
An Immunoinformatics Approach to Design a Potent Multi-Epitope Vaccine against Asia-1 Genotype of Crimean–Congo Haemorrhagic Fever Virus Using the Structural Glycoproteins as a Target
 , ,
, ,  , ,
, ,  , ,
, ,  ,
,  ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Sequences Retrieval and Multiple Sequence Alignment
2.2. Linear B-Cell Epitopes Prediction
2.3. Cytotoxic T-lymphocyte Epitopes Prediction
2.4. Antigenicity, Allergenicity, Toxicity, and Non-Homology Analysis of Epitopes
2.5. Construction of Multi-Epitope Chimeric Vaccine
2.6. Population Coverage Analysis
2.7. Evaluation of Immunological and Physicochemical Properties of the Chimeric Vaccine
2.8. Computational Immune Assay for the Chimeric Vaccine
2.9. Prediction, Refinement and Validation of Tertiary Structure of the Constructed Vaccine
2.10. Molecular Docking Analysis
2.10.1. Peptide Modelling and Docking Analysis of Predicted Epitopes with MHC-I
2.10.2. Molecular Docking of Multi-Epitope Vaccine Construct and TLRs
2.11. Molecular Dynamics Simulations
2.12. Analysis of MD Trajectories
2.13. Binding Free Energy Calculation
3. Results
3.1. Sequence Retrieval and Conservation
3.2. Linear B-Cell Epitopes Prediction
3.3. CTL Epitope Prediction
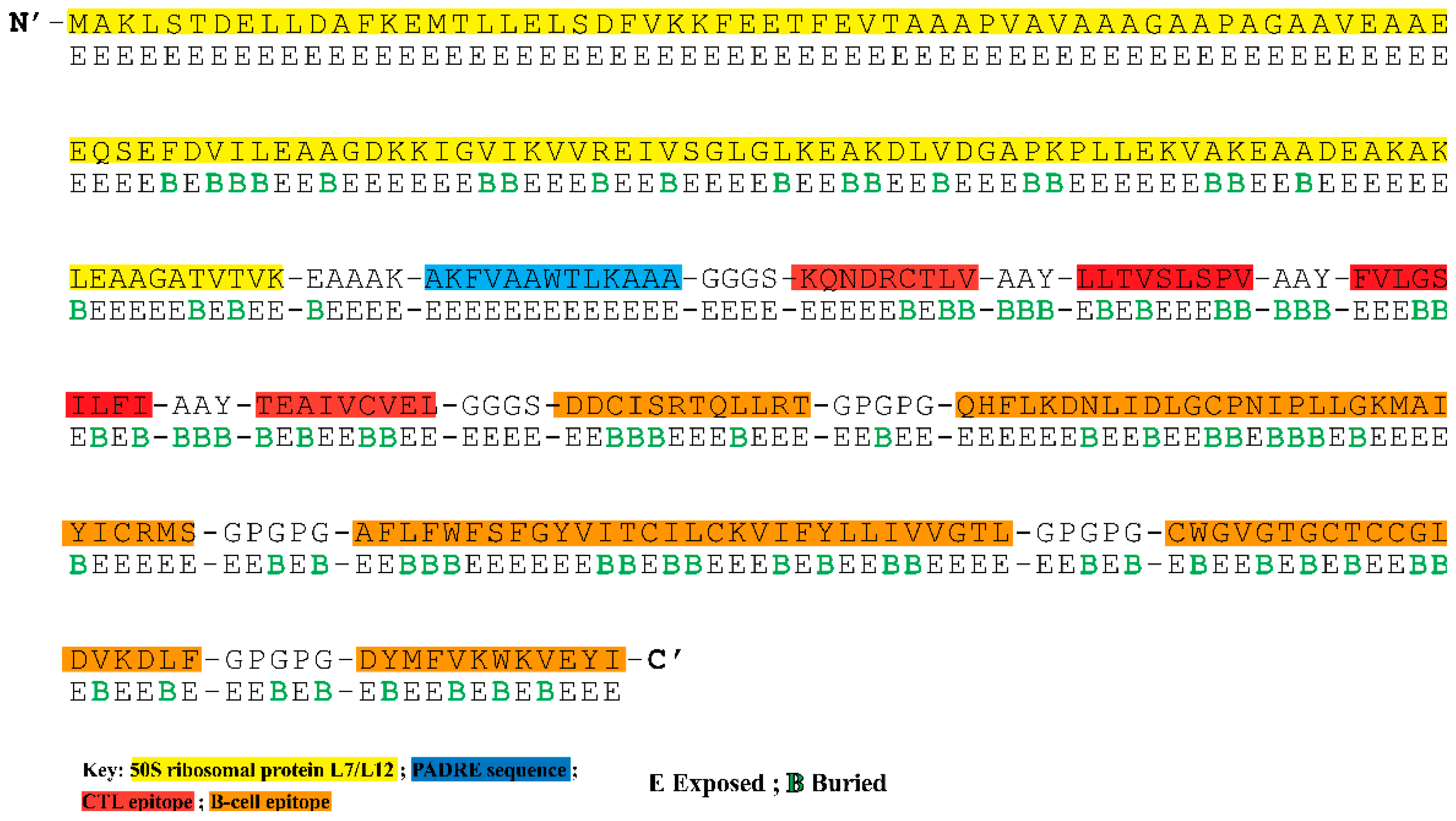
3.4. Construction of Multi-Epitope Vaccine Construct
3.5. Analysis of Surface Accessibility and Population Coverage of the Chimeric Construct
3.6. The Chimeric Vaccine Accumulates Features of Safety and Effective Antigen
3.7. Physicochemical Analysis Suggested Positive Parameters for Vaccine Production
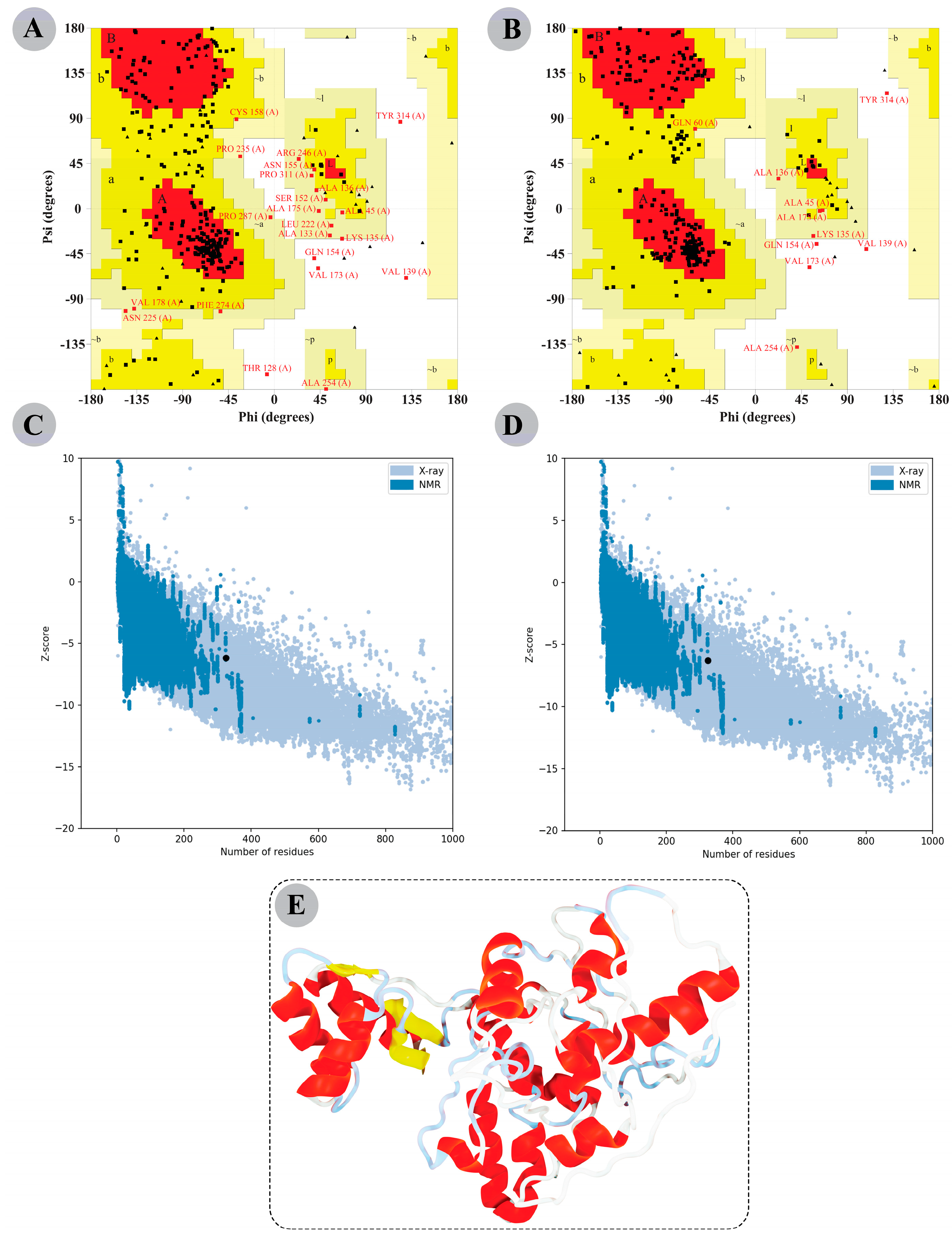
3.8. The Modelled Vaccine Attained a Desirable 3D Orientation
3.9. Molecular Docking Analysis
3.9.1. Peptide Modelling and Docking Analysis of Predicted Epitopes with MHC-I
