
Molecular Amplification and Cell Culturing Efficiency for Enteroviruses’ Detection in Cerebrospinal Fluids of Algerian Patients Suffering from Meningitis

 ,
,  ,
,  ,
,  and
and
Abstract
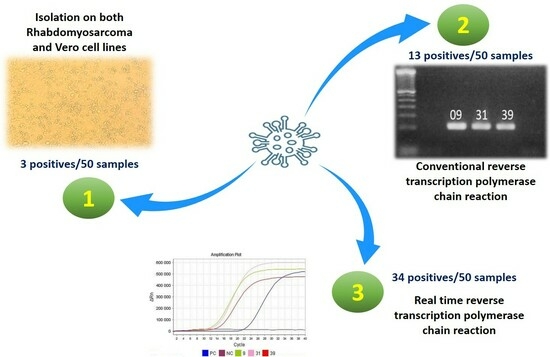
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Conventional and Real-Time RT-PCR
2.2.1. RNA Extraction
2.2.2. Conventional RT-PCR
2.2.3. Real-Time RT-PCR
2.2.4. PCR Product Analysis
2.3. Viral Serotype Identification
2.4. Sensitivity Evaluation
3. Results
3.1. Cell Culture
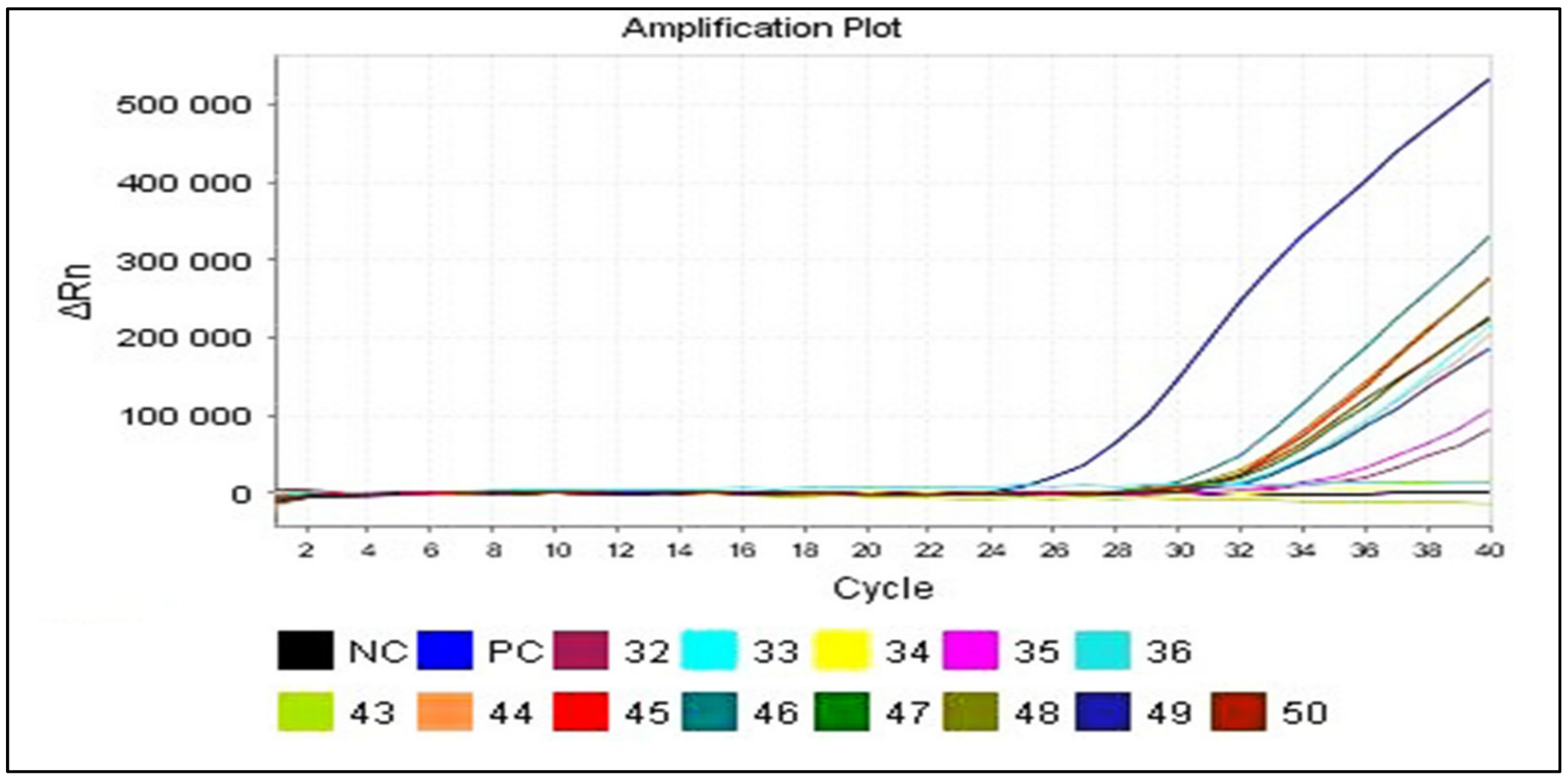
3.2. Gene Amplification
3.2.1. From Clinical Samples
3.2.2. From Positive Culture Supernatants



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | RD | Vero | PCR | Real T PCR | Samples | RD | Vero | PCR | Real T PCR |
|---|---|---|---|---|---|---|---|---|---|
| 01 | − | − | − | + | 26 | − | − | − | + |
| 02 | − | − | − | − | 27 | − | − | − | − |
| 03 | − | − | − | − | 28 | − | − | − | ++ |
| 04 | − | − | − | + | 29 | − | − | − | − |
| 05 | − | − | − | + | 30 | − | − | − | − |
| 06 | − | − | − | + | 31 | + | − | +* | +* |
| 07 | − | − | + | + | 32 | − | − | − | ++ |
| 08 | − | − | + | + | 33 | − | − | − | ++ |
| 09 | + | − | + | + | 34 | − | − | − | +/− |
| 10 | − | − | − | − | 35 | − | − | − | ++ |
| 11 | − | − | + | + | 36 | − | − | − | − |
| 12 | − | − | − | + | 37 | − | − | +/− | + |
| 13 | − | − | − | − | 38 | − | − | + | + |
| 14 | − | − | − | − | 39 | + | − | +* | +* |
| 15 | − | − | − | − | 40 | − | − | +/− | + |
| 16 | − | − | − | − | 41 | − | − | +/− | + |
| 17 | − | − | − | − | 42 | − | − | +/− | + |
| 18 | − | − | +/− | + | 43 | − | − | − | − |
| 19 | − | − | − | + | 44 | − | − | − | ++ |
| 20 | − | − | − | ++ | 45 | − | − | − | ++ |
| 21 | − | − | + | + | 46 | − | − | − | ++ |
| 22 | − | − | − | ++ | 47 | − | − | − | ++ |
| 23 | − | − | − | ++ | 48 | − | − | − | ++ |
| 24 | − | − | + | + | 49 | − | − | − | ++ |
| 25 | − | − | − | − | 50 | − | − | +/- | ++ |
3.3. Molecular Identification of the Cell Culture-Isolated Strains
3.4. Sensitivity Evaluation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Afifi, S.S.; Zaki, S.A.; Mohamed, A.F.; El-Hosseiny, H. Isolation and Identification of Non-Polio Enteroviruses from Children in Different Egyptian Governorates. AJBAS 2009, 3, 3230–3238. [Google Scholar]
