
From Clinical Specimen to Whole Genome Sequencing of A(H3N2) Influenza Viruses: A Fast and Reliable High-Throughput Protocol

 , , and
, , and
Abstract
:1. Introduction
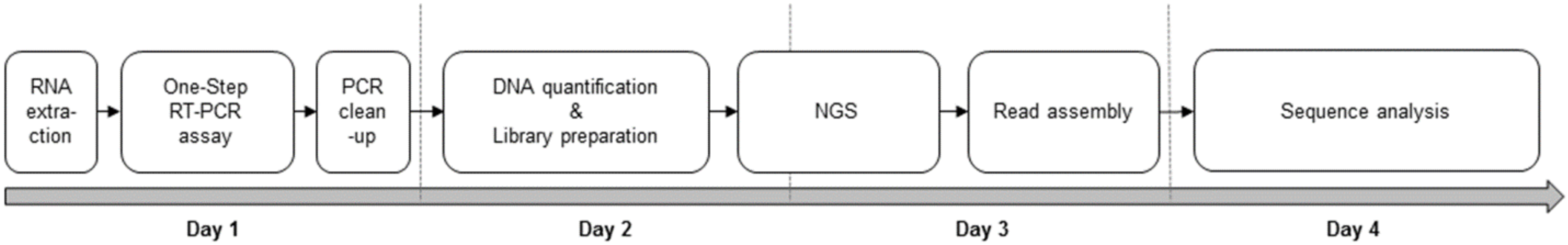
2. Experimental Design
3. Materials and Equipment
3.1. Clinical Specimens
3.2. Nucleic Acid Extraction
- -
- Invisorb® Spin Virus RNA Mini kit (Invitek Molecular GmbH, Berlin, Germany) for 250 viral RNA extractions, consisted of:
- Lysis Buffer RV, ready-to-use
- lyophilized Proteinase K, resuspended by adding 2 mL ddH2O to each tube
- lyophilized Carrier RNA, resuspended by adding 2 mL RNase free water (not provided) to each tube
- Binding Solution, obtained by filling 120 mL 99.7% isopropanol into the empty bottle at the first use.
- concentrate Wash Buffer R1, diluted by adding 80 mL 96–100% ethanol to each bottle at the first use.
- concentrate Wash Buffer R2, diluted by adding 160 mL 96–100% ethanol to each bottle at the first use.
- Elution Buffer, ready-to-use
- RTA spin filter
- 2 mL RTA receiver tubes
- 2 mL safe-lock tubes
- 1.5 mL elution microcentrifuge tubes
- -
- vortex mixer
- -
- thermomixer
- -
- high speed benchtop microcentrifuge
- (1)
- Pipet 600 µL of Lysis Buffer RV into a 2 mL safe-lock tube.
- (2)
- Transfer 20 µL of Proteinase K and 20 µL of Carrier RNA into the 2 mL safe-lock tube containing the lysis buffer.
- (3)
- Mix by pulse-vortexing the clinical sample and then add 200 µL of specimen to the lysis buffer-proteinase K-carrier RNA in the 2 mL safe-lock tube.
- (4)
- Mix by pulse-vortexing the lysis solution for 15 s and briefly centrifuge the tube to remove drops from the inside of the lid.
- (5)
- Incubate the tube into a thermomixer at 65 °C for 10 min.
- (6)
- Briefly centrifuge the tube to remove drops from the inside of the lid.
- (7)
- Add 400 μL Binding Solution to the lysate sample, and mix by pulse-vortexing for 20 s. After mixing, briefly centrifuge the tube to remove drops from inside the lid.
- (8)
- Carefully apply 650 μL of the solution from step 7 to the RTA spin filter (in a 2 mL RTA receiver tube) without wetting the rim. Close the cap, and incubate at room temperature for 1 min. Then, centrifuge at 8000 rpm for 1 min. Discard the filtrate and place the RTA spin filter back into its 2 mL RTA receiver tube.
- (9)
- Repeat step 8 to load onto the spin column the left volume (approximately 600 μL) of the lysate. Place the RTA spin filter into a clean 2 mL RTA receiver tube.
- (10)
- Add 600 μL Wash Buffer R1 into the RTA spin filter, close the cap, and centrifuge at 8000 rpm for 1 min. Place the RTA spin filter in a clean 2 mL RTA receiver tube, and discard the tube containing the filtrate.