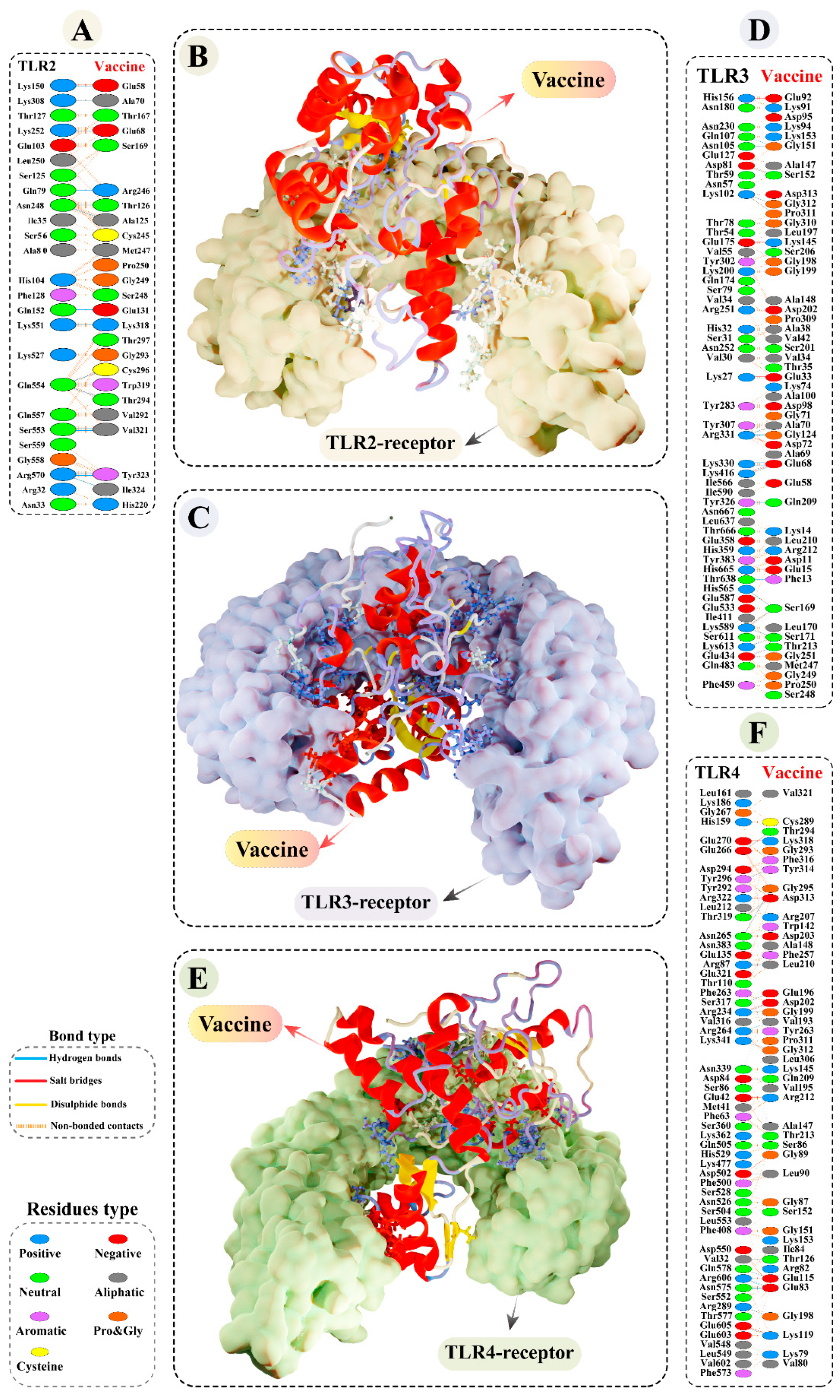
3.9.2. Molecular Docking of Modelled Vaccine with TLRs
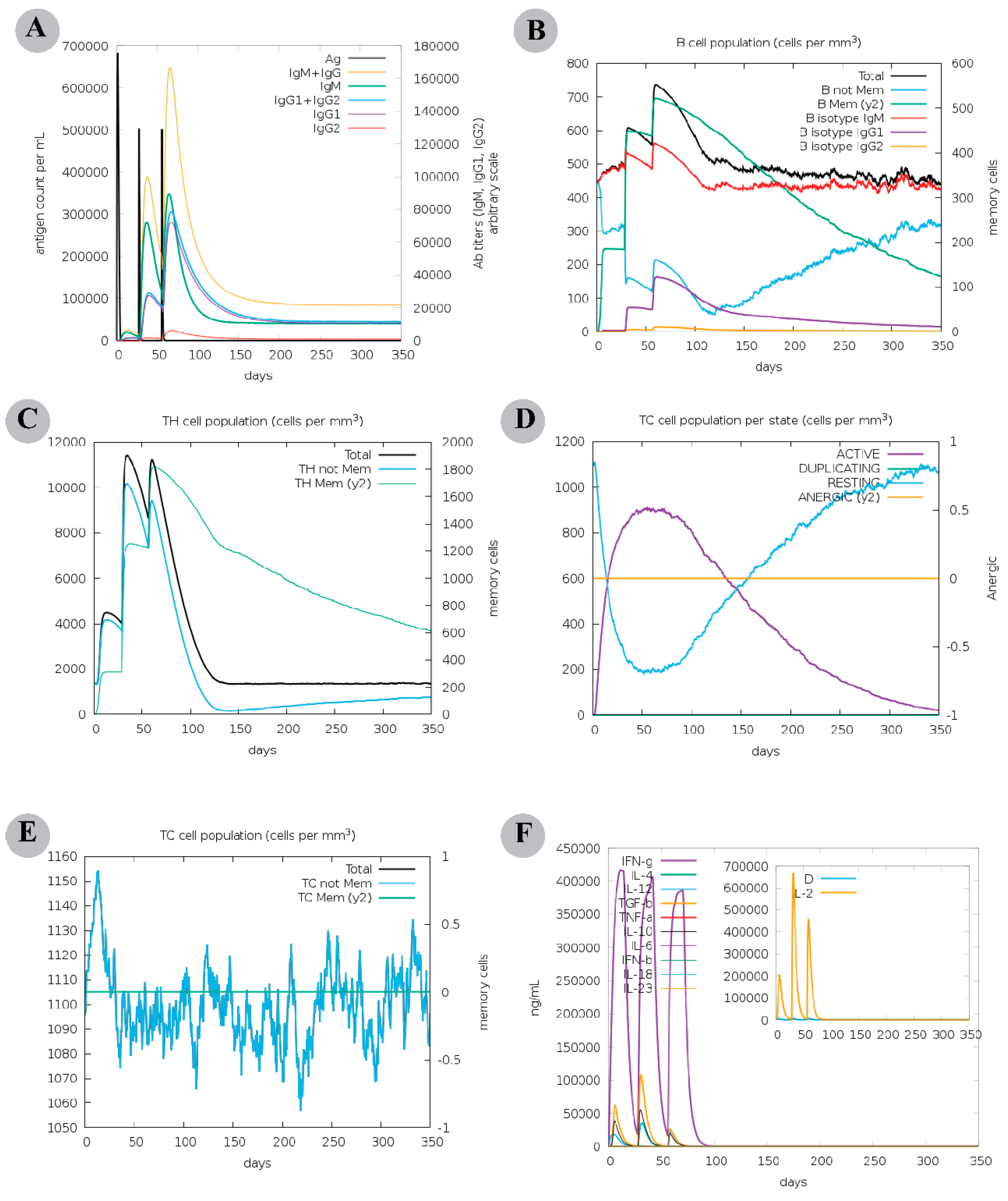
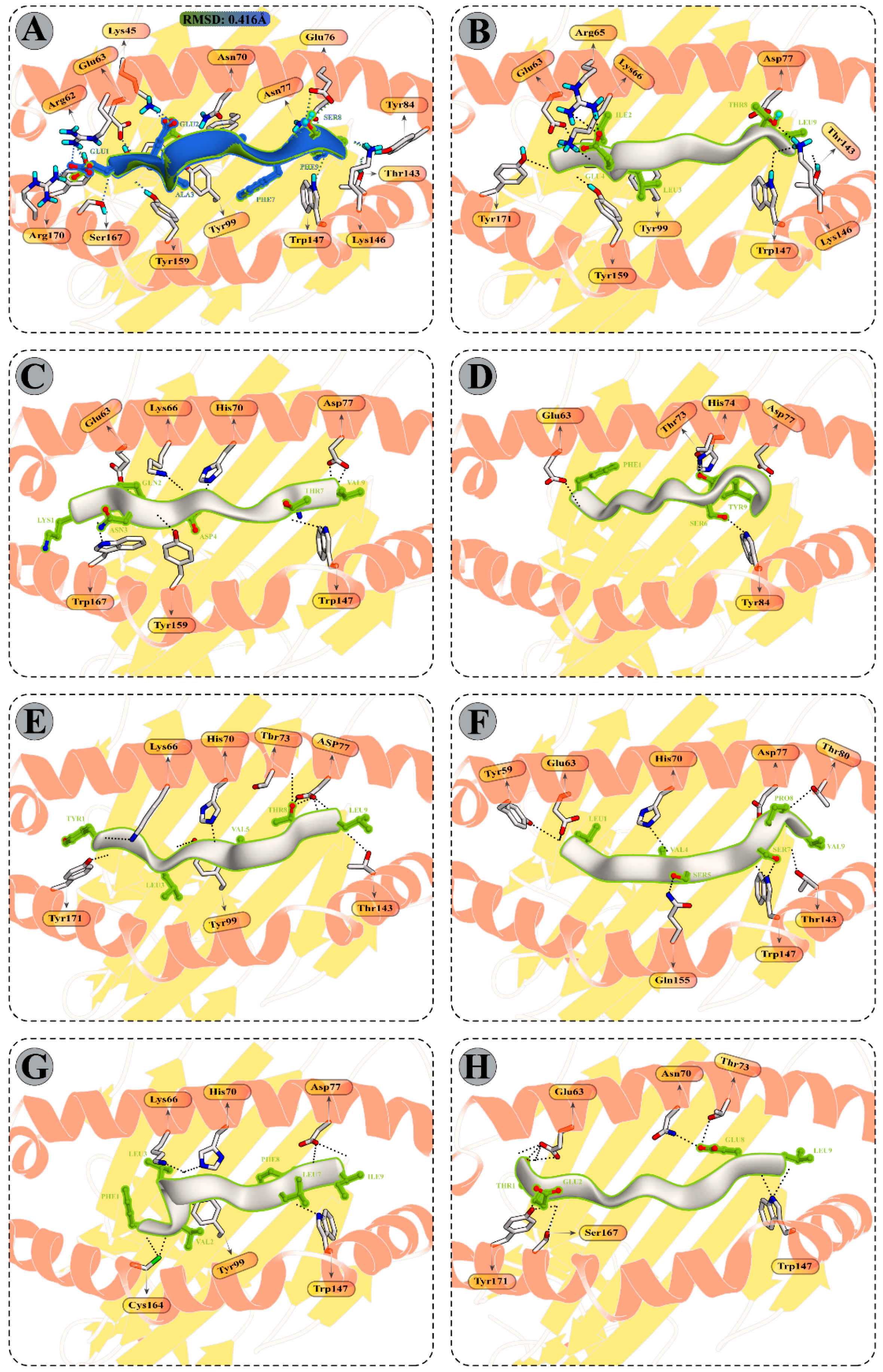
3.10. Molecular Dynamics Simulations of CTL Epitopes–HLA Complex
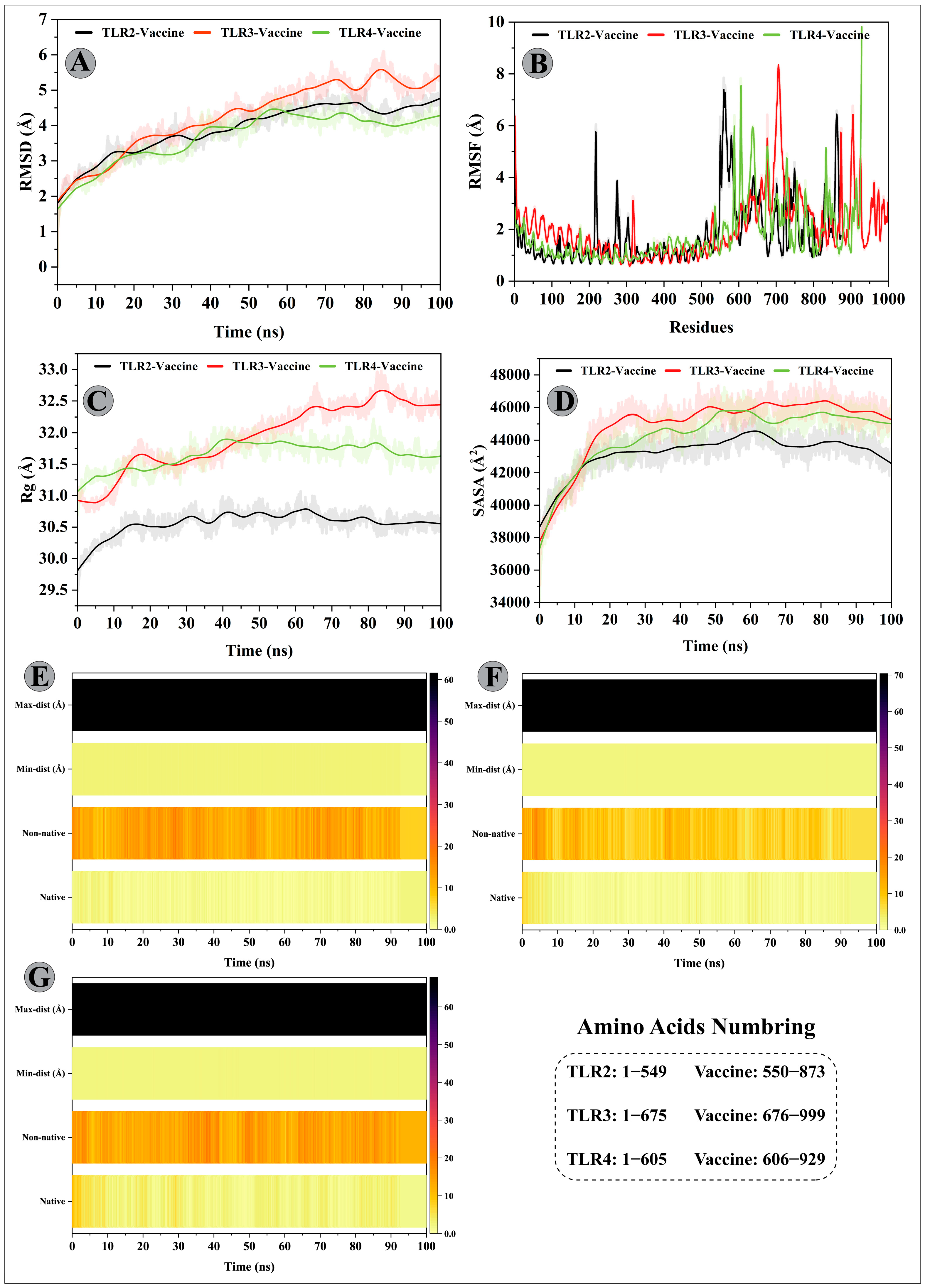
3.11. Molecular Dynamics Simulations of Modelled Vaccine–TLRs Complex
3.12. Binding Free Energy Calculation of Selected Peptide–HLA Molecule Complexes
3.13. Binding Free Calculation of Vaccine–TLRs Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Abri, S.S.; Abaidani, I.A.; Fazlalipour, M.; Mostafavi, E.; Leblebicioglu, H.; Pshenichnaya, N.; Memish, Z.A.; Hewson, R.; Petersen, E.; Mala, P.; et al. Current status of Crimean-Congo haemorrhagic fever in the World Health Organization Eastern Mediterranean Region: Issues, challenges, and future directions. Int. J. Infect Dis. 2017, 58, 82–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, R.M.; Pljusnin, A. Bunyaviridae: Molecular and Cellular Biology; Caister Acad. Press: Norfolk, UK, 2011. [Google Scholar]
- Deyde, V.M.; Khristova, M.L.; Rollin, P.E.; Ksiazek, T.G.; Nichol, S.T. Crimean-Congo hemorrhagic fever virus genomics and global diversity. J. Virol. 2006, 80, 8834–8842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, A.J.; Vincent, M.J.; Nichol, S.T. Characterization of the glycoproteins of Crimean-Congo hemorrhagic fever virus. J. Virol. 2002, 76, 7263–7275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haferkamp, S.; Fernando, L.; Schwarz, T.F.; Feldmann, H.; Flick, R. Intracellular localization of Crimean-Congo Hemorrhagic Fever (CCHF) virus glycoproteins. Virol. J. 2005, 2, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti-Ciarlet, A.; Smith, J.; Strecker, K.; Paragas, J.; Altamura, L.A.; McFalls, J.M.; Frias-Staheli, N.; Garcia-Sastre, A.; Schmaljohn, C.S.; Doms, R.W. Cellular localization and antigenic characterization of crimean-congo hemorrhagic fever virus glycoproteins. J. Virol. 