- Andreoletti, L.; Renois, F.; Jacques, J.; Leveque, N. Entérovirus non poliomyélitiques et pathologies respiratoires. Med. Sci. 2009, 25, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Moreni, G.; Wolthers, K.C.; Pajkrt, D. World-Wide Prevalence and Genotype Distribution of Enteroviruses. Viruses 2021, 13, 434. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, X.-Y.; Jin, W.-P.; Wang, C.; Qian, S.-S.; Wang, M.-J.; Wang, W.-H.; Meng, S.-L.; Guo, J.; Wang, Z.-J.; et al. Identification of a Conserved, Linear Epitope on VP3 of Enterovirus A Species Recognized by a Broad-Spectrum Monoclonal Antibody. Viruses 2023, 15, 1028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, F.; Chang, X.; Hu, J.; Zhang, Z.; Cui, X.; Zheng, X.; Wang, X. A Neonatal Murine Model for Caprine Enterovirus Infection and the Viral Tissue Tropism. Viruses 2023, 15, 475. [Google Scholar] [CrossRef] [PubMed]
- Sandoni, M.; Ciardo, L.; Tamburini, C.; Boncompagni, A.; Rossi, C.; Guidotti, I.; Garetti, E.; Lugli, L.; Iughetti, L.; Berardi, A. Enteroviral Infections in the First Three Months of Life. Pathogens 2022, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Wang, K.; Zhao, K.; Hua, S.C.; Du, J. The Structure, Function, and Mechanisms of Action of Enterovirus Non-structural Protein 2C. Front. Microbiol. 2020, 11, 615965. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hu, X.-Y.; Yu, X.-F. Current Understanding of Human Enterovirus D68. Viruses 2019, 11, 490. [Google Scholar] [CrossRef]
- Piralla, A.; Daleno, C.; Scala, A.; Greenberg, D.; Usonis, V.; Principi, N.; Baldanti, F.; Esposito, S.; CAP-PRI Study Group. Genome characterisation of enteroviruses 117 and 118: A new group within human enterovirus species C. PLoS ONE 2013, 8, e60641. [Google Scholar] [CrossRef]
- Renois, F.; Bouin, A.; Wehbe, M.; Leveque, N.; Andreoletti, L. Infections persistantes à entérovirus et pathologies humaines. Virologie 2014, 18, 306–311. [Google Scholar]
- Simmonds, P.; Gorbalenya, A.E.; Harvala, H.; Hovi, T.; Knowles, N.J.; Lindberg, A.M.; Oberste, M.S.; Palmenberg, A.C.; Reuter, G.; Skern, T.; et al. Recommendations for the nomenclature of enteroviruses and rhinoviruses. Arch. Virol. 2020, 165, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Mammette, A. Virologie Médicale; Presses Universitaires Lyon: Lyon, France, 2002; pp. 265–280. [Google Scholar]