- (11)
- Add 600 μL Wash Buffer R2 into the RTA spin filter, close the cap, and centrifuge at 8000 rpm for 1 min. Place the RTA spin filter in a clean 2 mL RTA receiver tube, and discard the tube containing the filtrate.
- (12)
- Repeat the washing step 11.
- (13)
- To eliminate possible Wash Buffer R2 carryover, centrifuge the RTA spin filter at full speed (around 12,000 rpm) for 4 min.
- (14)
- Place the RTA spin filter in a clean 1.5 mL elution microcentrifuge tube. Discard the old RTA receiver tube containing the filtrate. Carefully, add 100 μL Elution Buffer (previously heated at 80 °C) directly onto the RTA spin filter and incubate at room temperature for 3 min.
- (15)
- Centrifuge the microcentrifuge tube with the RTA spin filter at 8000 rpm for 1 min. Discard the RTA spin filter and store the microcentrifuge tube with the eluate for further bio-molecular analyses.
3.3. Viral Genome Amplification
- -
- Super Script™ III One-Step RT-PCR System with Platinum™ Taq DNA Polymerase (ThermoFisher Scientific, Waltham, MA, USA), consisted of:
- 2X Reaction Mix (with MgSO4 included at a final concentration of 1.6 mM)
- SuperScript™ III RT/Platinum™ Taq Mix
- -
- MBTuni-12 forward primer and MBTuni-13 reverse primer (working concentration 30 µM) [32]
- -
- DNase/RNase-free water (Sigma-Aldrich, St. Louis, MO, USA)
- -
- vortex mixer
- -
- benchtop microcentrifuge
- -
- thermal-cycler
- (1)
- Gently mix the 2X Reaction Mix, the SuperScript™ III RT/Platinum™ Taq Mix and the primer tubes.
- (2)
- In a sterile, nuclease-free microcentrifuge tube, combine the following components on ice: 25 µL of 2X Reaction Mix Buffer, 2 µL of forward (MBTuni-12) and reverse (MBTuni-13) primer, 2 µL of SuperScript™ III RT/Platinum™ Taq Mix and 14 µL of DNase/RNase-free water to reach a final volume of 45 µL.
- (3)
- Add 15 µL of extracted RNA to the reaction mixture.
- (4)
- Mix by vortexing and centrifuge briefly.
- (5)
- Place the reaction tube into a thermal-cycler programmed as follow: 42 °C for 1 h for the RT phase followed by 94 °C for 2 min for enzyme inactivation; 5 cycles at 94 °C for 30 s, 45 °C for 30 s and 68 °C for 3 min, 35 cycles at 94 °C for 30 s, 57 °C for 30 s, 68 °C for 3 min, followed by 1 cycle at 68 °C for 7 min.
3.4. Amplicon Purification
- -
- NucleoSpin Gel® and PCR Clean-up kit (Macherey-Nagel, Düren, Germany) for 250 preparations, consisted of:
- Buffer NTI, ready-to-use
- concentrated Buffer NT3, diluted by adding 200 mL 96–100% ethanol to each bottle at the first use.
- Buffer NE, ready-to-use
- NucleoSpin Gel® and PCR Clean-up Column
- 2 mL collection tube
- -
- dry block heating
- -
- high speed benchtop microcentrifuge
- (1)
- Mix 50 µL sample with 100 µL Buffer NTI (proportion 1:2) by pipetting up and down.
- (2)
- Load the solution (150 µL) on the NucleoSpin Gel® and PCR Clean-up Colum. Centrifuge the column for 30 s at 11,000× g. Discard flow-through and place the column back into the collection tube.
- (3)
- Add 650 µL Buffer NT3 to the NucleoSpin Gel® and PCR Clean-up Column. Centrifuge the column for 30 s at 11,000× g. Discard flow-through and place the column back into the collection tube.
- (4)
- Repeat the washing step 3.
- (5)
- Centrifuge the NucleoSpin Gel® and PCR Clean-up Column for 5 min at 12,000 rpm to remove Buffer NT3 completely.
- (6)
- Incubate the NucleoSpin Gel® and PCR Clean-up Column, with the lid opened, for 10 min at room temperature to remove any residual ethanol from Buffer NT3.
- (7)
- Place the NucleoSpin Gel® and PCR Clean-up Column into a new 1.5 mL microcentrifuge tube (not provided) and add 20 µL Buffer NE (previously heated at 70 °C). Incubate at room temperature for 1 min. Centrifuge for 5 min at 11,000× g.
