
Targeting Human Proteins for Antiviral Drug Discovery and Repurposing Efforts: A Focus on Protein Kinases


1
Department of Pharmacy, Faculty of Pharmacy, Al-Zaytoonah University of Jordan, P.O. Box 130, Amman 11733, Jordan
2
Laboratory for Molecular Modeling, Division of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, The University of North Carlina at Chapel Hill, Chapel Hill, NC 27599, USA
3
Jordan CDC, Amman 11118, Jordan
4
Department of Pharmaceutical Sciences, School of Pharmacy, University of Jordan, Amman 11942, Jordan
*
Author to whom correspondence should be addressed.
Viruses 2023, 15(2), 568; https://doi.org/10.3390/v15020568
Submission received: 11 January 2023
/
Revised: 7 February 2023
/
Accepted: 9 February 2023
/
Published: 19 February 2023
(This article belongs to the Special Issue Drug-Repositioning Opportunities for Antiviral Therapy: Volume 2)
Abstract
:Despite the great technological and medical advances in fighting viral diseases, new therapies for most of them are still lacking, and existing antivirals suffer from major limitations regarding drug resistance and a limited spectrum of activity. In fact, most approved antivirals are directly acting antiviral (DAA) drugs, which interfere with viral proteins and confer great selectivity towards their viral targets but suffer from resistance and limited spectrum. Nowadays, host-targeted antivirals (HTAs) are on the rise, in the drug discovery and development pipelines, in academia and in the pharmaceutical industry. These drugs target host proteins involved in the virus life cycle and are considered promising alternatives to DAAs due to their broader spectrum and lower potential for resistance. Herein, we discuss an important class of HTAs that modulate signal transduction pathways by targeting host kinases. Kinases are considered key enzymes that control virus-host interactions. We also provide a synopsis of the antiviral drug discovery and development pipeline detailing antiviral kinase targets, drug types, therapeutic classes for repurposed drugs, and top developing organizations. Furthermore, we detail the drug design and repurposing considerations, as well as the limitations and challenges, for kinase-targeted antivirals, including the choice of the binding sites, physicochemical properties, and drug combinations.
1. Introduction
Antiviral drugs could be targeted towards either viral proteins (e.g., polymerases, integrase, proteases, accessory proteins, and viral structural proteins), or host proteins that support the viral life cycle [1,2,3]. In 1963, idoxuridine was the first directly acting antiviral (DAA) drug to be approved, initiating a new era in fighting viral infections using drugs that directly target viral proteins [4]. Such drugs gained great attention due to the discovery and licensing of 80 DAA drugs [1]. These drugs were considered safe for humans since they target viral proteins which have no human homologs, except for viral polymerase, which does share some structural similarities with human polymerases, thus being a major reason for nucleoside-based antiviral toxicity [5].
Despite the initial success and low toxicity outcomes on humans, the DAA drug approach faced many hurdles, including the narrow spectrum of activity and the development of antiviral resistance. In fact, every virus has its own set of highly specialized proteins [5]; some proteins share homology among some viruses, but the majority are unique. As a result, broad-spectrum DAA drug discovery efforts often fail and there are only a few other broad-spectrum antiviral drugs in the market that are approved [5]. Therefore, older antivirals often fail to fight novel emerging viruses which threaten the human population by their ability to cause epidemics and pandemics [6]. Additionally, and despite initial success, DAA drugs usually lose their efficacy due to constant viral mutations.
Resistant viral variants emerge over time with different speeds for RNA versus DNA viruses [7]. RNA viruses have mutational rates reaching 10−4 (i.e., one mutation per 10,000 base replications), in comparison to mutational rates of 10−8 for DNA viruses [8]. Thus, extended exposure to viral infection and continuing viral replication are key factors in the development of antiviral resistance [7]. This led to new legislation for the clinical development of antiviral agents which require testing for viral resistance, mutations, and cross-resistance that persists with treatment [9]. Another hurdle facing the DAA drug approach is the limited number of potential viral proteins that could be targeted with drugs, especially for viruses with small genomes, such as human papillomavirus [7].
Targeting human proteins for antiviral drug discovery may result in the development and approval of broader spectrum antiviral drugs that are inherently less susceptible to viral resistance. Thus, host-targeted antivirals (HTAs) are considered promising therapeutic options for combating emerging novel pathogens; even before their genes and proteins are fully characterized [7,10]. Such drugs could potentially lead to universal antiviral agents. Herein, we provide a comprehensive overview of disease-causing viruses, their classification, their life cycle, and their reliance on host kinases to replicate and produce offspring. We also detail the drug discovery strategies that target host kinases to halt the viral life cycle that could potentially lead to universal antiviral agents.
2. Disease-Causing Viruses
Disease-causing viruses cause viral infections, which include any illness that is caused by a virus. A virion is the infectious form of the virus that is released from host cells after viral replication [11]. It protects the virus genome and facilitates its entry into specific host cells, which have the required receptors or proteins that facilitate its entry [11,12]. The virion contains the genome, and it is surrounded by a capsid, which is made up of proteins, to protect the genome [13]. Some viruses, such as those from the Pleolipoviridae family, do not have capsids [14]. Other viruses have an envelope, which encloses the capsid and is made up of a lipid bilayer embedded with virus-specific glycoproteins, derived from the host’s cell plasma membrane or an intracellular vesicle [13]. Depending on the virus type, other components of virions include mRNAs, proteins and enzymes, and polyamines [13].
2.1. Classification of Viruses
The International Committee on Taxonomy of Viruses (ICTV) system classifies viruses into different taxonomic levels, starting with realms and ending with species [15,16]. Currently, ICTV’s database of taxonomy shows that there are 10,434 species of viruses [17].
There are two different classification systems being adopted for viruses. The classification systems do not correspond to each other, and each is used separately. The first classifies viruses based on their genome; DNA or RNA [13]. They can be viewed in the most recent ICTV Report on Virus Classification and Taxon Nomenclature [18]. The second, which is the Baltimore Classes (BC) system [19,20,21,22], classifies viruses according to the path and process in which the genome is transcribed into an mRNA that is needed for a translation into proteins [13]. The production of mRNA from each type is discussed in the Virus Life Cycle section below. The classification systems are summarized in Table 1.
2.2. Virus Life Cycle
The life cycle of viruses comprises several basic steps starting with viral entry into the host cell and followed by gene expression, gene replication, and finally ending in assembly and viral egress to release new infectious viral particles [13]. However, there could be many differences in how each virus (or class of viruses) achieves these steps. To design effective treatments or preventive therapeutics for viral diseases, especially those targeting host proteins, we need to understand the similarities and differences in the life cycles of viruses [23].