- Wells, A.I.; Coyne, C.B. Enteroviruses: A Gut-Wrenching Game of Entry, Detection, and Evasion. Viruses 2019, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Sittikul, P.; Batty, E.M.; Yodsawat, P.; Nuanpirom, J.; Kosoltanapiwat, N.; Sangket, U.; Chatchen, S.; Day, N.P.J.; Thaipadungpanit, J. Diversity of Human Enterovirus Co-Circulations in Five Kindergartens in Bangkok between July 2019 and January 2020. Viruses 2023, 15, 1397. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Sadeuh-Mba, S.A.; Joffret, M.L.; Razafindratsimandresy, R.; Polston, P.; Volle, R.; Rakoto-Andrianarivelo, M.; Blondel, B.; Njouom, R.; Delpeyroux, F. Whole Genome Sequencing of Enterovirus species C Isolates by High-Throughput Sequencing: Development of Generic Primers. Front. Microbiol. 2016, 26, 1294. [Google Scholar] [CrossRef] [PubMed]
- Jacques, J.; Moret, H.; Minette, D.; Lévêque, N.; Jovenin, N.; Deslée, G.; Lebargy, F.; Motte, J.; Andréoletti, L. Epidemiological, molecular, and clinical features of enterovirus respiratory infections in French children between 1999 and 2005. J. Clin. Microbiol. 2008, 46, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Meyers, L.; Bard, J.D.; Galvin, B.; Nawrocki, J.; Niesters, H.G.M.; Stellrecht, K.A.; George, K.S.T.; Daly, J.D.; Blaschke, A.J.; Robinson, C.; et al. Enterovirus D68 outbreak detection through a syndromic disease epidemiology network. J. Clin. Virol. 2020, 124, 104262. [Google Scholar] [CrossRef] [PubMed]
- Chansaenroj, J.; Tuanthap, S.; Thanusuwannasak, T.; Duang-in, A.; Klinfueng, S.; Thaneskongtong, N.; Vutithanachot, V.; Vongpunsawad, S.; Poovorawan, Y. Human enteroviruses associated with and without diarrhea in Thailand between 2010 and 2016. PLoS ONE 2017, 12, e0182078. [Google Scholar] [CrossRef]
- Machado, R.S.; de Sousa, I.P.; Monteiro, J.C.; Ferreira, J.L.; Dos Santos Alves, J.C.; Tavares, F.N. Detection and identification of enteroviruses circulating in children with acute gastroenteritis in Pará State, Northern Brazil (2010–2011). Virol. J. 2020, 17, 156. [Google Scholar] [CrossRef]
- Chen, B.S.; Lee, H.C.; Lee, K.M.; Gong, Y.N.; Shih, S.R. Enterovirus and Encephalitis. Front. Microbiol. 2020, 11, 261. [Google Scholar] [CrossRef]
- Piralla, A.; Pellegrinelli, L.; Giardina, F.; Galli, C.; Binda, S.; Pariani, E.; Baldanti, F. Contribution of Enteroviruses to Acute Central Nervous System or Systemic Infections in Northern Italy (2015–2017): Is It Time to Establish a National Laboratory-Based Surveillance System? BioMed Res. Int. 2020, 2020, 9393264. [Google Scholar] [CrossRef]