- (8)
- Repeat twice the elution step 7 to have a final elution volume of 60 µL.
- (9)
- Discard the NucleoSpin Gel® and PCR Clean-up Column and store the microcentrifuge tube with the eluate for further bio-molecular analyses.
3.5. Library Preparation for Whole Genome Sequencing Using NGS Technology
- -
- Nextera-XT DNA sample preparation kit (Illumina Inc., San Diego, CA, USA) for 96 samples, consisted of:
- Amplicon Tagment Mix (ATM)
- Tagment DNA Buffer (TD)
- Nextera PCR Master Mix (NPM)
- Resuspension Buffer (RSB)
- Library Normalization Additives (LNA1)
- Library Normalization Wash (LNW1)
- Hybridization Buffer (HT1)
- Neutralize Tagment Buffer (NT)
- Library Normalization Beads 1 (LNB1)
- Library Normalization Storage Buffer 1 (LNS1)
- -
- Nextera-XT DNA sample preparation index kit (Illumina Inc., San Diego, CA, USA) for 96 samples, consisted of tubes with index primers 1 and index primers 2.
- -
- Agencourt AMPure XP 60 mL kit (Beckman Coulter Life Sciences, Brea, CA, USA), consisted of AMPure XP Beads.
- -
- 80% ethanol
- -
- 0.1 N of NaOH
- -
- vortex mixer
- -
- shaker
- -
- 96-plate magnetic stand
- -
- 96-plate benchtop centrifuge
- -
- benchtop microcentrifuge
- -
- thermal-cycler
- -
- dry block heater
- (1)
- Mix gently and centrifuge briefly the reagents ATM and TD.
- (2)
- Transfer 10 µL TD to each well of a 96-plate (plate A) according to the number of samples to be processed.
- (3)
- Add 5 µL DNA and 5 µL ATM in each well and mix by pipetting up and down.
- (4)
- Seal and centrifuge the plate A for 1 min at 1200 rpm.
- (5)
- Place the plate A into a thermal-cycler programmed as follows: 55 °C for 5 min and hold at 10 °C.
- (6)
- Remove the plate A from the thermal-cycler and add 5 µL NT in each well; mix the solution by pipetting up and down.
- (7)
- Seal and centrifuge the plate A for 1 min at 1200 rpm.
- (8)
- Incubate the plate A for 5 min at room temperature.
- (1)
- Mix gently and centrifuge briefly the reagent NPM and the index primers.
- (2)
- Transfer 15 µL NPM in each well of the plate A according to the number of samples to be processed.
- (3)
- Add 5 µL of a specific index primer 1 and 5 µL of a specific index primer 2 in each well; mix by pipetting up and down.
- (4)
- Seal and centrifuge the plate A for 1 min at 1200 rpm.
- (5)
- Place the plate A into a thermal-cycler programmed as follows: 1 cycle at 72 °C for 3 min, followed by 95 °C for 30 s; 12 cycles at 95 °C for 10 s, 55 °C for 30 s and 72 °C for 3 s, followed by 1 cycle at 72 °C for 5 min and hold at 10 °C.
- (1)
- Remove the plate A from the thermal-cycler and transfer 50 µL of each well of the plate A to a well of a new plate (plate B).
- (2)
- Add 30 µL of AMPure XP Beads in each well.
- (3)
- Seal and shake the plate B for 2 min at 1800 rpm; then, incubate for 5 min at room temperature.
- (4)
- Place the plate B to a magnetic stand and wait until the liquid is clear (approximately 2 min).
- (5)
- Keep the plate B on magnetic stand and remove the supernatant.
- (6)
- Add 200 µL of 80% ethanol in each well and incubate for 30 s at room temperature.
- (7)
- Keep the plate B on magnetic stand and remove the ethanol.
- (8)
- Repeat step 6 and 7.
- (9)
- Keep the plate B on magnetic stand and wait 15 min to eliminate residual ethanol.
- (10)
- Remove the plate B from the magnetic stand and add 52.5 µL RSB in each well.
- (11)
- Seal and shake the plate B for 2 min at 1800 rpm. Then, incubate for 2 min at room temperature.
- (12)
- Place the plate B to the magnetic stand and wait until the liquid is clear (approximately 2 min).