2005, 79, 6152–6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, E.; Cihan, A.Ç.; Aligholipour, T.; Foldes, K.; Engin, E.D.; Özkul, A. Assessment of the Immunogenicity and Protective Aspects of a DNA Vaccine Targeting Crimean Congo Hemorrhagic Fever Virus Glycoprotein Gc. Duzce Med. J. 2021, 23, 66–75. [Google Scholar] [CrossRef]
- Buttigieg, K.R.; Dowall, S.D.; Findlay-Wilson, S.; Miloszewska, A.; Rayner, E.; Hewson, R.; Carroll, M.W. A novel vaccine against Crimean-Congo Haemorrhagic Fever protects 100% of animals against lethal challenge in a mouse model. PLoS ONE 2014, 9, e91516. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, J.; Vloet, R.P.; McAuley, A.J.; Shen, X.; Bosch, B.J.; de Vries, L.; Moormann, R.J.; Bente, D.A. Crimean-Congo Hemorrhagic Fever Virus Subunit Vaccines Induce High Levels of Neutralizing Antibodies But No Protection in STAT1 Knockout Mice. Vector Borne Zoonotic Dis. 2015, 15, 759–764. [Google Scholar] [CrossRef] [Green Version]
- Chumakov, M.P. Studies of virus haemorrhagic fevers. J. Hyg. Epidemiol. Microbiol. Immunol. 1963, 7, 125–135. [Google Scholar]
- Ergönül, Ö. Crimean-Congo haemorrhagic fever. Lancet Infect. Dis. 2006, 6, 203–214. [Google Scholar] [CrossRef]
- Hoogstraal, H. Review Article 1: The Epidemiology of Tick-Borne Crimean-Congo Hemorrhagic Fever in Asia, Europe, and Africa 2 3. J. Med. Entomol. 1979, 15, 307–417. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.C.; Papa, A. Crimean-Congo hemorrhagic fever: Risk for emergence of new endemic foci in Europe? Travel Med. Infect. Dis. 2010, 8, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Oany, A.R.; Ahmad, S.A.I.; Hossain, M.U.; Jyoti, T.P. Identification of highly conserved regions in L-segment of Crimean–Congo hemorrhagic fever virus and immunoinformatic prediction about potential novel vaccine. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, F.J.; Swanepoel, R.; Shieh, W.J.; Smith, J.F.; Leman, P.A.; Greer, P.W.; Coffield, L.M.; Rollin, P.E.; Ksiazek, T.G.; Peters, C.J.; et al. Immunohistochemical and in situ localization of Crimean-Congo hemorrhagic fever (CCHF) virus in human tissues and implications for CCHF pathogenesis. Arch. Pathol. Lab. Med. 1997, 121, 839–846. [Google Scholar] [PubMed]
- Peyrefitte, C.N.; Perret, M.; Garcia, S.; Rodrigues, R.; Bagnaud, A.; Lacote, S.; Crance, J.M.; Vernet, G.; Garin, D.; Bouloy, M.; et al. Differential activation profiles of Crimean-Congo hemorrhagic fever virus- and Dugbe virus-infected antigen-presenting cells. J. Gen. Virol. 2010, 91, 189–198. [Google Scholar] [CrossRef]
- Connolly-Andersen, A.M.; Douagi, I.; Kraus, A.A.; Mirazimi, A. Crimean Congo hemorrhagic fever virus infects human monocyte-derived dendritic cells. Virology 2009, 390, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Connolly-Andersen, A.M.; Moll, G.; Andersson, C.; Akerstrom, S.; Karlberg, H.; Douagi, I.; Mirazimi, A. Crimean-Congo hemorrhagic fever virus activates endothelial cells. J. Virol. 2011, 85, 7766–7774. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.A.; Mirazimi, A. Molecular biology and pathogenesis of Crimean–Congo hemorrhagic fever virus. Future Virol. 2010, 5, 469–479. [Google Scholar] [CrossRef]
- Duygu, F.; Kaya, T.; Baysan, P. Re-evaluation of 400 Crimean-Congo hemorrhagic fever cases in an endemic area: Is ribavirin treatment suitable? Vector-Borne Zoonotic Dis. 2012, 12, 812–816. [Google Scholar] [CrossRef] [Green Version]