- O’Neal, A.J.; Hanson, M.R. The Enterovirus Theory of Disease Etiology in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Critical Review. Front. Med. 2021, 8, 688486. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, H.; Tang, J.; He, Y.; Xiong, T.; Li1, W.; Qu, Y.; Mu, D. Clinical characteristics of severe neonatal enterovirus infection: A systematic review. BMC Pediatr. 2021, 21, 127. [Google Scholar] [CrossRef] [PubMed]
- Ratuszny, D.; Sühs, K.-W.; Novoselova, N.; Kuhn, M.; Kaever, V.; Skripuletz, T.; Pessler, F.; Stangel, M. Identification of Cerebrospinal Fluid Metabolites as Biomarkers for Enterovirus Meningitis. Int. J. Mol. Sci. 2019, 20, 337. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.M.; Mulkey, S.B.; Campos, J.M.; DeBiasi, R.L. Laboratory diagnosis of CNS infections in children due to emerging and re-emerging neurotropic viruses. Pediatr. Res. 2023. epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dahee, J.; Heo, T.H.; Byeon, J.H.; Kim, J.H.; Kim, M.K.; Eun, S.H.; Eun, B.L. Analysis of clinical information and rRT-PCR for early diagnosis of enteroviral meningitis. Korean J. Pediatr. 2015, 58, 446. [Google Scholar] [CrossRef]
- Huang, H.I.; Shih, S.R. Neurotropic Enterovirus Infections in the Central Nervous System. J. Viruses 2015, 7, 6051–6053. [Google Scholar] [CrossRef]
- Opanda, S.M.; Wamunyokoli, F.; Khamadi, S.; Coldren, R.; Bulimo, W.D. Genotyping of enteroviruses isolated in Kenya from pediatric patients using partial VP1 region. Springerplus 2016, 5, 158. [Google Scholar] [CrossRef]
- Harvala, H.; Brobergb, E.; Benschopc, K.; Bergincd, N.; Ladhanie, S.; Susif, P.; Christianseng, C.; McKennah, J.; Allene, D.; Makiellok, P.; et al. Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J. Clin. Virol. 2018, 101, 11–17. [Google Scholar] [CrossRef]
- Apostol, L.N.; Suzuki, A.; Bautista, A.; Galang, H.; Paladin, F.J.; Fuji, N.; Lupisan, S.; Olveda, R.; Oshitani, H. Detection of Non-Polio Enteroviruses From 17 Years of Virological Surveillance of Acute Flaccid Paralysis in the Philippines. J. Med. Virol. 2012, 84, 624–626. [Google Scholar] [CrossRef]
- Zhu, R.; Cheng, T.; Yin, Z.; Liu, D.; Xu, L.; Li, Y.; Wang, W.; Liu, J.; Que, Y.; Ye, X.; et al. Serological survey of neutralizing antibodies to eight major enteroviruses among healthy population. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Couderé, K.; van der Straten, K.; Brouwer, L.; Koen, G.; van Eijk, H.; Pajkrt, D.; Murk, J.L.; Wolthers, K.C. Neutralising Antibodies against Enterovirus and Parechovirus in IVIG Reflect General Circulation: A Tool for Sero-Surveillance. Viruses 2021, 13, 1028. [Google Scholar] [CrossRef] [PubMed]