2.2.1. Virus Entry and Uncoating
The entry of viruses into the host cell comprises two stages: attachment and penetration. Entry is later followed by uncoating. These stages would differ between enveloped and non-enveloped viruses. As for enveloped viruses, their membrane would initially fuse with the host cell membrane via the binding of specific viral envelope glycoproteins (also called fusion proteins) to host cell receptors [24]. Consequently, a pore opening (fusion pore) [24,25,26], which might be either enlarged or not [25,27,28,29], allows the passage of the virus core into the host cell cytoplasm [26]. A different mechanism involves the uptake of the virus into an endocytic vesicle followed by the fusion of the viral envelope with the vesicle membrane and releasing the capsid [13]. The fusion mechanisms are either pH-dependent or pH-independent [26].
Non-enveloped viruses attach themselves to host cells via either a single protein or multiple protein structures [30,31]. Then, the virus is internalized into the cell by an endocytic vesicle formed via receptor-mediated uptake, followed by the release of the virus and genome into the host cell cytoplasm [13]. Other viruses can inject their genome directly into the host cell cytoplasm across the host cell membrane [13].
Viral attachment to host cells is followed by uncoating, which is the process of releasing the viral genome either into the cytoplasm or directly into the host cell nucleus via the nuclear pores after breaking down viral capsids [13,32]. This step is essential for starting the virus life cycle and permitting the virus to replicate its genetic material.
2.2.2. Gene Expression and Replication
Viral gene expression involves mRNA synthesis (transcription) followed by protein synthesis (translation) inside host cells. Transcription is accomplished via the host and/or viral enzymes, while translation is accomplished via host ribosomes. All viral genomes, irrespective of their type, would be transcribed into mRNA. Gene expression differences do exist between viruses based on the viral genome type. Gene replication, in which a new viral genome is produced to be incorporated into new virions, occurs inside host cells as well. Some viruses undergo replication processes of the genetic material before their genome is eventually transcribed, such as BCII, BCVI, and BCVII viruses [13,22]. On the other hand, BCI, BCIII, and BCV viruses would be transcribed directly, while BCIV viruses are directly translated [13,22].
BCI double-stranded DNA (dsDNA) and BCII single-stranded DNA (ssDNA) viruses, except for poxviruses, have their genome-containing capsids or nucleoprotein complexes moved to the nuclear pores, which permit viral genome entry into the nucleus where gene expression and replication occur [13]. The gene expression of BCIV ((+) ssRNA) viruses starts directly after viral uncoating in the cytoplasm, where viral genomes associate with host ribosomes to start the translation process of viral proteins [13]. However, the genomes of BCIII (dsRNA) and BCV ((−) ssRNA) viruses are associated with the viral RNA-dependent RNA polymerase (RdRp) [13]. Reverse transcribing viruses, BCVI ((+) ssRNA) viruses and BCVII (dsDNA) viruses, associate with the viral reverse transcriptase enzyme (RTz) [13]. A summary of the mechanisms involved in the gene expression and replication of several classes of viruses is presented in Table 2.
2.2.3. Assembly and Egress
The assembly of virions usually takes place at the site of genome replication [13]. Most RNA virions are assembled in the cytoplasm, while most DNA viruses at least start their assembly inside the nucleus. The egress (release) of new virions depends on their type in terms of non-enveloped or enveloped virions [13]. Non-enveloped virions are released after the lysis of the host cell, whereas enveloped virions are released via budding from the host cell and acquiring an envelope from a particular cellular membrane. The envelope could be acquired from the plasma membrane in the final step or could be from the nuclear membrane, Golgi, or other organelle membranes. The new virion is transported via vesicle to the plasma membrane, in which the vesicle fuses with the plasma membrane and release the virion.
3. Antiviral Drugs in Different Stages of Clinical Development
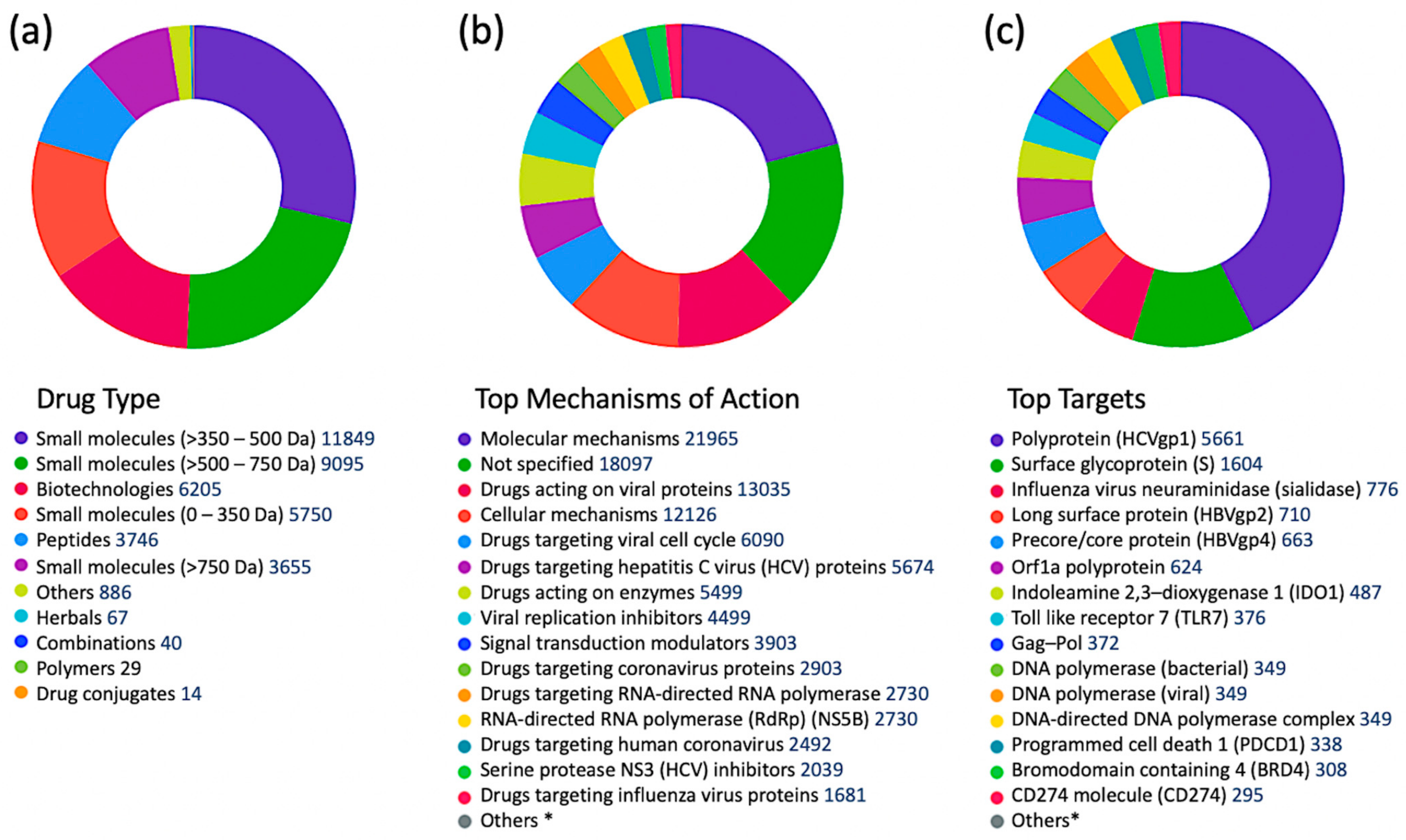
According to the Cortellis Drug Discovery Intelligence (CDDI) database [37], there are 41,321 records for drugs in different stages of clinical development, classified as antiviral agents based on “therapeutic group” designation. Among these, 13,027 drugs and biologics target viral proteins (31.5%), while the rest target non-viral proteins [38,39]. The type of drugs, top mechanisms of action, and top targets of antiviral drugs are shown in Figure 1. The top six drug targets are all viral proteins, but host proteins, including indolamine 2,3-dioxygenase 1 (IDO1), toll-like receptor (TLR) 7, programmed cell death 1 (PDCD1), bromodomain containing 4 (BRD4), and a cluster of differentiation 274 (CD274) (also known as Programmed death-ligand 1(PD-L1)) are included. The top non-viral mechanism of action was signal transduction modulation [38].
