815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, N.; Gomez, G.B.; Bozzani, F.; Chatterjee, S.; Foster, N.; Baena, I.G.; Laurence, Y.V.; Qiang, S.; Siroka, A.; Sweeney, S.; et al. Cost-effectiveness and resource implications of aggressive action on tuberculosis in China, India, and South Africa: A combined analysis of nine models. Lancet Glob. Health 2016, 4, e816–e826. [Google Scholar] [CrossRef] [PubMed]
- Arregui, S.; Iglesias, M.J.; Samper, S.; Marinova, D.; Martin, C.; Sanz, J.; Moreno, Y. Data-driven model for the assessment of Mycobacterium tuberculosis transmission in evolving demographic structures. Proc. Natl. Acad. Sci. USA 2018, 115, E3238–E3245. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Eisenhut, M.; Harris, R.J.; Rodrigues, L.C.; Sridhar, S.; Habermann, S.; Snell, L.B.; Mangtani, P.; Adetifa, I.; Lalvani, A.; et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: Systematic review and meta-analysis. BMJ 2014, 349, g4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Meeren, O.; Hatherill, M.; Nduba, V.; Wilkinson, R.J.; Muyoyeta, M.; Van Brakel, E.; Ayles, H.M.; Henostroza, G.; Thienemann, F.; Scriba, T.J.; et al. Phase 2b Controlled Trial of M72/AS01(E) Vaccine to Prevent Tuberculosis. N. Engl. J. Med. 2018, 379, 1621–1634. [Google Scholar] [CrossRef]
- Tait, D.R.; Hatherill, M.; Van Der Meeren, O.; Ginsberg, A.M.; Van Brakel, E.; Salaun, B.; Scriba, T.J.; Akite, E.J.; Ayles, H.M.; Bollaerts, A.; et al. Final Analysis of a Trial of M72/AS01E Vaccine to Prevent Tuberculosis. N. Engl. J. Med. 2019, 381, 2429–2439. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Aguilo, N.; Marinova, D.; Gonzalo-Asensio, J. Update on TB Vaccine Pipeline. Appl. Sci. 2020, 10, 2632. [Google Scholar] [CrossRef] [Green Version]
- Lewinsohn, D.A.; Lewinsohn, D.M.; Scriba, T.J. Polyfunctional CD4(+) T Cells as Targets for Tuberculosis Vaccination. Front. Immunol. 2017, 8, 1262. [Google Scholar] [CrossRef] [Green Version]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; E Shea, J.; McClain, J.B.; Hussey, G.D.; A Hanekom, W.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.L.; Smith, M.T.; Yu, K.K.Q.; Luedemann, C.; Suscovich, T.J.; Grace, P.S.; Cain, A.; Yu, W.H.; McKitrick, T.R.; Lauffenburger, D.; et al. IFN-γ-independent immune markers of Mycobacterium tuberculosis exposure. Nat. Med. 2019, 25, 977–987. [Google Scholar] [CrossRef]
- Smith, C.M.; Proulx, M.K.; Lai, R.; Kiritsy, M.C.; Bell, T.A.; Hock, P.; de Villena, F.P.-M.; Ferris, M.T.; Baker, R.E.; Behar, S.M.; et al. Functionally Overlapping Variants Control Tuberculosis Susceptibility in Collaborative Cross Mice. mBio 2019, 10, e02791-19. [Google Scholar] [CrossRef] [Green Version]
- Behar, S.M. Antigen-specific CD8(+) T cells and protective immunity to tuberculosis. Adv. Exp. Med. Biol. 2013, 783, 141–163. [Google Scholar] [CrossRef]