- Dolskiy, A.A.; Grishchenko, I.V.; Yudkin, D.V. Cell Cultures for Virology: Usability, Advantages, and Prospects. Int. J. Mol. Sci. 2020, 21, 7978. [Google Scholar] [CrossRef]
- Liu, B.; Forman, M.; Valsamakis, A. Optimization and evaluation of a novel real-time RT-PCR test for detection of parechovirus in cerebrospinal fluid. J. Virol. Methods 2019, 272, 113690. [Google Scholar] [CrossRef] [PubMed]
- Artika, I.M.; Dewi, Y.P.; Nainggolan, I.M.; Siregar, J.E.; Antonjaya, U. Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis. Genes 2022, 13, 2387. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; Van Maarseveen, N.; Shuuman, R.; Verkuijlen, S.; De Vos, M.; Hendriksen, K.; Van Loon, A.M. Rapid and Sensitive Routine Detection of All Members of the Genus Enterovirus in different Clinical specimens by Real-Time PCR. J. Clin. Microbiol. 2002, 40, 3666–3670. [Google Scholar] [CrossRef]
- Graham, A.K.; Murdoch, D.R. Association between Cerebrospinal Fluid Pleocytosis and Enteroviral Meningitis. J. Clin. Microbiol. 2005, 43, 1491. [Google Scholar] [CrossRef]
- Norder, H.; Bjerregaard, L.; Magnius, L.; Lina, B.; Aymard, M.; Chomel, J.J. Sequencing of ‘untypable’ enteroviruses reveals two new types, EV-77 and EV-78, within human enterovirus type B and substitutions in the BC loop of the VP1 protein for known types. J. Gen. Virol. 2003, 84, 827–836. [Google Scholar] [CrossRef]
- Oberste, M.S.; Nix, W.A.; Maher, K.; Pallansch, M.A. Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J. Clin. Microbiol. 2003, 26, 375–376. [Google Scholar] [CrossRef]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef]
- Shabani, A.; Makvandi, M.; Samarbafzadeh, A.; Teimoori, A.; Rasti, M.; Karami, C.; Rastegarvand, N.; Nikfar, R.; Shamsizadeh, A.; Salehi, A.; et al. Echovirus 30 and coxsackievirus A9 infection among young neonates with sepsis in Iran. Iran. J. Microbiol. 2018, 10, 259–263. [Google Scholar]
- Marchini, A.; Petrillo, M.; Parrish, A.; Buttinger, G.; Tavazzi, S.; Querci, M.; Betsou, F.; Elsinga, G.; Medema, G.; Abdelrahman, T.; et al. New RT-PCR Assay for the Detection of Current and Future SARS-CoV-2 Variants. Viruses 2023, 15, 206. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Polio Laboratory Manual; No. WHO/IVB/04.10; WHO: Geneva, Switzerland, 2004; Available online: https://apps.who.int/iris/handle/10665/68762 (accessed on 10 February 2019).