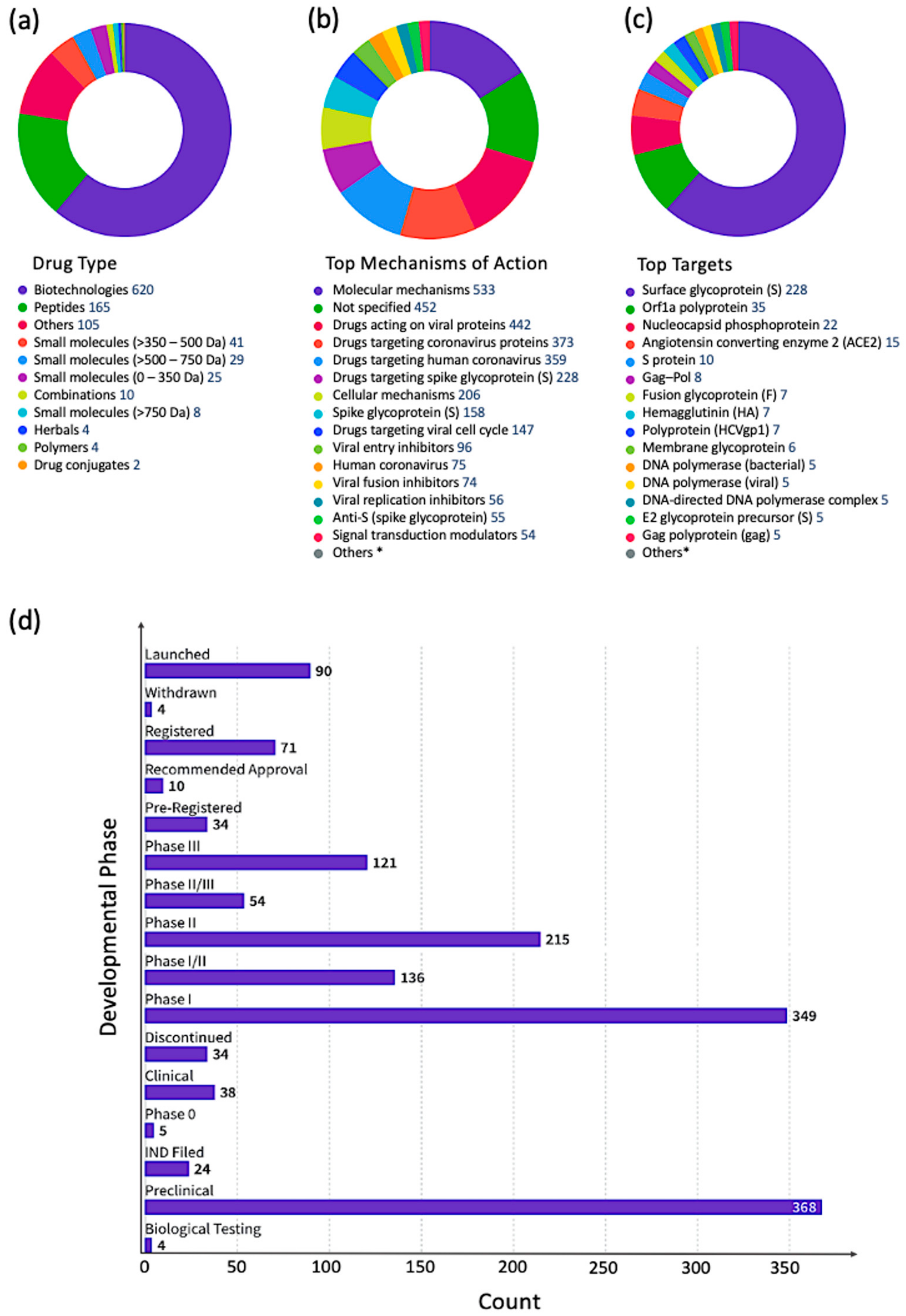
Filtering the complete set of 41,321 antiviral drugs by the following development status criteria: “under active development = yes” and “condition = infection”, resulted in 1013 antiviral drugs (Figure 2), of which 442 drugs are targeting viral proteins (43.6%). The top five infections targeted by these drugs were: coronavirus infections (507 drugs), influenza (162 drugs), herpes (63), respiratory syncytial virus infections (47 drugs), and papillomavirus infections (39).