- Behar, S.M.; Woodworth, J.S.; Wu, Y. Next generation: Tuberculosis vaccines that elicit protective CD8+ T cells. Expert Rev. Vaccines 2007, 6, 441–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boom, W.H. New TB vaccines: Is there a requirement for CD8 T cells? J. Clin. Investig. 2007, 117, 2092–2094. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Flynn, J. CD8+ T cells in tuberculosis. Am. J. Respir. Crit. Care Med. 2002, 166, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, D.A.; Swarbrick, G.M.; Park, B.; Cansler, M.E.; Null, M.D.; Toren, K.G.; Baseke, J.; Zalwango, S.; Mayanja-Kizza, H.; Malone, L.L.; et al. Comprehensive definition of human immunodominant CD8 antigens in tuberculosis. Npj. Vaccines 2017, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.L.; Flynn, J.L. CD8 T cells and Mycobacterium tuberculosis infection. Semin. Immunopathol. 2015, 37, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Kamath, A.B.; Woodworth, J.; Xiong, X.; Taylor, C.; Weng, Y.; Behar, S.M. Cytolytic CD8+ T cells recognizing CFP10 are recruited to the lung after Mycobacterium tuberculosis infection. J. Exp. Med. 2004, 200, 1479–1489. [Google Scholar] [CrossRef] [Green Version]
- Jeyanathan, M.; Mu, J.; McCormick, S.; Damjanovic, D.; Small, C.-L.; Shaler, C.R.; Kugathasan, K.; Xing, Z. Murine airway luminal antituberculosis memory CD8 T cells by mucosal immunization are maintained via antigen-driven in situ proliferation, independent of peripheral T cell recruitment. Am. J. Respir. Crit. Care Med. 2010, 181, 862–872. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Klein, M.R.; Malin, A.S.; Sillah, J.; Huygen, K.; Andersen, P.; McAdam, K.P.W.J.; Dockrell, H.M. Human CD8(+) T cells specific for Mycobacterium tuberculosis secreted antigens in tuberculosis patients and healthy BCG-vaccinated controls in The Gambia. Infect. Immun. 2000, 68, 7144–7148. [Google Scholar] [CrossRef] [Green Version]
- Tascon, R.E.; Stavropoulos, E.; Lukacs, K.V.; Colston, M.J. Protection against Mycobacterium tuberculosis infection by CD8+ T cells requires the production of gamma interferon. Infect. Immun. 1998, 66, 830–834. [Google Scholar] [CrossRef] [Green Version]
- Müller, I.; Cobbold, S.P.; Waldmann, H.; Kaufmann, S.H. Impaired resistance to Mycobacterium tuberculosis infection after selective in vivo depletion of L3T4+ and Lyt-2+ T cells. Infect. Immun. 1987, 55, 2037–2041. [Google Scholar] [CrossRef] [PubMed]
- Orme, I.M.; Collins, F.M. Adoptive protection of the Mycobacterium tuberculosis-infected lung. Dissociation between cells that passively transfer protective immunity and those that transfer delayed-type hypersensitivity to tuberculin. Cell. Immunol. 1984, 84, 113–120. [Google Scholar] [CrossRef]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Mehra, V.; Thoma-Uszynski, S.; Stenger, S.; Serbina, N.; Mazzaccaro, R.J.; Flynn, J.L.; Barnes, P.F.; Southwood, S.; Celis, E.; et al. Antimicrobial activity of MHC class I-restricted CD8+ T cells in human tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 12210–12215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbina, N.V.; Liu, C.C.; Scanga, C.A.; Flynn, J.L. CD8+ CTL from lungs of Mycobacterium tuberculosis-infected mice express perforin in vivo and lyse infected macrophages. J. Immunol. 2000, 165, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbina, N.V.; Flynn, J.L. CD8(+) T cells participate in the memory immune response to Mycobacterium tuberculosis. Infect. Immun. 2001, 69, 4320–4328. [Google Scholar] [CrossRef] [Green Version]