- Casas, I.; Powell, L.; Klapper, P.E.; Cleator, G.M. New method for the extraction of viral RNA and DNA from cerebro spinal fluid for use in the polymerase chain reaction assay. J. Virol. Methods 1995, 53, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Rotbart, H.A.; Sawyer, M.H.; Fast, S.; Lewinski, C.; Murphy, N.; Keyser, E.F.; Spadoro, J.; Kao, S.Y.; Loeffelholz, M. Diagnosis of Enteroviral Meningitis by Using PCR with a Colorimetric Microwell Detection Assay. J. Clin. Microbiol. 1994, 32, 2590–2591. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, W.A.; Kuhn, S.; Kockx, M.M.; Van De Vyvere, M.E.; Mertens, A.H. Rapid detection of enterovirus RNA in cerebrospinal fluid specimens with a novel single-tube real-time reverse transcription-PCR assay. J. Clin. Microbiol. 2001, 39, 4093–4096. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, T.F.; Rahman, S.N.; Agudelo, C.W.; Wein, A.J.; Lazar, J.M.; Everaert, K.; Dmochowski, R.R. Foundational Statistical Principles in Medical Research: Sensitivity, Specificity, Positive Predictive Value, and Negative Predictive Value. Medicina 2021, 57, 503. [Google Scholar] [CrossRef]
- Hamza, I.A.; Jurzik, L.; Uberla, K.; Wilhelm, M. Methods to detect infectious human enteric viruses in environmental water samples. Int. J. Hyg. Environ. Health 2011, 214, 226. [Google Scholar] [CrossRef]
- Melnick, J.L. My role in the discovery and classification of the enteroviruses. Annu. Rev. Microbiol. 1996, 50, 2–18. [Google Scholar] [CrossRef]
- Johnston, S.L.G.; Siegel, C.S. Presumptive Identification of Enteroviruses with RD, HEp-2, and RMK Cell Lines. J. Clin. Microbiol. 1990, 28, 1049–1050. [Google Scholar] [CrossRef]
- De Crom, S.C.M.; Obihara, C.C.; Van Loon, A.M.; Argilagos-Alvarez, A.A.; Peeters, M.F.; Van Furth, A.M.; Rossen, J.W.A. Detection of enterovirus RNA in cerebrospinal fluid: Comparaison of two molecular assays. J. Virol. Methods 2011, 179, 104–107. [Google Scholar] [CrossRef]
- Gorgievski-Hrisoho, M.; Schumacher, J.D.; Vilimonovic, N.; Germann, D.; Matter, L. Detection by PCR of Enteroviruses in Cerebrospinal Fluid during a Summer Outbreak of Aseptic Meningitis in Switzerland. J. Clin. Microbiol. 1998, 36, 2408–2412. [Google Scholar] [CrossRef] [PubMed]
- Read, S.J.; Jeffery, K.J.; Bangham, C.R. Aseptic Meningitis and Encephalitis: The Role of PCR in the Diagnostic Laboratory. J. Clin. Microbiol. 1997, 35, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Volle, R.; Bailly, J.L.; Mirand, A.; Pereira, B.; Marque, J.S.; Chambon, M.; Regagnon, C.; Brebion, A.; Henquell, C.; Peigue, L.H.; et al. Variations in cerebrospinal fluid viral loads among Enterovirus genotypes in patients hospitalized with laboratory-confirmed meningitis due to Enterovirus. J. Infect. Dis. 2014, 210, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Menegus, M.A.; Hollick, G.E. Increased Efficiency of Group B Coxsackievirus Isolation from Clinical Specimens by Use of BGM Cells. J. Clin. Microbiol. 1982, 15, 946–948. [Google Scholar] [CrossRef]