4. Targeting Host Proteins for Antiviral Drug Discovery
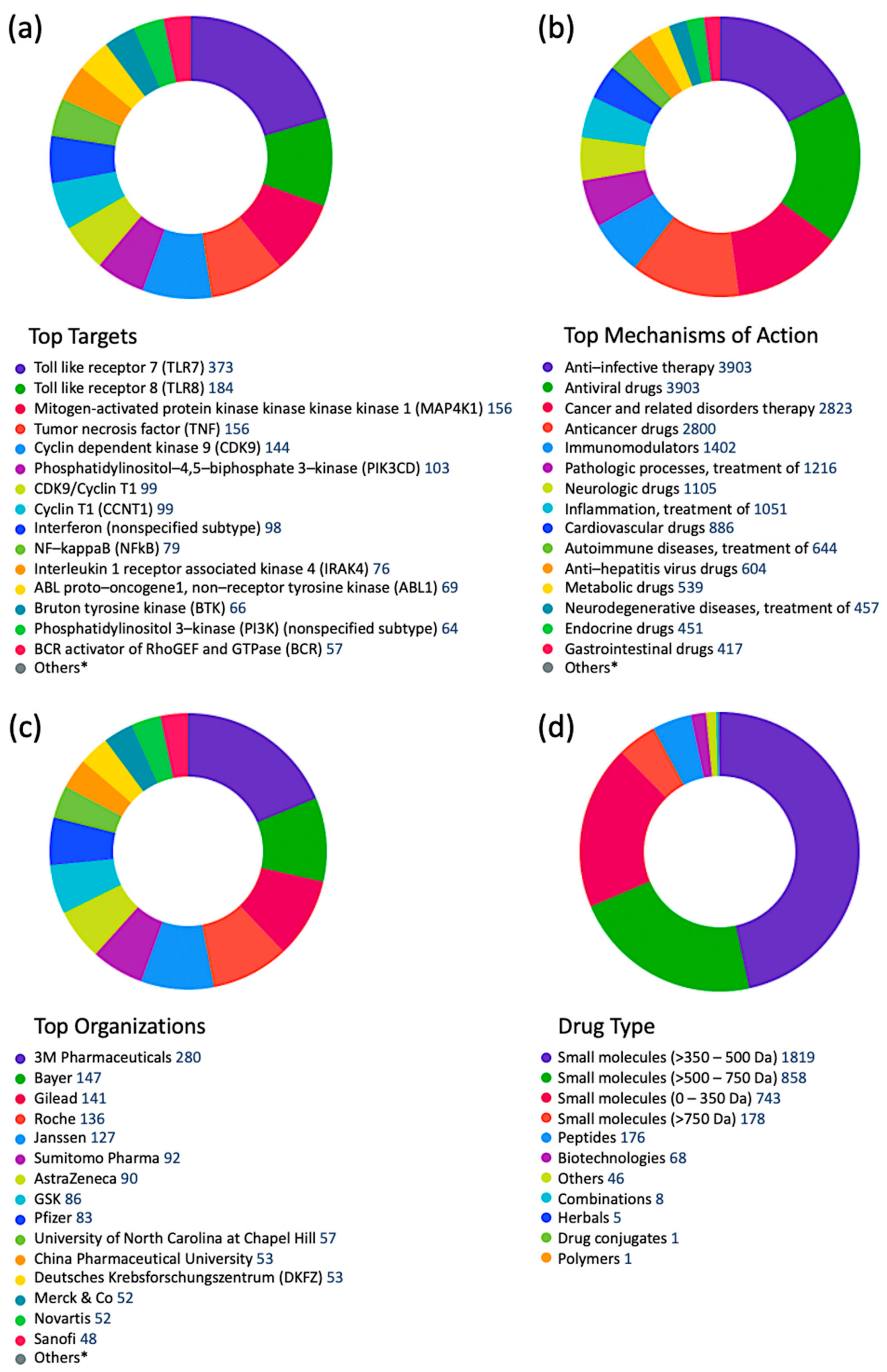
Mining the CDDI drug intelligence database indicated that pharmaceutical and biotechnology companies are largely investigating host proteins as targets for antiviral drug discovery efforts [37]. In fact, antiviral drugs targeting viral proteins constituted less than one-third of all antiviral drugs in different stages of drug development. Signal transduction modulators were the most important group of drugs targeting non-viral proteins in the antiviral drug development pipeline. From 41,321 antiviral drug records in different stages of clinical development, there are 3903 signal transduction modulators targeting human proteins, including TLRs (e.g., TLR7, TLR8), kinases (e.g., MAP4K1, CDK9, PIK3, IRAK4, BTK), tumor necrosis factor (TNF), cyclins (e.g., CCNT1), interferons (IFNs), and others. Drugs targeting TLRs and kinases were the most abundant indicating the importance of these two target families in modulating signal transduction in response to viral infections. Figure 3 summarizes signal transduction modulators according to top targets (Figure 3a), top therapeutic groups (Figure 3b), top organizations developing these drugs (Figure 3c), and drug types (Figure 3d).
5. Targeting Human Protein Kinases for Antiviral Drug Discovery
Kinase enzymes catalyze the phosphorylation reaction of a broad range of substrates, including lipids, carbohydrates, proteins, and nucleic acids. They are widely present in nature and are involved in a multitude of biological processes [40]. They play key roles in regulating important cellular functions, including metabolism, cell cycle regulation, survival, and differentiation [41]. Considering their vital physiological role, extensive efforts have been directed toward the identification of activity-modulating ligands, such as agonists or antagonists [42]. There are 518 kinases encoded by nearly 2% of the human genome [43]. Interestingly, about half of the kinases are largely uncharacterized with poorly understood roles [40], which may provide a window for the emergence of significant innovational therapies based on the design and development of novel inhibitors of the orphan protein kinases. The recent advancements in science and biotechnology, especially in bioassay design, have enabled the assessment of kinase inhibition in living cells [44], which may offer a valuable tool for shifting from chemical probes to drug candidates.
Evidently, protein kinases are upregulated in numerous pathological states, and hence their inhibition presents a therapeutic solution for many disorders, including cancer and inflammation [45]. In addition, evidence has begun to accumulate supporting the relationship between kinase inhibition and antiviral activities [46,47]. In fact, host protein kinases are known for their association with the virus life cycle [7,48]. Viruses of both types, DNA or RNA, are known to develop several manipulation mechanisms against the host’s innate and acquired immune mechanisms to control host cellular activities and to create a favorable environment for their replication [48,49,50]. Among these cellular activities is the host’s cell cycle, in which many kinases are involved. Both DNA and RNA viruses manipulate host cell cycle proteins for their own replication advantage. Non-cell cycle kinases also get involved in the progression of viral infections. Most DNA viruses replicate their genetic material inside host cellular nuclei, in the S-phase of the host cell cycle or in arrested cells, such as neurons [48]. Viruses replicating at the S-phase either use the host’s DNA replication proteins that are active at S-phase or require host cellular factors that are activated in the S-phase to express viral proteins required for their DNA replication [48]. Likewise, RNA viruses, replicating in the cytoplasm or nucleus of a host cell, manipulate cell-cycle proteins to replicate their own genetic material [48,50]. RNA viruses activate the IFN-induced double-stranded RNA-dependent protein kinase, known as protein kinase RNA-activated (PKR), which is a member of a small family of serine-threonine kinases that are activated by extracellular stresses [51,52]. The classical activator of PKR is dsRNA, which directly binds PKR and triggers PKR kinase activity [53].
6. Kinases as Validated Biomarkers for Viral Infections
To get a better idea about which kinases have any potential for antiviral drug discovery, we mined all drugs and biologics in the CDDI database using the following search criteria: “condition = viral infection” and “mechanism = kinase”. These search criteria allow the retrieval of all drugs and biologics that have been linked to antiviral activity and can also modulate a kinase. Our search resulted in 1251 drugs and biologics, of which 1204 drugs are still in the biological testing phase and have not progressed in the drug development pipeline yet. In fact, 1224 drugs and biologics were coming from patents and could progress in the drug development pipeline soon. The top 10 kinase targets that have drugs “under active development” status for viral infections is protein kinase C (PKC), AXL receptor tyrosine kinase (AXL), Casein kinase II (CK2), mitogen-activated protein kinase (MAPK), mitogen-activated protein kinase kinase (MAP2K; MAPKK; MEK), extracellular signal-regulated kinase (ERK), MAPK p38, cyclin-dependent kinase (CDK) 1 (CDK1), TEK receptor tyrosine kinase, and protein kinase B (PKB; Akt).