- (13)
- Transfer 50 µL the supernatant of each well of the plate B to a well of a new plate (plate C).
- (1)
- Mix by vortexing the reagents LNA1 and LNB1.
- (2)
- In a 50 mL tube, transfer 45.8 µL LNA1 per number of samples and add 8.3 µL LNB1 per number of samples. Invert the tube to mix the solution.
- (3)
- Transfer 20 µL of the liquid of each well of plate C to a well of a new plate (plate D).
- (4)
- Add 45 µL LNA1/LNB1 solution in each well of the plate D according to the number of samples to be processed.
- (5)
- Seal and shake the plate D for 30 min at 1800 rpm.
- (6)
- Place the plate D to the magnetic stand and wait until the liquid is clear (approximately 2 min).
- (7)
- Keep the plate D on magnetic stand and remove the supernatant.
- (8)
- Remove the plate D from the magnetic stand and add 45 µL LNW1 in each well.
- (9)
- Seal and shake the plate D for 5 min at 1800 rpm.
- (10)
- Place the plate D to the magnetic stand and wait until the liquid is clear (approximately 2 min).
- (11)
- Keep the plate D on magnetic stand and remove the supernatant.
- (12)
- Repeat steps 8–11.
- (13)
- Remove the plate D from the magnetic stand and add 30 µL NaOH 0.1 N in each well.
- (14)
- Seal and shake the plate D for 5 min at 1800 rpm.
- (15)
- Place the plate D to the magnetic stand and wait until the liquid is clear (approximately 2 min).
- (16)
- Keep the plate D on the magnetic stand and transfer 30 µL of the supernatant of each well to a well of a new plate (plate E).
- (17)
- Add 30 µL LNS1 into each well of the plate E according to the number of samples to be processed.
- (18)
- Seal the plate E and centrifuge for 1 min at 1000× g.
- (1)
- Transfer 5 µL of each library into a 1.5 mL microcentrifuge tube (tube A). Then, transfer 24 µL of the pooled library (tube A) into a new 1.5 mL microcentrifuge tube (tube B).
- (2)
- Add 576 µL HT1 into the tube B and mix by pipetting up and down.
- (3)
- Vortex the tube B and incubate for 2 min at 96 °C.
- (4)
- Invert twice the tube B and cool on ice for 5 min.
- (5)
- Load the library of the tube B into the MiSeq Reagent Cartridge.
- (6)
- Place the MiSeq Reagent Cartridge into the Load Sample reservoir and start the run by using the Illumina MiSeq Instrument (Illumina Inc., San Diego, CA, USA).
4. Detailed Procedure
5. Results
6. Discussion
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Quiñones-Mateu, M.E.; Avila, S.; Reyes-Teran, G.; Martinez, M.A. Deep sequencing: Becoming a critical tool in clinical virology. J. Clin. Virol. 2014, 61, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vemula, S.V.; Zhao, J.; Liu, J.; Wang, X.; Biswas, S.; Hewlett, I. Current Approaches for Diagnosis of Influenza Virus Infections in Humans. Viruses 2016, 8, 96. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, Y.; Luo, J.; Pang, K.; Xu, X.; Wu, J.; Li, X.; Jin, S. Next-Generation Sequencing Reveals the Progression of COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 632490. [Google Scholar] [CrossRef] [PubMed]
- John, G.; Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Williams, C.; Chaubey, A.; Rojiani, A.M.; Kolhe, R. Next-Generation Sequencing (NGS) in COVID-19: A Tool for SARS-CoV-2 Diagnosis, Monitoring New Strains and Phylodynamic Modeling in Molecular Epidemiology. Curr. Issues Mol. Biol. 2021, 43, 845–867. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef]
- Leung, P.; Eltahla, A.A.; Lloyd, A.R.; Bull, R.A.; Luciani, F. Understanding the complex evolution of rapidly mutating viruses with deep sequencing: Beyond the analysis of viral diversity. Virus Res. 2017, 239, 43–54. [Google Scholar] [CrossRef]
- Beerenwinkel, N.; Günthard, H.F.; Roth, V.; Metzner, K.J. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front. Microbiol. 2012, 3, 329. [Google Scholar] [CrossRef]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, K.M.; Sharma, P.; Volkening, J.D.; Goraichuk, I.V.; Wajid, A.; Rehmani, S.F.; Basharat, A.; Shittu, I.; Joannis, T.M.; Miller, P.J.; et al. A robust and cost-effective approach to sequence and analyze complete genomes of small RNA viruses. Virol. J. 2017, 14, 72. [Google Scholar] [CrossRef]
- Kustin, T.; Ling, G.; Sharabi, S.; Ram, D.; Friedman, N.; Zuckerman, N.; Bucris, E.D.; Glatman-Freedman, A.; Stern, A.; Mandelboim, M. A method to identify respiratory virus infections in clinical samples using next-generation sequencing. Sci. Rep. 2019, 9, 2606. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012; pp. 749–761. [Google Scholar]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and Swine: Proposal for a new genus in the Orthomyxoviridae family. MBio 2014, 5, e00031-14. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Wallis, T.R.; Harmon, M.W.; Rota, J.S.; Kendal, A.P.; Nerome, K. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 1990, 175, 59–68. [Google Scholar] [CrossRef]
- Medina, R.A.; García-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Influenza (Flu) Viruses. Available online: https://www.cdc.gov/flu/about/viruses/index.htm (accessed on 10 August 2022).