- Heim, A. From poliovirus surveillance to enterovirus surveillance: A complete picture? J. Med. Microbiol. 2005, 54, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Piqueur, M.A.C.; Verstrepen, W.A.; Bruynseels, P.; Mertens, A.H. Improvement of a real time RT-PCR assay for the detection of enterovirus RNA. Virol. J. 2009, 6, 95. [Google Scholar] [CrossRef]
- Kares, S.; Lönnrot, M.; Vuorinen, P.; Oikarinen, S.; Taurianen, S.; Hyöty, H. Real-time PCR for rapid diagnosis of entero- and rhinovirus infections using Light Cycler. J. Clin. Virol. 2004, 29, 99–104. [Google Scholar] [CrossRef]
- Archimbaud, C.; Mirand, A.; Chambon, M.; Regagnon, C.; Bailly, J.L.; Lafeuille, H.P.; Henquell, C. Improved Diagnosis on a Daily Basis of Enterovirus Meningitis Using a One-Step Real-Time RT-PCR Assay. J. Med. Virol. 2004, 74, 604–610. [Google Scholar] [CrossRef]
- Relova, D.; Rios, L.; Acevedo, A.M.; Coronado, L.; Perera, C.L.; Pérez, L.J. Impact of RNA Degradation on Viral Diagnosis: An Understated but Essential Step for the Successful Establishment of a Diagnosis Network. Vet. Sci. 2018, 5, 19. [Google Scholar] [CrossRef]
- Saeedinia, A.; Shamsara, M.; Zeinoddini, M.; Sadeghi, V.; Maghsoudi, N. Evaluation of nucleic acid sequence-based amplification (NASBA) and Reverse Transcription Polymerase Chain Reaction for detection of coxsackievirus B3 in cell culture and animal tissue samples. Iranian J. Biotechnol. 2008, 6, 222–228. [Google Scholar]
- Caro, V.; Guillot, S.; Delpeyroux, F.; Crainic, R. Molecular strategy for ‘serotyping’ of human enteroviruses. J. Gen. Virol. 2001, 82, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, W. Mise au Point et Evaluation D’une Technique de PCR Permettant la Détection et le Typage des Entérovirus Directement à Partir de Produits Pathologiques ou D’échantillons Environnementaux. Ph.D. Thesis, Faculté de Médecine Jacques Lisfranc, Université Jean Monnet de Saint-Etienne, Saint-Priest-en-Jarez, France, 2014. [Google Scholar]
- Michos, A.G.; Syriopoulou, V.P.; Hadjichristodoulou, C.; Daikos, G.L.; Lagona, E.; Douridas, P.; Mostrou, G.; Theodoridou, M. Aseptic Meningitis in Children: Analysis of 506 Cases. PLoS ONE 2007, 2, e674. [Google Scholar] [CrossRef] [PubMed]
- Moreni, G.; van Eijk, H.; Koen, G.; Johannesson, N.; Calitz, C.; Benschop, K.; Cremer, J.; Pajkrt, D.; Sridhar, A.; Wolthers, K. Non-Polio Enterovirus C Replicate in Both Airway and Intestine Organotypic Cultures. Viruses 2023, 15, 1823. [Google Scholar] [CrossRef] [PubMed]