- Brookes, R.H.; Pathan, A.A.; McShane, H.; Hensmann, M.; Price, D.A.; Hill, A.V.S. CD8+ T cell-mediated suppression of intracellular Mycobacterium tuberculosis growth in activated human macrophages. Eur. J. Immunol. 2003, 33, 3293–3302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Thorson, L.; Stokes, R.W.; Santosuosso, M.; Huygen, K.; Zganiacz, A.; Hitt, M.; Xing, Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J. Immunol. 2004, 173, 6357–6365. [Google Scholar] [CrossRef] [Green Version]
- Darrah, P.A.; Zeppa, J.J.; Maiello, P.; Hackney, J.A.; Ii, M.H.W.; Hughes, T.K.; Pokkali, S.; Ii, P.A.S.; Grant, N.L.; Rodgers, M.A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roordink, D.; Williams, A.; Fritzell, B.; Laddy, D.J.; Gerdil, E.; Graffin, A.M.; Tait, D.; van der Pol, L.; van den Brink, I.; Holleman, M.; et al. The TB vaccine development pathway—An innovative approach to accelerating global TB vaccine development. Tuberculosis 2021, 126, 102040. [Google Scholar] [CrossRef]
- Whitlow, E.; Mustafa, A.S.; Hanif, S.N.M. An Overview of the Development of New Vaccines for Tuberculosis. Vaccines 2020, 8, 586. [Google Scholar] [CrossRef]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. The dawn of mRNA vaccines: The COVID-19 case. J. Control. Release 2021, 333, 511–520. [Google Scholar] [CrossRef]
- Xue, T.; Stavropoulos, E.; Yang, M.; Ragno, S.; Vordermeier, M.; Chambers, M.; Hewinson, G.; Lowrie, D.B.; Colston, M.J.; Tascon, R.E. RNA encoding the MPT83 antigen induces protective immune responses against Mycobacterium tuberculosis infection. Infect. Immun. 2004, 72, 6324–6329. [Google Scholar] [CrossRef] [Green Version]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [Green Version]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef]
- Ljungberg, K.; Liljeström, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Leitner, W.W.; Hwang, L.N.; Deveer, M.J.; Zhou, A.; Silverman, R.H.; Williams, B.; Dubensky, T.W.; Ying, H.; Restifo, N.P. Alphavirus-based DNA vaccine breaks immunological tolerance by activating innate antiviral pathways. Nat. Med. 2003, 9, 33–39. [Google Scholar] [CrossRef]
- Bertholet, S.; Ireton, G.C.; Ordway, D.J.; Windish, H.P.; Pine, S.O.; Kahn, M.; Phan, T.; Orme, I.M.; Vedvick, T.S.; Baldwin, S.L.; et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci. Transl. Med. 2010, 2, 53ra74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.E.; Baldwin, S.L.; Orr, M.T.; Reese, V.A.; Pecor, T.; Granger, B.; Cauwelaert, N.D.; Podell, B.K.; Coler, R.N. Enhanced Anti-Mycobacterium tuberculosis Immunity over Time with Combined Drug and Immunotherapy Treatment. Vaccines 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coler, R.N.; Bertholet, S.; Moutaftsi, M.; Guderian, J.A.; Windish, H.P.; Baldwin, S.L.; Laughlin, E.M.; Duthie, M.; Fox, C.; Carter, D.; et al. Development and characterization of synthetic glucopyranosyl lipid adjuvant system as a vaccine adjuvant. PLoS ONE 2011, 6, e16333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.E.; Reese, V.A.; Pecor, T.; Berube, B.J.; Cooper, S.K.; Brewer, G.; Ordway, D.; Henao-Tamayo, M.; Podell, B.K.; Baldwin, S.L.; et al. Subunit vaccine protects against a clinical isolate of Mycobacterium avium in wild type and immunocompromised mouse models. Sci. Rep. 2021, 11, 9040. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Ireton, G.C.; Beebe, E.A.; Huang, P.-W.D.; Reese, V.A.; Argilla, D.; Coler, R.N.; Reed, S.G. Immune subdominant antigens as vaccine candidates against Mycobacterium tuberculosis. J. Immunol. 2014, 193, 2911–2918. [Google Scholar] [CrossRef] [Green Version]
- Bertholet, S.; Ireton, G.C.; Kahn, M.; Guderian, J.; Mohamath, R.; Stride, N.; Laughlin, E.M.; Baldwin, S.L.; Vedvick, T.S.; Coler, R.N.; et al. Identification of human T cell antigens for the development of vaccines against Mycobacterium tuberculosis. J. Immunol. 2008, 181, 7948–7957. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.T.; Ireton, G.C.; Beebe, E.A.; Huang, P.-W.D.; Reese, V.A.; Argilla, D.; Coler, R.N.; Reed, S.G. Elimination of the cold-chain dependence of a nanoemulsion adjuvanted vaccine against tuberculosis by lyophilization. J. Control. Release 2014, 177, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; da Silva Antunes, R.; Sidney, J.; Lindestam Arlehamn, C.S.; Grifoni, A.; Dhanda, S.K.; Paul, S.; Peters, B.; Weiskopf, D.; Sette, A. A Review on T Cell Epitopes Identified Using Prediction and Cell-Mediated Immune Models for Mycobacterium tuberculosis and Bordetella pertussis. Front. Immunol. 2018, 9, 2778. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Lewinsohn, D.; Sette, A.; Lewinsohn, D. Antigens for CD4 and CD8 T cells in tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 4, a018465. [Google Scholar] [CrossRef] [Green Version]
- Gagneux, S.; Small, P.M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 2007, 7, 328–337. [Google Scholar] [CrossRef]
- Day, T.A.; Penn-Nicholson, A.; Luabeya, A.K.K.; Fiore-Gartland, A.; Du Plessis, N.; Loxton, A.G.; Vergara, J.; Rolf, T.A.; Reid, T.D.; Toefy, A.; et al. Safety and immunogenicity of the adjunct therapeutic vaccine ID93 + GLA-SE in adults who have completed treatment for tuberculosis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet Respir. Med. 2020, 9, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Ching, L.K.; Pine, S.O.; Moutaftsi, M.; Lucas, E.; Vallur, A.; Orr, M.T.; Bertholet, S.; Reed, S.G.; Coler, R.N. Protection against tuberculosis with homologous or heterologous protein/vector vaccine approaches is not dependent on CD8+ T cells. J. Immunol. 2013, 191, 2514–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.-J.; Barreira-Silva, P.; Boyce, S.; Powers, J.; Cavallo, K.; Behar, S.M. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021, 36, 109696. [Google Scholar] [CrossRef]
- Irvine, E.B.; O’Neil, A.; Darrah, P.A.; Shin, S.; Choudhary, A.; Li, W.; Honnen, W.; Mehra, S.; Kaushal, D.; Gideon, H.P.; et al. Robust IgM responses following intravenous vaccination with Bacille Calmette-Guérin associate with prevention of Mycobacterium tuberculosis infection in macaques. Nat. Immunol. 2021, 22, 1515–1523. [Google Scholar] [CrossRef]