- World Health Organization (WHO). Influenza (Seasonal). Available online: http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed on 10 August 2022).
- Webster, R.G.; Govorkova, E.A. Continuing challenges in influenza. Ann. N. Y. Acad. Sci. 2014, 1323, 115–139. [Google Scholar] [CrossRef]
- Allen, J.D.; Ross, T.M. H3N2 influenza viruses in humans: Viral mechanisms, evolution, and evaluation. Hum. Vaccin Immunother. 2018, 14, 1840–1847. [Google Scholar] [CrossRef]
- Taubenberger, J.K.; Morens, D.M. Influenza viruses: Breaking all the rules. MBio 2013, 4, e00365-13. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Influenza Surveillance and Response System (GISRS). Available online: https://www.who.int/initiatives/global-influenza-surveillance-and-response-system (accessed on 10 August 2022).
- World Health Organization (WHO). Global Influenza Surveillance Network. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf?sequence=1&isAllowed=y (accessed on 10 August 2022).
- Centers for Disease Control and Prevention (CDC). Influenza Virus Genome Sequencing and Genetic Characterization. Available online: https://www.cdc.gov/flu/about/professionals/genetic-characterization.htm#:~:text=Methods%20of%20Flu%20Genome%20Sequencing,-One%20influenza%20sample&text=Traditionally%2C%20scientists%20have%20used%20a,found%20in%20a%20virus%20sample (accessed on 10 August 2022).
- Istituto Superiore di Sanità (ISS). InfluNet: Rete Italiana Sorveglianza Influenza. Available online: https://w3.iss.it/site/RMI/influnet/Default.aspx (accessed on 10 August 2022).
- Piralla, A.; Daleno, C.; Pariani, E.; Conaldi, P.; Esposito, S.; Zanetti, A.; Baldanti, F. Virtual quantification of influenza A virus load by real-time RT-PCR. J. Clin. Virol. 2013, 56, 65–68. [Google Scholar] [CrossRef]
- Medical Wire. Sigma Virocult® MW951S. 2022. Available online: https://www.mwe.co.uk/microbiology-lab-supplies/culture-swabs-liquid/sigma-virocult-mini-mw951s/ (accessed on 10 August 2022).
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Geneious 11.1. Available online: https://assets.geneious.com/manual/2021.1/index.html (accessed on 10 August 2022).
- Rutvisuttinunt, W.; Chinnawirotpisan, P.; Simasathien, S.; Shrestha, S.K.; Yoon, I.K.; Klungthong, C.; Fernandez, S. Simultaneous and complete genome sequencing of influenza A and B with high coverage by Illumina MiSeq Platform. J. Virol. Methods 2013, 193, 394–404. [Google Scholar] [CrossRef]
- The Francis Crick Institute. Report Prepared for the WHO Annual Consultation on the Composition of Influenza Vaccines for the Southern Hemisphere 2019. 24th–26th September 2018. Available online: https://www.crick.ac.uk/sites/default/files/2018-10/September%202018%20interim%20report_opt.pdf (accessed on 10 August 2022).