A more targeted search of the CDDI database was performed using the developmental status condition as follows: “development status condition = infection, viral” and “mechanism of action = drugs targeting kinases”. Imposing these filtering criteria ensure that the retrieved drugs and biologics have antiviral effects and are being developed precisely to combat viral infections by targeting kinases. This search resulted in seven drugs and biologics (Table 3). The drug targets of these drugs include casein kinase 2 (CK2), MAP2K, MAPK, MAPK p38, and CDK1. Five of the drugs have the “under active development (UAD)” label, indicating that products are actively moving through the drug research and development (R&D) pipeline from preclinical stages through registration. The launched drug 3-Angeloylingenol is not being investigated for new conditions, and therefore it is not considered UAD. In 2020, this drug was withdrawn from the market in Canada, the European Union, and the United Kingdom.
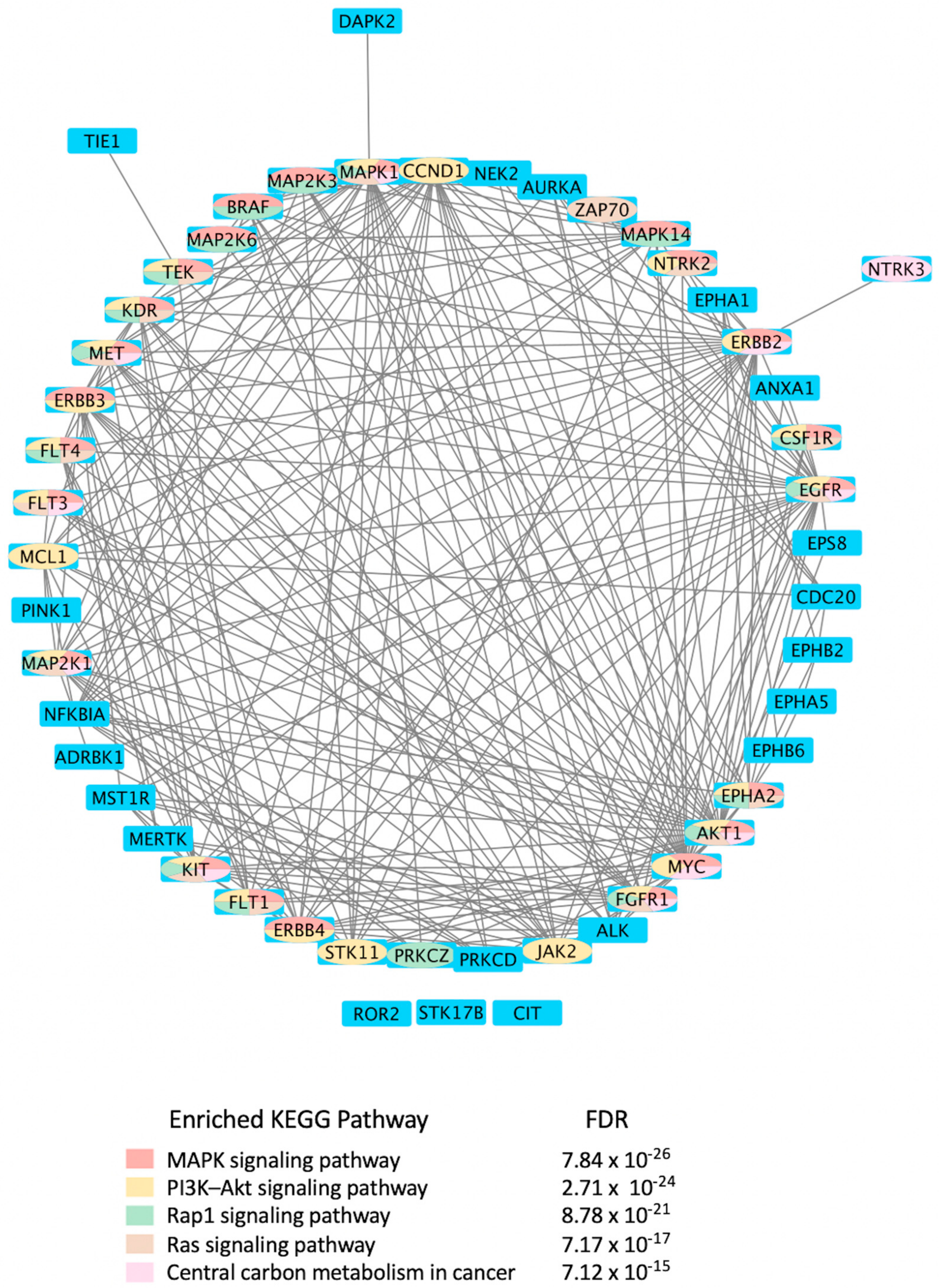
Mining CDDI for diagnostic and prognostic biomarkers aimed at viral infections resulted in 2994 biomarker records as hits. Among these, 1021 genomic and proteomic biomarkers have reached high validity levels (i.e., were either approved, recommended, or in clinical studies) according to the CDDI database [37]. Kinases make up 4.9% of these 1021 biomarkers, which highlights key roles in the pathogenesis of viral diseases. To get a better idea about the kinase biomarkers for viral infections, we generated a direct protein-protein interactions network using 51 kinases (resulting from the above data-mining effort in CDDI) as nodes (Figure 4). Network edges represented relationships between nodes based on validated experimental protein-protein interaction data extracted from the STRING [54] database. Network generation and visualization were performed in Cytoscape version 3.9.1 [55]. Network generation was followed by a functional enrichment analysis which highlighted the following pathways as top enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways: (1) MAPK signaling pathway; (2) phosphatidylinositol 3-kinases (PI3K)-Akt signaling pathway; (3) Ras-associated protein 1 (Rap1) signaling pathway; (4) RAS signaling pathway; and central carbon metabolism in cancer. The complete enrichment results are provided in Supporting Information (Supplementary Table S1).
Notably, the top enriched pathways are directly linked to the virus life cycle and the biological processes that allow the virus to replicate and produce infectious progeny. For example, the MAPK signaling pathway can be activated by a diverse group of viruses [56], and it is involved in the replication of their genetic material. The information in the MAPK pathway is transmitted from one protein to another by phosphorylating serine and threonine residues in a diverse group of proteins leading to a multitude of cellular responses. Furthermore, ERK/MAPK and PI3K/Akt/mTOR signaling responses play a critical role in the pathogenesis of many viral diseases. In fact, viruses such as the Middle East respiratory syndrome coronavirus (MERS-CoV) inhibit these pathways [56], among others.