- Umair, M.; Khurshid, A.; Alam, M.M.; Akhtar, R.; Salman, M.; Ikram, A. Genetic diversity and phylogenetic analysis of Crimean-Congo Hemorrhagic Fever viruses circulating in Pakistan during 2019. PLoS Negl. Trop. Dis. 2020, 14, e0008238. [Google Scholar] [CrossRef]
- Alam, M.M.; Khurshid, A.; Sharif, S.; Shaukat, S.; Rana, M.S.; Angez, M.; Zaidi, S.S. Genetic analysis and epidemiology of Crimean Congo Hemorrhagic fever viruses in Baluchistan province of Pakistan. BMC Infect. Dis. 2013, 13, 201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amanna, I.J.; Slifka, M.K. Contributions of humoral and cellular immunity to vaccine-induced protection in humans. Virology 2011, 411, 206–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabel, G.J. HIV vaccine strategies. Vaccine 2002, 20, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Sweredoski, M.J.; Baldi, P. COBEpro: A novel system for predicting continuous B-cell epitopes. Protein Eng. Des. Sel. 2009, 22, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Duffy, L.; O’Reilly, S.C. Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. Immunotargets Ther. 2016, 5, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Lu, Y.; Chen, Y. Bioinformatics analysis of SARS-CoV M protein provides information for vaccine development. Prog. Nat. Sci. 2003, 13, 844–847. [Google Scholar] [CrossRef]
- Sedeyn, K.; Schepens, B.; Saelens, X. Respiratory syncytial virus nonstructural proteins 1 and 2: Exceptional disrupters of innate immune responses. PLoS Pathog. 2019, 15, e1007984. [Google Scholar] [CrossRef] [Green Version]
- Hall, T. BioEdit version 7.0.0. 2004. Available online: www.mbio.ncsu.edu/BioEdit/bioedit.html (accessed on 10 October 2022).