- Thieulent, C.J.; Carossino, M.; Peak, L.; Wolfson, W.; Balasuriya, U.B.R. Multiplex One-Step RT-qPCR Assays for Simultaneous Detection of SARS-CoV-2 and Other Enteric Viruses of Dogs and Cats. Viruses 2023, 15, 1890. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Celma, C.; Pegg, E.; Polra, K.; Dunning, J.; Martin, J. Detection and Typing of Human Enteroviruses from Clinical Samples by Entire-Capsid Next Generation Sequencing. Viruses 2021, 13, 641. [Google Scholar] [CrossRef] [PubMed]
- Słomka, A.; Kowalewski, M.; Żekanowska, E. Coronavirus Disease 2019 (COVID–19): A Short Review on Hematological Manifestations. Pathogens 2020, 9, 493. [Google Scholar] [CrossRef]
- Elmakaty, I.; Ferih, K.; Karen, O.; Ouda, A.; Elsabagh, A.; Amarah, A.; Malki, M.I. Clinical Implications of COVID-19 Presence in CSF: Systematic Review of Case Reports. Cells 2022, 11, 3212. [Google Scholar] [CrossRef]
- Bogs, T.; Saleh, N.; Yavuz, S.T.; Fazeli, W.; Ganschow, R.; Schreiner, F. Aseptic Meningitis, Mucocutaneous Lesions and Arthritis after COVID-19 Vaccination in a 15-Year-Old Boy. Vaccines 2022, 10, 325. [Google Scholar] [CrossRef]



| Primers | Genome Region | Sequence | Location | Reference |
|---|---|---|---|---|
| EV2 | 5′NC | 5′-TCCGGCCCCTGAATGCGGCTAATCC-3′ | 446–470 | |
| EV1 | 5′NC | 5′-ACACGGACACCCAAAGTAGTCGGTCC-3′ | 559–533 | [45] |
| Primers | Genome Region | Sequence | Location | Reference |
|---|---|---|---|---|
| Vrp F | 5′NC | 5′-CCCTGAATGCGGCTAATCC-3′ | ||
| Vrp R | 5′NC | 5′-ATTGTCACCATAAGCAGCCA-3′ | 452–596 | [46] |
| Probe | 5′NC | 5′-AACCGACTACTTTGGGTGTCCGTGTTTC-3′ |
| Primer | Genome Region | Sequence | Location | Reference |
|---|---|---|---|---|
| AN89 | VP1 | CCAGCACTGACAGCAGYNGARAYNGG | 2602-2627 | |
| AN88 | VP1 | TACTGGACCACCTGGNGGNAYRWACAT | 2977-2951 | |
| AN232 | VP1 | CCAGCACTGACAGCA | 2602-2616 | |
| AN233 | VP1 | TACTGGACCACCTGG | 2977-2963 | [40] |
| Conventional RT-PCR | |||||
| Positive | Negative | Total | |||
| Real-time RT-PCR | Positive | 13 | + | 21 | 50 |
| + | + | ||||
| Negative | 0 | + | 16 | ||
| Total | 50 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, A.; Ammi, Z.; Anes-Boulahbal, D.L.; Assadi, A.A.; Amrane, A.; Baaloudj, O.; Mouni, L. Molecular Amplification and Cell Culturing Efficiency for Enteroviruses’ Detection in Cerebrospinal Fluids of Algerian Patients Suffering from Meningitis. Viruses 2024, 16, 170. https://doi.org/10.3390/v16020170
Rai A, Ammi Z, Anes-Boulahbal DL, Assadi AA, Amrane A, Baaloudj O, Mouni L. Molecular Amplification and Cell Culturing Efficiency for Enteroviruses’ Detection in Cerebrospinal Fluids of Algerian Patients Suffering from Meningitis. Viruses. 2024; 16(2):170. https://doi.org/10.3390/v16020170
Chicago/Turabian StyleRai, Abdelwahab, Zohra Ammi, Dahbia Leila Anes-Boulahbal, Aymen Amin Assadi, Abdeltif Amrane, Oussama Baaloudj, and Lotfi Mouni. 2024. "Molecular Amplification and Cell Culturing Efficiency for Enteroviruses’ Detection in Cerebrospinal Fluids of Algerian Patients Suffering from Meningitis" Viruses 16, no. 2: 170. https://doi.org/10.3390/v16020170