- Lee, H.K.; Tang, J.W.; Kong, D.H.; Loh, T.P.; Chiang, D.K.; Lam, T.T.; Koay, E.S. Comparison of mutation patterns in full-genome A/H3N2 influenza sequences obtained directly from clinical samples and the same samples after a single MDCK passage. PLoS ONE 2013, 8, e79252. [Google Scholar] [CrossRef]
- Chen, H.; Deng, Q.; Ng, S.H.; Lee, R.T.; Maurer-Stroh, S.; Zhai, W. Dynamic Convergent Evolution Drives the Passage Adaptation across 48 Years’ History of H3N2 Influenza Evolution. Mol. Biol. Evol. 2016, 33, 3133–3143. [Google Scholar] [CrossRef]
- Galli, C.; Orsi, A.; Pariani, E.; Lai, P.L.; Guarona, G.; Pellegrinelli, L.; Ebranati, E.; Icardi, G.; Panatto, D. In-depth phylogenetic analysis of the hemagglutinin gene of influenza A(H3N2) viruses circulating during the 2016–2017 season revealed egg-adaptive mutations of vaccine strains. Expert Rev. Vaccines 2020, 19, 115–122. [Google Scholar] [CrossRef]
- Roy, S.; Hartley, J.; Dunn, H.; Williams, R.; Williams, C.A.; Breuer, J. Whole-genome Sequencing Provides Data for Stratifying Infection Prevention and Control Management of Nosocomial Influenza A. Clin. Infect. Dis. 2019, 69, 1649–1656. [Google Scholar] [CrossRef]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Methods 2014, 195, 194–204. [Google Scholar] [CrossRef]
- Kohl, C.; Brinkmann, A.; Dabrowski, P.W.; Radonić, A.; Nitsche, A.; Kurth, A. Protocol for metagenomic virus detection in clinical specimens. Emerg. Infect. Dis. 2015, 21, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Barbezange, C.; Jones, L.; Blanc, H.; Isakov, O.; Celniker, G.; Enouf, V.; Shomron, N.; Vignuzzi, M.; van der Werf, S. Seasonal Genetic Drift of Human Influenza A Virus Quasispecies Revealed by Deep Sequencing. Front. Microbiol. 2018, 9, 2596. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Tamura, K.; Tanigaki, T.; Takizawa, M.; Nakayama, E.; Taniguchi, T.; Okamoto, M.; Nishiyama, Y.; Tarumoto, N.; Mitsutake, K.; et al. Whole Genome Sequencing of Influenza A and B Viruses with the MinION Sequencer in the Clinical Setting: A Pilot Study. Front. Microbiol. 2018, 9, 2748. [Google Scholar] [CrossRef]
- Alnaji, F.G.; Holmes, J.R.; Rendon, G.; Vera, J.C.; Fields, C.J.; Martin, B.E.; Brooke, C.B. Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses. J. Virol. 2019, 93, e00354-19. [Google Scholar] [CrossRef]
- Fischer, N.; Indenbirken, D.; Meyer, T.; Lütgehetmann, M.; Lellek, H.; Spohn, M.; Aepfelbacher, M.; Alawi, M.; Grundhoff, A. Evaluation of Unbiased Next-Generation Sequencing of RNA (RNA-seq) as a Diagnostic Method in Influenza Virus-Positive Respiratory Samples. J. Clin. Microbiol. 2015, 53, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Pichon, M.; Valette, M.; Burfin, G.; Richard, M.; Lina, B.; Josset, L. Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season. Viruses 2019, 11, 108. [Google Scholar] [CrossRef]
- Seong, M.W.; Cho, S.I.; Park, H.; Seo, S.H.; Lee, S.J.; Kim, E.C.; Park, S.S. Genotyping Influenza Virus by Next-Generation Deep Sequencing in Clinical Specimens. Ann. Lab. Med. 2016, 36, 255–258. [Google Scholar] [CrossRef]
- Viljoen, G.J.; Nel, L.H.; Ceowther, J.R. Molecular Diagnostic PCR Handbook; Springer: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three next generation sequencing platforms: Comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom. 2012, 13, 341. [Google Scholar] [CrossRef]
- McGinnis, J.; Laplante, J.; Shudt, M.; George, K.S. Next generation sequencing for whole genome analysis and surveillance of influenza A viruses. J. Clin. Virol. 2016, 79, 44–50. [Google Scholar] [CrossRef]