Rap1 signaling regulates T-cell and antigen-presenting cell (APC) interactions and modulates T-cell responses to pathogens, such as viruses [57]. In fact, the activation state of Rap1 determines T-cell responses to antigens. Rap1 is also a target for the TLRs, which are key regulators of the immune response to viral infections [58]. Ras signaling was found important for the life cycle of some viruses, including the reovirus. Evidence showed that Ras-transformation affects viral uncoating and disassembly, PKR-induced translational inhibition, generation of viral progeny, the release of progeny, and viral spread [59].
It is also known that viruses hijack host metabolic resources and induce a plethora of metabolic alterations in host-cell including host central carbon metabolism. In fact, viral replication relies on extracellular carbon sources, such as glucose and glutamine [60]. The PI3K/Akt/mTOR and HIF-1 signaling pathways regulate glycolysis. Thus, targeting them with inhibitors, such as MK2206 (an Akt inhibitor) or 2-deoxy-D-glucose (2-DG, glycolysis inhibitor), can lower the viral burden in the cells in vitro [61,62,63,64].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 3.
Antiviral drugs targeting human kinases in advanced stages of clinical development.
| Compound (Route of Administration) | Highest Phase (Condition) | * UAD | Target | Side Effects |
|---|---|---|---|---|
 3-Angeloylingenol (Topical) | Launched (Actinic Keratosis) | No | PKC | Local skin reactions at the application site, headache, periorbital edema, nasopharyngitis [1,65]. In 2020, the European Medicines Agency (EMA) recommended the suspension of the product in the EU and EEA as a precautionary measure. Later this year, the marketing authorization was withdrawn by the EMA as the product may increase the risk of skin cancer and the risks outweigh its benefits [37]. |
 CIGB-300 P15-Tat (Topical) | Phase II (Genital warts) | Yes | CK2 | Local adverse events at the injection site including pain, bleeding, hematoma and erythema. Systemic adverse events include: rash, facial edema, itching, hot flashes, and localized cramps [66,67]. |
 Zapnometinib (Oral) | Phase II/Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (COVID-19) | Yes | MEK | Adverse event profile is still unknown. Studies are ongoing [68]. Other MEK inhibitors caused cardiac and ophthalmologic side effects, rash, diarrhea, peripheral edema, fatigue, and dermatitis acneiform [68]. |
 Trans-resveratrol (Topical) | Phase II (Herpes labialis) | Yes | MAPK | Headache, abdominal pain, gastrointestinal problems, urinary tract infections, falls and dizziness [69]. |
 Terameprocol | Phase I (Prevention of HIV transmission) | Yes | CDK1 | Ileus, constitutional symptoms, interstitial nephritis, dyspnea and hypoxia, constipation, anorexia [70,71]. |
| POLB-001 | Phase I (Influenza) | Yes | MAPK p38 | Available clinical data indicated that the drug was tolerated at all tested drug doses with no serious adverse events or trial volunteer withdrawals [10]. It did not elicit liver or cardiac toxicities which could result from the polypharmacological effects of some p38 MAPK inhibitors due to modulating other kinases [72]. |
* UAD: under active development indicates, indicating that products are actively moving through the drug research and development (R&D) pipeline from preclinical stages through registration.
7. The Most Frequently Targeted Kinases for Antiviral Drug Development
7.1. Cyclin-Dependent Kinases (CDKs) and Cyclins
More than 20 CDKs have been identified in humans so far. These kinases are involved in cell-cycle progression (e.g., CDK1 to CDK6) and gene expression regulation (e.g., CDK7 to CDK12) [50]. CDKs are controlled by several mechanisms, such as binding to cyclins (ligands), phosphorylation, or binding to CDK inhibitors (CDKis) [50]. CDK inhibitor gene families in mammals include INK4 and Cip/Kip. The INK4 family proteins (p16INK4a, p15INK4b, p18INK4c, and p19INK4g) inhibit CDK4/6 activity, whereas the Cip/Kip family members (p21Cip1, p27Kip1, and p57Kip2) modulate the function of several cyclins and CDKs [74,75].
DNA and RNA viruses modify CDKs functions to aid their replication by either dysregulating the host’s cell-cycle progression or overcoming restriction factors [76]. These modifications are achieved via specific viral proteins interfering with cyclin/CDKs complexes interactions, phosphorylation of CDKs, or expression of cyclins and CDKis along with other cell cycle proteins, as extensively summarized by Gutierrez-Chamorro et al. [50].
It is known that dysregulation of cyclin/CDK functions can lead to uncontrolled cell division and proliferation and thus leading to cancer [77]. Hence, several pharmacological CDKis have been developed and tested clinically for the potential use as cancer therapeutic options [78,79], with only three selective pharmacological CDKis on CDK4/6, namely palbociclib, ribociclib, and abemaciclib, being approved for the treatment of metastatic breast cancer along with hormone therapy by the United States Food and Drug Administration (FDA) and by the European Medicines Agency [50,80]. However, there are currently no approvals for pharmacological CDKis as potential treatment options for viral infections, although there are several promising results in in vitro studies, such as the use of roscovitine, alsterpaullone, dinaciclib, PHA-690509, FIT-039, and abemaciclib [81,82,83,84,85,86]. Moreover, other FDA-approved pharmacological CDKis, such as palbociclib and flavopiridol, were identified as possible treatment options against viruses via simulations or the Library of Integrated Network-Based Cellular Signatures [85,86].
Cyclins could also be targeted to affect the viral life cycle. CCNT1 cyclin T1 gene encodes one of the highly conserved cyclin C proteins. The encoded protein CycT1 is a core subunit of positive-transcription elongation factor b and is highly associated with CDK9. The CycT1-CDK9 complex acts as a cofactor of the HIV-1 Tat protein and is crucial for the amplification of HIV-1 genome RNA.
In addition, this complex is involved in activating transcript elongation via the phosphorylation of Ser2 of the heptad repeats in the carboxy-terminal domain (CTD) of RNA polymerase II (RNApol-II), and subsequently, the phosphorylated RNApol-II initiates the elongation of HIV-1 transcripts [87,88,89,90].
Hence, targeting this family of proteins may interfere with the virus life cycle in terms of RNA synthesis for replication or transcription processes [91].
7.2. Mitogen-Activated Protein Kinases (MAPKs)
MAPKs are among the cell signaling pathways which are stimulated by extracellular stimuli [92,93,94,95], such as cytokine, hormone, growth factors, pathogens with viruses as an example, osmotic stress, heat, oxidative stress, and microtubule disorganization, resulting in multiple cellular responses [96,97,98]. Among the responses being regulated by MAPKs are gene expression, cell differentiation, cell survival, mitosis, apoptosis, and metabolism [99,100]. The three major MAPK signaling pathways present in mammals are ERK (also known as p42/44 MAPK), Janus kinase (JNK) (also known as stress-activated protein kinase-1 (SAPK1)), and MAPK p38 (also known as SAPK2/RK) [101,102]. RNA and DNA viruses [56] can activate MAPK signaling pathways through their interactions with cell surface receptors known as receptor tyrosine kinases (RTKs), G-protein coupled receptors [95] and integrins [95]. Depending on the type of virus, MAPK pathways could either enhance or hamper virus replication [103] by regulating single or multiple steps in the viral life cycle [104], including viral entry [105,106,107,108], assembly, maturation, release [109,110,111,112], protein synthesis [101], RNA synthesis [108,112], membrane fusion, and cell-to-cell spreading of viruses [111]. In fact, the sustained activation of the MAPK pathway can be achieved by viral secretory proteins, such as the vaccinia virus growth factor (VGF) on the ERK1/2 signaling MAPK pathway [104]. Other effects of MAPK signaling pathways are summarized by Kumar et al. [56].