- Kolaskar, A.S.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Sami, S.A.; Marma, K.K.S.; Mahmud, S.; Khan, M.A.N.; Albogami, S.; El-Shehawi, A.M.; Rakib, A.; Chakraborty, A.; Mohiuddin, M.; Dhama, K.; et al. Designing of a Multi-epitope Vaccine against the Structural Proteins of Marburg Virus Exploiting the Immunoinformatics Approach. ACS Omega 2021, 6, 32043–32071. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Valkenburg, S.A.; Josephs, T.M.; Clemens, E.B.; Grant, E.J.; Nguyen, T.H.; Wang, G.C.; Price, D.A.; Miller, A.; Tong, S.Y.; Thomas, P.G.; et al. Molecular basis for universal HLA-A*0201-restricted CD8+ T-cell immunity against influenza viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 4440–4445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodossis, A.; Guillonneau, C.; Welland, A.; Ely, L.K.; Clements, C.S.; Williamson, N.A.; Webb, A.I.; Wilce, J.A.; Mulder, R.J.; Dunstone, M.A. Constraints within major histocompatibility complex class I restricted peptides: Presentation and consequences for T-cell recognition. Proc. Natl. Acad. Sci. USA 2010, 107, 5534–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Operating Environment (MOE), 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2022.
- Blender, O. Blender—A 3D modelling and rendering package. Retrieved. represents the sequence of Constructs1 to. 2018; 4. [Google Scholar]
- Case, D.A.; Belfon, H.M.A.K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A., III; Cruzeiro, V.W.D.; Darden, T.A.; et al. Amber2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Press, W.H.; Flannery, B.P.; Teukolsky, S.A.; Vetterling, W.T.; Kramer, P.B. Numerical recipes: The art of scientific computing. Phys. Today 1987, 40, 120. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Kim, S.; Voter, A.F.; Roitberg, A.E. Bad Seeds Sprout Perilous Dynamics: Stochastic Thermostat Induced Trajectory Synchronization in Biomolecules. J. Chem. Theory Comput. 2009, 5, 1624–1631. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wu, J.C.; Yan, C.; Wang, Y.; Luo, R.; Gonzales, M.B.; Dalby, K.N.; Ren, P. Virtual screening using molecular simulations. Proteins 2011, 79, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Chu, Z.; Yu, F.; Zha, Y. Contriving Multi-Epitope Subunit of Vaccine for COVID-19: Immunoinformatics Approaches. Front. Immunol. 2020, 11, 1784. [Google Scholar] [CrossRef]
- Jyotisha; Singh, S.; Qureshi, I.A. Multi-epitope vaccine against SARS-CoV-2 applying immunoinformatics and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2022, 40, 2917–2933. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.A.; Mimi, N.J.; Chowdhury, M.M.; Eva, T.A.; Nainu, F.; Paul, A.; Shahriar, A.; Tareq, A.M.; Emon, N.U.; et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput. Biol. Med. 2020, 124, 103967. [Google Scholar] [CrossRef]
- Sharma, R.; Rajput, V.S.; Jamal, S.; Grover, A.; Grover, S. An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins. Sci. Rep. 2021, 11, 13836. [Google Scholar] [CrossRef]
- Yu, M.; Zhu, Y.; Li, Y.; Chen, Z.; Li, Z.; Wang, J.; Li, Z.; Zhang, F.; Ding, J. Design of a recombinant multivalent epitope vaccine based on SARS-CoV-2 and its variants in Immunoinformatics approach. Front. Immunol. 2022, 13, 2116. [Google Scholar]
- Guo, L.; Yin, R.; Liu, K.; Lv, X.; Li, Y.; Duan, X.; Chu, Y.; Xi, T.; Xing, Y. Immunological features and efficacy of a multi-epitope vaccine CTB-UE against H. pylori in BALB/c mice model. Appl. Microbiol. Biotechnol. 2014, 98, 3495–3507. [Google Scholar] [CrossRef]
- Cao, Y.; Li, D.; Fu, Y.; Bai, Q.; Chen, Y.; Bai, X.; Jing, Z.; Sun, P.; Bao, H.; Li, P.; et al. Rational design and efficacy of a multi-epitope recombinant protein vaccine against foot-and-mouth disease virus serotype A in pigs. Antivir. Res. 2017, 140, 133–141. [Google Scholar] [CrossRef]
- Lennerz, V.; Gross, S.; Gallerani, E.; Sessa, C.; Mach, N.; Boehm, S.; Hess, D.; von Boehmer, L.; Knuth, A.; Ochsenbein, A.F.; et al. Immunologic response to the survivin-derived multi-epitope vaccine EMD640744 in patients with advanced solid tumors. Cancer Immunol. Immunother. 2014, 63, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Slingluff, C.L.; Lee, S.; Zhao, F.; Chianese-Bullock, K.A.; Olson, W.C.; Butterfield, L.H.; Whiteside, T.L.; Leming, P.D.; Kirkwood, J.M. A Randomized Phase II Trial of Multiepitope Vaccination with Melanoma Peptides for Cytotoxic T Cells and Helper T Cells for Patients with Metastatic Melanoma (E1602) Multipeptide Vaccine for Advanced Melanoma. Clin. Cancer Res. 2013, 19, 4228–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, H.; Baly, A.; Castro, O.; Resik, S.; Laferte, J.; Rolo, F.; Navea, L.; Lobaina, L.; Cruz, O.; Miguez, J.; et al. A phase I clinical trial of a multi-epitope polypeptide TAB9 combined with Montanide ISA 720 adjuvant in non-HIV-1 infected human volunteers. Vaccine 2001, 19, 4328–4336. [Google Scholar] [CrossRef] [PubMed]