- MacFadden, D.R.; McGeer, A.; Athey, T.; Perusini, S.; Olsha, R.; Li, A.; Eshaghi, A.; Gubbay, J.B.; Hanage, W.P. Use of Genome Sequencing to Define Institutional Influenza Outbreaks, Toronto, Ontario, Canada, 2014–2015. Emerg. Infect. Dis. 2018, 24, 492–497. [Google Scholar] [CrossRef]
- Ghedin, E.; Sengamalay, N.A.; Shumway, M.; Zaborsky, J.; Feldblyum, T.; Subbu, V.; Spiro, D.J.; Sitz, J.; Koo, H.; Bolotov, P.; et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 2005, 437, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.; Barr, I.; Cox, N.; Donis, R.O.; Siddhivinayak, H.; Jernigan, D.; Katz, J.; McCauley, J.; Motta, F.; Odagiri, T.; et al. Improving the selection and development of influenza vaccine viruses—Report of a WHO informal consultation on improving influenza vaccine virus selection, Hong Kong SAR, China, 18–20 November 2015. Vaccine 2017, 35, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Dinis, J.M.; Florek, N.W.; Fatola, O.O.; Moncla, L.H.; Mutschler, J.P.; Charlier, O.K.; Meece, J.K.; Belongia, E.A.; Friedrich, T.C. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J. Virol. 2016, 90, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Meinel, D.M.; Heinzinger, S.; Eberle, U.; Ackermann, N.; Schönberger, K.; Sing, A. Whole genome sequencing identifies influenza A H3N2 transmission and offers superior resolution to classical typing methods. Infection 2018, 46, 69–76. [Google Scholar] [CrossRef]
- Van Poelvoorde, L.A.E.; Saelens, X.; Thomas, I.; Roosens, N.H. Next-Generation Sequencing: An Eye-Opener for the Surveillance of Antiviral Resistance in Influenza. Trends Biotechnol. 2020, 38, 360–367. [Google Scholar] [CrossRef]


{kind=link}
| Influenza A Virus Genome | ||||||||
|---|---|---|---|---|---|---|---|---|
| Segment 1 | Segment 2 | Segment 3 | Segment 4 | Segment 5 | Segment 6 | Segment 7 | Segment 8 | |
| Polymerase Basic 2 (PB2) | Polymerase Basic 1 (PB1) | Polymerase Acid (PA) | Hemagglutinin (HA) | Nucleoprotein (NP) | Neuraminidase (NA) | Matrix 1 (M1) | Non Structural (NS) | |
| 2341 bps | 2341 bps | 2233 bps | 1778 bps | 1565 bps | 1413 bps | 1027 bps | 890 bps | |
| Mean N. of mappedbases | 4,754,119.20 | 2,596,691.75 | 5,418,931.93 | 6,023,123.21 | 6,450,089.98 | 6,612,054.32 | 10,126,540.35 | 9,175,172.23 |
| Mean N. of mapped reads | 33,164.68 | 37,966.80 | 38,286.33 | 43,176.47 | 44,939.80 | 44,097.74 | 73,918.02 | 64,266.64 |
| Mean coverage depth | 2154.15 | 1101.45 | 2694.11 | 3745.52 | 3521.58 | 3933.64 | 10,475.41 | 8348.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galli, C.; Ebranati, E.; Pellegrinelli, L.; Airoldi, M.; Veo, C.; Della Ventura, C.; Seiti, A.; Binda, S.; Galli, M.; Zehender, G.; et al. From Clinical Specimen to Whole Genome Sequencing of A(H3N2) Influenza Viruses: A Fast and Reliable High-Throughput Protocol. Vaccines 2022, 10, 1359. https://doi.org/10.3390/vaccines10081359
Galli C, Ebranati E, Pellegrinelli L, Airoldi M, Veo C, Della Ventura C, Seiti A, Binda S, Galli M, Zehender G, et al. From Clinical Specimen to Whole Genome Sequencing of A(H3N2) Influenza Viruses: A Fast and Reliable High-Throughput Protocol. Vaccines. 2022; 10(8):1359. https://doi.org/10.3390/vaccines10081359
Chicago/Turabian StyleGalli, Cristina, Erika Ebranati, Laura Pellegrinelli, Martina Airoldi, Carla Veo, Carla Della Ventura, Arlinda Seiti, Sandro Binda, Massimo Galli, Gianguglielmo Zehender, and et al. 2022. "From Clinical Specimen to Whole Genome Sequencing of A(H3N2) Influenza Viruses: A Fast and Reliable High-Throughput Protocol" Vaccines 10, no. 8: 1359. https://doi.org/10.3390/vaccines10081359