- Orr, M.T.; Beebe, E.A.; Hudson, T.E.; Argilla, D.; Huang, P.-W.D.; Reese, V.A.; Fox, C.B.; Reed, S.G.; Coler, R.N. Mucosal delivery switches the response to an adjuvanted tuberculosis vaccine from systemic TH1 to tissue-resident TH17 responses without impacting the protective efficacy. Vaccine 2015, 33, 6570–6578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilkington, E.H.; Suys, E.J.; Trevaskis, N.L.; Wheatley, A.K.; Zukancic, D.; Algarni, A.; Al-Wassiti, H.; Davis, T.P.; Pouton, C.W.; Kent, S.J.; et al. From influenza to COVID-19: Lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater. 2021, 131, 16–40. [Google Scholar] [CrossRef]
- Mulligan, M.J.; Lyke, K.E.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Raabe, V.; Bailey, R.; Swanson, K.A.; et al. Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature 2020, 586, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool 1 | 43 | AAAYETAYRLTVPPP | 86 | PGLPVEYLQVPSPSM | |
| 1 | HMTINYQFGDVDAHG | 44 | YRLTVPPPVIAENRT | 87 | LQVPSPSMGRDIKVQ |
| 2 | FGDVDAHGAMIRAQA | 45 | PVIAENRTELMTLTA | 88 | MGRDIKVQFQSGGNN |
| 3 | GAMIRAQAGSLEAEH | 46 | TELMTLTATNLLGQN | 89 | QFQSGGNNSPAVYLL |
| 4 | AGSLEAEHQAIISDV | 47 | ATNLLGQNTPAIEAN | 90 | NSPAVYLLDGLRAQD |
| 5 | HQAIISDVLTASDFW | 48 | NTPAIEANQAAYSQM | Pool 10 | |
| 6 | VLTASDFWGGAGSAA | 49 | NQAAYSQMWGQDAEA | 91 | LDGLRAQDDYNGWDI |
| 7 | WGGAGSAACQGFITQ | 50 | MWGQDAEAMYGYAAT | 92 | DDYNGWDINTPAFEW |
| 8 | ACQGFITQLGRNFQV | Pool 6 | 93 | INTPAFEWYYQSGLS | |
| 9 | QLGRNFQVIYEQANA | 51 | AMYGYAATAATATEA | 94 | WYYQSGLSIVMPVGG |
| 10 | VIYEQANAHGQKVQA | 52 | TAATATEALLPFEDA | 95 | SIVMPVGGQSSFYSD |
| Pool 2 | 53 | ALLPFEDAPLITNPG | 96 | GQSSFYSDWYSPACG | |
| 11 | AHGQKVQAAGNNMAQ | 54 | APLITNPGGLLEQAV | 97 | DWYSPACGKAGCQTY |
| 12 | AAGNNMAQTDSAVGS | 55 | GGLLEQAVAVEEAID | 98 | GKAGCQTYKWETFLT |
| 13 | QTDSAVGSSWADDID | 56 | VAVEEAIDTAAANQL | 99 | YKWETFLTSELPQWL |
| 14 | SSWADDIDWDAIAQC | 57 | DTAAANQLMNNVPQA | 100 | TSELPQWLSANRAVK |
| 15 | DWDAIAQCESGGNWA | 58 | LMNNVPQALQQLAQP | Pool 11 | |
| 16 | CESGGNWAANTGNGL | 59 | ALQQLAQPAQGVVPS | 101 | LSANRAVKPTGSAAI |
| 17 | AANTGNGLYGGLQIS | 60 | PAQGVVPSSKLGGLW | 102 | KPTGSAAIGLSMAGS |
| 18 | LYGGLQISQATWDSN | Pool 7 | 103 | IGLSMAGSSAMILAA | |
| 19 | SQATWDSNGGVGSPA | 61 | SSKLGGLWTAVSPHL | 104 | SSAMILAAYHPQQFI |
| 20 | NGGVGSPAAASPQQQ | 62 | WTAVSPHLSPLSNVS | 105 | AYHPQQFIYAGSLSA |
| Pool 3 | 63 | LSPLSNVSSIANNHM | 106 | IYAGSLSALLDPSQG | |
| 21 | AAASPQQQIEVADNI | 64 | SSIANNHMSMMGTGV | 107 | ALLDPSQGMGPSLIG |
| 22 | QIEVADNIMKTQGPG | 65 | MSMMGTGVSMTNTLH | 108 | GMGPSLIGLAMGDAG |
| 23 | IMKTQGPGAWPKCSS | 66 | VSMTNTLHSMLKGLA | 109 | GLAMGDAGGYKAADM |
| 24 | GAWPKCSSCSQGDAP | 67 | HSMLKGLAPAAAQAVE | 110 | GGYKAADMWGPSSDP |
| 25 | SCSQGDAPLGSLTHI | 68 | PAAAQAVETAAENGV | Pool 12 | |
| 26 | PLGSLTHILTFLAAE | 69 | ETAAENGVWAMSSLG | 111 | MWGPSSDPAWERNDP |
| 27 | ILTFLAAETGGCSGS | 70 | VWAMSSLGSQLGSSL | 112 | PAWERNDPTQQIPKL |
| 28 | ETGGCSGSRDDVVDF | Pool 8 | 113 | PTQQIPKLVANNTRL | |