- Gadelha, C.; Zhang, W.; Chamberlain, J.W.; Chait, B.T.; Wickstead, B.; Field, M.C. Architecture of a host–parasite interface: Complex targeting mechanisms revealed through proteomics. Mol. Cell. Proteom. 2015, 14, 1911–1926. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Solberg, O.D.; Mack, S.J.; Lancaster, A.K.; Single, R.M.; Tsai, Y.; Sanchez-Mazas, A.; Thomson, G. Balancing selection and heterogeneity across the classical human leukocyte antigen loci: A meta-analytic review of 497 population studies. Hum. Immunol. 2008, 69, 443–464. [Google Scholar] [CrossRef] [Green Version]
- Athanasiou, E.; Agallou, M.; Tastsoglou, S.; Kammona, O.; Hatzigeorgiou, A.; Kiparissides, C.; Karagouni, E. A Poly(Lactic-co-Glycolic) Acid Nanovaccine Based on Chimeric Peptides from Different Leishmania infantum Proteins Induces Dendritic Cells Maturation and Promotes Peptide-Specific IFNgamma-Producing CD8(+) T Cells Essential for the Protection against Experimental Visceral Leishmaniasis. Front. Immunol. 2017, 8, 684. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Majumder, K.; Gutierrez, G.J.; Gupta, D.; Mittal, B. Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Goboza, M.; Klein, A.; Madiehe, A.M.; Meyer, M. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci. Rep. 2021, 11, 19707. [Google Scholar] [CrossRef]
- Rouzbahani, A.K.; Kheirandish, F.; Hosseini, S.Z. Design of a multi-epitope-based peptide vaccine against the S and N proteins of SARS-CoV-2 using immunoinformatics approach. Egypt. J. Med. Hum. Genet. 2022, 23, 1–18. [Google Scholar] [CrossRef]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 4409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolenc, I.; Seemuller, E.; Baumeister, W. Decelerated degradation of short peptides by the 20S proteasome. FEBS Lett. 1998, 434, 357–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Sun, W.; Guo, J.; Zhao, G.; Sun, S.; Yu, H.; Guo, Y.; Li, J.; Jin, X.; Du, L.; et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccin. Immunother. 2015, 11, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Livingston, B.; Crimi, C.; Newman, M.; Higashimoto, Y.; Appella, E.; Sidney, J.; Sette, A. A rational strategy to design multiepitope immunogens based on multiple Th lymphocyte epitopes. J. Immunol. 2002, 168, 5499–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanches, R.C.; Tiwari, S.; Ferreira, L.C.; Oliveira, F.M.; Lopes, M.D.; Passos, M.J.; Maia, E.H.; Taranto, A.G.; Kato, R.; Azevedo, V.A. Immunoinformatics design of multi-epitope peptide-based vaccine against Schistosoma mansoni using transmembrane proteins as a target. Front. Immunol. 2021, 12, 621706. [Google Scholar] [CrossRef] [PubMed]
- Apostolico Jde, S.; Lunardelli, V.A.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, Modus Operandi, and Licensing. J. Immunol. Res. 2016, 2016, 14593–14594. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Shin, S.J.; Lee, M.H.; Lee, M.G.; Kang, T.H.; Park, W.S.; Soh, B.Y.; Park, J.H.; Shin, Y.K.; Kim, H.W.; et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS ONE 2014, 9, e104351. [Google Scholar] [CrossRef]
- Droppa-Almeida, D.; Franceschi, E.; Padilha, F.F. Immune-Informatic Analysis and Design of Peptide Vaccine From Multi-epitopes Against Corynebacterium pseudotuberculosis. Bioinform. Biol. Insights 2018, 12, 1177932218755337. [Google Scholar] [CrossRef] [Green Version]
- Rekik, I.; Chaabene, Z.; Grubb, C.D.; Drira, N.; Cheour, F.; Elleuch, A. In silico characterization and Molecular modeling of double-strand break repair protein MRE11 from Phoenix dactylifera v deglet nour. Theor. Biol. Med. Model. 2015, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carty, M.; Bowie, A.G. Recent insights into the role of Toll-like receptors in viral infection. Clin. Exp. Immunol. 2010, 161, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Xu, Q.; Zhang, Y.; Gao, W.; Zhang, M.; Zhai, W.; Rajkumar, R.S.; Liu, Z. Toll-like receptor-mediated innate immunity against herpesviridae infection: A current perspective on viral infection signaling pathways. Virol. J. 2020, 17, 1–15. [Google Scholar]
- Compton, T.; Kurt-Jones, E.A.; Boehme, K.W.; Belko, J.; Latz, E.; Golenbock, D.T.; Finberg, R.W. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 2003, 77, 4588–4596. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, E.; Staal, J.; Beyaert, R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 2008, 21, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Li, C.; Pan, H.; Wang, M.; Wang, X.; Lin, Q. Preparation and pore-forming mechanism of hydrogen bond and ionic bond double-driven chitosan-based mesoporous carbon. Int. J. Biol. Macromol. 2021, 179, 519–531. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Lippert, T.; Rarey, M. Fast automated placement of polar hydrogen atoms in protein-ligand complexes. J. Cheminform. 2009, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Eagen, R.; Kim, S.H.; Kronstad, J.W.; Breuil, C. An hydroxynaphtalene reductase gene from the wood-staining fungus Ophiostoma floccosum complements the buff phenotype in Magnaporthe grisea. Mycol. Res. 2001, 105, 461–469. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Start | End | Peptide | Length | VexiJen | Percent Identity |