MAP4K1, also known as hematopoietic progenitor kinase 1, is a member of MAPKs that negatively regulate cellular antiviral responses [112]. Eventually, this will promote the genome replication of viruses and reduce the host’s antiviral responses [112]. Briefly, pathogens, including viruses, can be detected by pattern recognition receptors (PRRs) in a host cell, such as retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs) [113] that are a member of the RLR signaling pathway. Subsequently, this will trigger the transcription of downstream regulatory proteins involving TANK-binding kinase 1 (TBK1)/IκB kinase-ε (IKKε), which will mediate the production of IFN-type I (IFN-I) and proinflammatory cytokines to stop the spreading of viruses. MAP4K1 has been shown to promote the degradation of TBK1/IKKε and hence, the inhibition of the cellular antiviral responses.
As discussed above, the MAPK pathway affects the viral life cycle in different ways, either enhancing or hampering their replication. As potential antiviral agents, there are several kinase inhibitors that target MAPK pathways [56]. However, they are still not approved. Examples of such inhibitors include vemurafenib, U0126, Cl-1040 (PD184352), FR180204, Ag-126, SB203580, SB 202190, AS601245, and SP600125.
7.3. KRR1 Small Subunit Processome Component Homolog
KRR-R motif-containing protein 1 (KRR1) small subunit processome component homolog (hereafter Rev) has been previously known as human immunodeficiency virus 1 (HIV-1) Rev-Binding Protein 2, HRB2, RIP-1, or KRR-R Motif-Containing Protein 1 [114,115]. Rev is one of the HIV-1 viral proteins that are responsible for the nucleocytoplasmic transport of HIV mRNAs as it binds to Rev-response elements (RRE) [116,117,118,119] inside the nucleolus of the host cell [120]. Rev-RRE ribonucleoprotein complexes are then exported to the cytoplasm. This eventually leads to the accumulation of intron-containing unspliced and partially spliced HIV transcripts in the cytoplasm for viral protein expression and viral assembly [121,122]. This nucleolar pathway could be the basis for a novel target for potential treatments against HIV-1 infections [123].
7.4. Receptor Interacting Protein Kinases (RIPKs)
Receptor-interacting protein kinases (PIPKs), i.e., RIP kinases, are a family of serine/threonine kinases that play a wide variety of functional roles in cellular signaling during pathogen infection. Several RIPKs are involved in necroptosis, which is a form of inflammatory programmed cell death that plays a role in fighting viral infections [124]. Necroptosis leads to cell membrane rupture and leakage of cellular contents [125], thus initiating inflammation and adaptive immunity against viruses [126]. Necroptosis can be achieved through different biological pathways involving different receptors, such as RIPK1, RIPK3, and mixed lineage kinase-like protein (MLKL), which are induced by TNF-alpha (TNF-α) [127,128,129]. Other pathways involve the binding of PRRs, such as DNA-induced activator of IFN (DAI, DLM, or ZBP1), TLRs, and RLRs, directly with RIPK homotypic interaction motif (RHIM) of RIPK3 [130,131,132].
Viruses can inhibit the necroptosis process from facilitating their replication. For example, murine cytomegalovirus and herpes simplex virus-1 (HSV-1) encode viral proteins with inhibitory RHIM that target RIPK1 and ZBP1 [133,134]. Vaccinia virus is sensitive to necroptosis mediated by TNF-α and RIPK1 [128,131]. Necroptosis also limits Vaccinia virus replication, and RIPK3 deletion in mice caused an increase in virus replication [135]. Furthermore, a study involving the use of swine kidney cell line (PK-15) and pseudorabies virus (PRV) [124] reported the induction of necroptosis by PRV via the RIPK3 pathway. Although PRV has induced necroptosis via the RIPK3 pathway in PK-15 cells, the study has shown that RIPK3 and MLKL knockdown had reduced necroptosis and enhanced PRV replication [124]. Hence, drugs targeting necroptotic pathways to interfere with viral replication and spreading are worth investigating.
7.5. Receptor Tyrosine Kinases (RTKs)
7.5.1. Epidermal Growth Factor Receptor (EGFR)
Epidermal Growth Factor Receptor (EGFR) is among the RTKs that are responsible for the regulation of variable cellular processes, such as cell proliferation, differentiation, division, survival, migration, metabolism, and cell cycle control [63,136,137].
Several viruses make use of the EGFR for their own replication and spreading. For example, the Vaccinia virus encodes for VGF, which is a homologue of EGF that activates EGFR [138,139,140,141]. EGFR activation is involved in cell motility, a process that is essential for viral infection spreading [142], including the Vaccinia virus, as it enhances cell-to-cell contact between infected cells and uninfected cells [143]. Gefitinib, an EGFR tyrosine kinase inhibitor, inhibited the spreading of poxvirus [144]. Hence, targeting EGFR to stop the spreading of the virus is worth further investigation.
Sodium taurocholate cotransporting polypeptide (NTCP) is a transporter expressed in the liver, which is responsible for bile acid uptake, and was found to be involved in hepatitis B virus (HBV) internalization into hepatocytes [145]. HBV internalization is achieved via NTCP oligomerization, a process that is inhibited by troglitazone and NTCP peptide [146,147]. In addition, EFGR interacts with NTCP allowing for HBV internalization [146]. Although gefitinib has no inhibitory effects on NTCP oligomerization, it has interfered with HBV internalization [147].
Respiratory viruses, such as influenza virus and Rhinovirus (RV), induce EGFR activation, which its activation inhibits IFN innate antiviral response in airway epithelium [147]. This is achieved via the suppression of IFN regulatory factor (IRF) 1–induced IFN-λ production that is considered the most significant IFN in mucosal antiviral response, and hence, increased viral infection [147].
Moreover, the selective EGFR tyrosine kinase inhibitor, AG 1478, has increased IRF1 and IFN-λ levels and decreased viral titers in vitro and in vivo.
Fucoidan KW (hereafter KW), an agent derived from brown algae Kjellmaniella crassifolia, interfered with influenza A virus (IAV) infection in vitro and in vivo [148]. KW interfered with EGFR activation and may inhibit the cellular EGFR pathway as it inhibited IAV endocytosis and EGFR-mediated internalization in IAV-infected cells [148].