| 29 | SRDDVVDFGALPPEI | 71 | GSQLGSSLGSSGLGA | 114 | LVANNTRLWVYCGNG |
| 30 | FGALPPEINSARMYA | 72 | LGSSGLGAGVAANLG | 115 | LWVYCGNGTPNELGG |
| Pool 4 | 73 | AGVAANLGRAASVGS | 116 | GTPNELGGANIPAEF | |
| 31 | INSARMYAGPGSASL | 74 | GRAASVGSLSVPPAW | 117 | GANIPAEFLENFVRS |
| 32 | AGPGSASLVAAAKMW | 75 | SLSVPPAWAAANQAV | 118 | FLENFVRSSNLKFQD |
| 33 | LVAAAKMWDSVASDL | 76 | WAAANQAVTPAARAL | 119 | SSNLKFQDAYNAAGG |
| 34 | WDSVASDLFSAASAF | 77 | VTPAARALPLTSLTS | 120 | DAYNAAGGHNAVFNF |
| 35 | LFSAASAFQSVVWGL | 78 | LPLTSLTSAAQTAPG | Pool 13 | |
| 36 | FQSVVWGLTVGSWIG | 79 | SAAQTAPGHMLGGLP | 121 | GHNAVFNFPPNGTHS |
| 37 | LTVGSWIGSSAGLMA | 80 | GHMLGGLPLGHSVNA | 122 | FPPNGTHSWEYWGAQ |
| 38 | GSSAGLMAAAASPYV | Pool 9 | 123 | SWEYWGAQLNAMKGD | |
| 39 | AAAASPYVAWMSVTA | 81 | PLGHSVNAGSGINNA | 124 | QLNAMKGDLQSSLGA |
| 40 | VAWMSVTAGQAQLTA | 82 | AGSGINNALRVPARA | 125 | AMKGDLQSSLGAGKL |
| Pool 5 | 83 | ALRVPARAYAIPRTP | |||
| 41 | AGQAQLTAAQVRVAA | 84 | AYAIPRTPAAGFSRP | ||
| 42 | AAQVRVAAAAYETAY | 85 | PAAGFSRPGLPVEYL | ||
| Candidate Regimen | CFU Log10 ± SEM | CFU Log10 Reduction $ | Log10 ID91 Total IgG EPT $ | % ID91 CD4+ T cells # | % ID91 CD8+ T cells # | |||
|---|---|---|---|---|---|---|---|---|
| Prime | Boost | Lung | Spleen | Lung | Spleen | |||
| Saline | Saline | 5.36 ± 0.09 | 4.08 ± 0.48 | - | - | 2.18 | 0.398 | 0.304 |
| ID91 + GLA-SE | ID91 + GLA-SE | 4.61 ± 0.15 | 3.52 ± 0.63 | 0.743 ** p = 0.0075 | 0.557 p = 0.9638 | 8.25 **** p < 0.0001 | 1.209 p = 0.3415 | 1.198 p = 0.8055 |
| ID91-RNA + NLC | ID91-RNA + NLC | 5.05 ± 0.017 | 4.51 ± 0.74 | 0.306 p = 0.4731 | −0.427 p = 0.9883 | 4.98 **** p < 0.0001 | 0.212 p = 0.9944 | 1.512 p = 0.9952 |
| ID91 + GLA-SE | ID91-RNA + NLC | 4.93 ± 0.11 | 4.58 ± 0.39 | 0.428 p = 0.1920 | −0.501 p = 0.9765 | 6.09 **** p < 0.0001 | 0.748 p = 0.9216 | 0.994 p = 0.9573 |
| ID91-RNA + NLC | ID91 + GLA-SE | 4.51 ± 0.18 | 3.72 ± 0.57 | 0.847 ** p = 0.0021 | 0.358 p = 0.9947 | 6.87 **** p < 0.0001 | 1.088 p = 0.4890 | 1.110 p = 0.9945 |
| Combination | Combination | 4.54 ± 0.13 | 2.66 ± 0.76 | 0.809 ** p = 0.0047 | 1.419 p = 0.4273 | 7.45 **** p < 0.0001 | 1.711 * p = 0.0487 | 3.048 * p = 0.0226 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, S.E.; Erasmus, J.H.; Reese, V.A.; Pecor, T.; Archer, J.; Kandahar, A.; Hsu, F.-C.; Nicholes, K.; Reed, S.G.; Baldwin, S.L.; et al. An RNA-Based Vaccine Platform for Use against Mycobacterium tuberculosis. Vaccines 2023, 11, 130. https://doi.org/10.3390/vaccines11010130
Larsen SE, Erasmus JH, Reese VA, Pecor T, Archer J, Kandahar A, Hsu F-C, Nicholes K, Reed SG, Baldwin SL, et al. An RNA-Based Vaccine Platform for Use against Mycobacterium tuberculosis. Vaccines. 2023; 11(1):130. https://doi.org/10.3390/vaccines11010130
Chicago/Turabian StyleLarsen, Sasha E., Jesse H. Erasmus, Valerie A. Reese, Tiffany Pecor, Jacob Archer, Amit Kandahar, Fan-Chi Hsu, Katrina Nicholes, Steven G. Reed, Susan L. Baldwin, and et al. 2023. "An RNA-Based Vaccine Platform for Use against Mycobacterium tuberculosis" Vaccines 11, no. 1: 130. https://doi.org/10.3390/vaccines11010130