|---|---|---|---|---|---|
| G1 protein | |||||
| 285 | 296 | DDCISRTQLLRT | 12 | 0.42 | 50.00% (XP_006711865.1) |
| 415 | 444 | QHFLKDNLIDLGCPNIPLLGKMAIYICRMS | 30 | 0.42 | 37.50% (XP_047304331.1) |
| 452 | 481 | AFLFWFSFGYVITCILCKVIFYLLIVVGTL | 30 | 0.57 | 43.75% (XP_047276342.1) |
| G2 protein | |||||
| 22 | 40 | CWGVGTGCTCCGLDVKDLF | 19 | 1.37 | 60.00% (NP_001305465.1) |
| 42 | 53 | DYMFVKWKVEYI | 12 | 1.00 | 83.33% (CAA69330.1) |
| Position | Peptide Sequence | Predicted MHC Binding Affinity | Binding Affinity Rescale Score | C-Terminal Cleavage Affinity | Transport Affinity | Prediction Score | VexiJen | BLASTp % Identity (Accession No.) |
|---|---|---|---|---|---|---|---|---|
| GP 1 protein | ||||||||
| 234 | KQNDRCTLV | 0.5317 | 0.7926 | 0.8507 | 0.5440 | 0.9474 | 1.5407 | 66.67% (MBY87633.1) |
| 453 | FLFWFSFGY | 0.4787 | 0.7136 | 0.9715 | 3.1150 | 1.0151 | 1.1001 | 77.78% (NP_114125.1) |
| 473 | YLLIVVGTL | 0.5793 | 0.8636 | 0.9660 | 0.8670 | 1.0518 | 0.8774 | 63.64% (XP_047286234.1) |
| 589 | LLTVSLSPV | 0.6810 | 1.0151 | 0.9670 | 0.3490 | 1.1776 | 1.3397 | 87.50% (AAL65133.2) |
| 623 | FVLGSILFI | 0.8208 | 1.2236 | 0.5436 | 0.6440 | 1.3373 | 0.6538 | 57.14% (NP_001308089.1) |
| G2 protein | ||||||||
| 55 | TEAIVCVEL | 0.4966 | 1.2306 | 0.9690 | 0.9160 | 1.4217 | 1.1768 | 75.00% (KAI2525983.1) |
| Property | Result | Indication |
|---|---|---|
| No. of amino acid | 324 | |
| Sol-Pro | 0.733538 | Soluble |
| Protein Sol | 0.490 | Soluble |
| Molecular weight | 33888.65 Da | Suitable |
| Formula | C1435H2228N374O428S13 | |
| Theoretical pI | 5.01 | Acidic |
| Instability index | 24.69 | (Stable) |
| Aliphatic index | 102.75 | (Thermostable) |
| Total number of negatively charged residues (Asp + Glu) | 37 | |
| Total number of positively charged residues (Arg + Lys) | 30 | |
| Half-Life | 30 h (mammalian reticulocytes, in vitro), >20 h (yeast, in vivo), >10 h (Escherichia coli, in vivo). | |
| Allergenicity | AllerTOP v.2.0 (Non-allergen), AllergenFP v.1.0 (Non-allergen) | Non-allergen |
| Antigenicity | VexiJen v2.0 (0.5163), ANTIGENpro (0.84) | Antigen |
| TM helices | 0 | Suitable |
| Peptide | ∆VDW (kcal/mol) | ∆EEL (kcal/mol) | ∆EGB (kcal/mol) | ∆ESURF (kcal/mol) | ∆G TOTAL (kcal/mol) |
|---|---|---|---|---|---|
| 3L3D | −78.36 ± 0.15 | −347.03 ± 1.02 | 384.95 ± 0.87 | −15.03 ± 0.01 | −55.48 ± 0.17 |
| 5HHP | −88.17 ± 0.11 | −357.42 ± 0.83 | 389.09 ± 0.69 | −13.87 ± 0.09 | −45.37 ± 0.17 |
| KQNDRCTLV | −70.69 ± 0.14 | −335.63 ± 0.90 | 367.02 ± 0.88 | −10.77 ± 0.01 | −50.08 ± 0.16 |
| FLFWFSFGY | −82.87 ± 0.22 | −196.62 ± 0.85 | 234.37 ± 0.80 | −12.37 ± 0.02 | −57.50 ± 0.23 |
| YLLIVVGTL | −84.64 ± 0.14 | −199.13 ± 0.65 | 228.07 ± 0.58 | −13.13 ± 0.02 | −68.84 ± 0.20 |
| LLTVSLSPV | −71.58 ± 0.19 | −132.33 ± 0.61 | 168.95 ± 0.58 | −10.39 ± 0.02 | −45.36 ± 0.19 |
| FVLGSILFI | −71.97 ± 0.18 | −154.77 ± 1.36 | 193.63 ± 1.31 | −10.58 ± 0.02 | −43.70 ± 0.24 |
| TEAIVCVEL | −78.05 ± 0.12 | −127.94 ± 1.19 | 163.53 ± 1.11 | −12.74 ± 0.01 | −55.21 ± 0.18 |
| Complex Name | ∆VDW (kcal/mol) | ∆EEL (kcal/mol) | ∆EGB (kcal/mol) | ∆ESURF (kcal/mol) | ∆G TOTAL (kcal/mol) |
|---|---|---|---|---|---|
| Modelled vaccine–TLR2 | −150.27 ± 0.19 | −466.56 ± 1.53 | 556.00 ± 1.50 | −19.95 ± 0.02 | −80.79 ± 0.17 |
| Modelled vaccine–TLR3 | −128.81 ± 0.24 | −376.39 ± 1.40 | 442.65 ± 1.37 | −16.87 ± 0.03 | −79.43 ± 0.20 |
| Modelled vaccine–TLR4 | −189.68 ± 0.22 | 172.25 ± 1.70 | −40.30 ± 1.60 | −26.51 ± 0.02 | −84.24 ± 0.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, S.Z.; Jabbar, B.; Mirza, M.U.; Waqas, M.; Aziz, S.; Halim, S.A.; Ali, A.; Rafique, S.; Idrees, M.; Khalid, A.; et al. An Immunoinformatics Approach to Design a Potent Multi-Epitope Vaccine against Asia-1 Genotype of Crimean–Congo Haemorrhagic Fever Virus Using the Structural Glycoproteins as a Target. Vaccines 2023, 11, 61. https://doi.org/10.3390/vaccines11010061
Shah SZ, Jabbar B, Mirza MU, Waqas M, Aziz S, Halim SA, Ali A, Rafique S, Idrees M, Khalid A, et al. An Immunoinformatics Approach to Design a Potent Multi-Epitope Vaccine against Asia-1 Genotype of Crimean–Congo Haemorrhagic Fever Virus Using the Structural Glycoproteins as a Target. Vaccines. 2023; 11(1):61. https://doi.org/10.3390/vaccines11010061
Chicago/Turabian StyleShah, Syed Zawar, Basit Jabbar, Muhammad Usman Mirza, Muhammad Waqas, Shahkaar Aziz, Sobia Ahsan Halim, Amjad Ali, Shazia Rafique, Muhammad Idrees, Asaad Khalid, and et al. 2023. "An Immunoinformatics Approach to Design a Potent Multi-Epitope Vaccine against Asia-1 Genotype of Crimean–Congo Haemorrhagic Fever Virus Using the Structural Glycoproteins as a Target" Vaccines 11, no. 1: 61. https://doi.org/10.3390/vaccines11010061