7.5.2. AXL Receptor Tyrosine Kinase
AXL is a member of the subfamily Tyro-Axl-Mer (TAM) receptor tyrosine kinases. It is one of the phosphatidylserine (PS) receptors present in phagocytes, which also include the T-cell immunoglobulin mucin (TIM) family in addition to TYRO3 and MER tyrosine kinase (MERTK). Generally, PS receptors can clear apoptotic cells, but they may allow viral entry into target cells [147,148,149,150,151,152]. These receptors recognize PS on enveloped viruses and mediate the attachment and internalization of the virus into the target cell via a process called “viral apoptotic mimicry” [83]. The recognition of viral PS is achieved through a PS-binding protein known as growth arrest-specific gene 6 (GAS6) protein, which in turn binds to AXL, and the virus becomes internalized, hence increasing viral infectiousness [153]. For example, AXL promotes the entry of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) into human cells [83]. Through in vitro experiments, AXL overexpression enhanced SARS-CoV-2 entry, while its downregulation significantly reduced SARS-CoV-2 infection to pulmonary cells [83]. Hence, targeting AXL receptor kinases may interfere with viral entry and infectivity of other viruses as well.
7.5.3. Janus Kinase–Signal Transducer and Activator of Transcription (JAK-STAT)
Proteins
Janus kinase/signal transducer and activator of transcription (JAK-STAT) proteins are intracellular molecules with key roles in signaling pathways modulated by growth factors and cytokines and involved in innate immunity. In fact, innate immunity is the primary defense line for detecting and clearing intruding viruses. Upon viral entry into host cells, viral components will be recognized by PRRs resulting in IFN production. IFNs bind to their respective receptors and activate the JAK-STAT pathway [154], which will result in the production of proinflammatory cytokines and the activation of numerous downstream antiviral IFN-stimulated genes [155]. This process generates an antiviral state that hinders viral replication and provokes the adaptive immune response [155].
The JAK-STAT signaling pathway encompasses three members: tyrosine kinase-related receptors, JAKs, and STATs [156]. Tyrosine kinase-related receptors are transmembrane cytokine receptors that are classified according to the specific cytokine families to which they bind [157]. Recognition and binding of cytokines to their specific receptors result in molecular conformation changes of JAKs (JAK1, JAK2, JAK3, and Tyk2), causing its autophosphorylation or transphosphorylation [158]. Phosphorylated JAKs are activated and may initiate consequent phosphorylation of the receptors and successive docking and phosphorylation of STATs [159]. Phosphorylated STATs form homodimers or heterodimers that are translocated to the nucleus, bind to DNA sequences, and regulate immune-related gene transcription [160,161].
Since the activation of the JAK-STAT pathway stimulates the upregulation of immune-related genes against various infections, JAKs were often recognized as potential therapeutic targets. Several JAK inhibitors (JAKis) are permitted for different clinical indications, including the treatment of rheumatologic, dermatologic, hematologic, and gastrointestinal, along with an emergency authorization for Coronavirus disease 2019 (COVID-19) [162]. In addition, ruxolitinib, tofacitinib, baricitinib, and filgocitinib have been recently approved as antiviral agents against HIV, regardless of their original clinical indication [163].
7.5.4. Proto-Oncogene Tyrosine-Protein Kinase MER
The proto-oncogene tyrosine-protein kinase MER, encoded by the MERTK gene, belongs to the TAM (TYRO3, AXL, and MERTK) receptor protein tyrosine kinases that regulate several cellular processes, including cell proliferation, cell adhesion, blood clot stabilization, and regulation of inflammatory cytokine release [164]. The TAM receptors are generally expressed by macrophages, dendritic cells, and natural killer cells and are known to antagonize the host’s innate immune responses by a negative feedback loop that hinders the innate immune responses started by TLR and IFN-I signaling pathway [165]. Several reports have demonstrated that the AXL and TYRO3 of the TAM receptors could advance the infection of different viruses in numerous mechanisms [150]. Nonetheless, little is known about the role of MERTK in viral infections. Adomati et al. reported that deletion of MERTK in MERTK knockout mice suppressed the release of IL-10 and TGF-β, resulting in the abolition of innate energy, enhanced viral replication, and poor post-infection survival [166]. Recently, Zheng et al. demonstrated that MERTK associates with E2 viral protein to facilitate virus entry. After virus entry, MERTK suppresses mRNA expression of IFN-β and promotes viral infection [167]. Hence, this family of kinases could be exploited for the development of novel antiviral drugs.
7.6. Cyclin G Associated Kinase (GAK)
Cyclin G-associated kinase (GAK) is a serine/threonine kinase. It plays a major role in clathrin-mediated endocytosis, which is dependent on oligomeric clathrin and adaptor protein complexes [168]. For example, Hepatitis C virus entry and assembly are driven via GAK [169,170]. Other viruses that use this mechanism to infect cells include influenza, dengue, Hantaan virus, Junin arenavirus, HIV, Ebola, and Zika [171,172,173,174]. There are several studies reporting the investigational use of several compounds as antiviral agents via targeting the GAK pathway [170,175,176,177,178].
7.7. Leucine Rich Repeat Kinase 2 (LRRK2)
The leucine-rich repeat kinase 2 (LRRK2) protein is a large multidomain protein. It belongs to the ROCO superfamily defined by the presence of tandem Ras of complex (Roc) G-domain and a kinase domain linked by a carboxy-terminal of Roc (COR) sequence. It is expressed in several cells, including immune cells, microglia, and neurons [179,180]. LRRK2 activation is linked to Parkinson’s disease (PD) pathogenesis [181]. Also, HIV-1 infection results in HIV-1-associated neurocognitive disorders (HAND), despite the medication [182,183,184], which shares disease characteristics with other neurodegenerative diseases including PD. HAND is driven by the HIV-1 Tat protein since current medications fail to stop its synthesis [185,186]. Puccini et al. [187] have reported that LRRK2 is a modulator of neuroinflammation and neurotoxicity that results in neuronal damage and causes HAND. LRRK2 kinase inhibitor (LRRK2-IN-1) [188] decreased microglial activation during HAND [189], hence the potential use of LRRK2 inhibitors as antiviral agents is worth further investigation.
7.8. Interleukin-1 Receptor-Associated Kinase 4 (IRAK4)
IRAK4 is an important signal transducer downstream of interleukin (IL)-1 receptor (IL-1R), IL-18R, and TLRs. TLRs perform an essential function in triggering the host’s innate immune systems as they take part in the activation of transcription factors and the production of inflammatory cytokines [190].
Upon activation, TLRs commence two major pathways that are essentially reliant on the adaptors: myeloid differentiation primary response 88 (MyD88) and TIR domain-containing adapter-inducing interferon. Myddosome, a bulk oligomeric signaling complex enclosing molecules of MyD88 and members of the IRAK family, forms upon the initiation of MyD88-dependent signal transduction [191,192,193]. Myddosome formation results in IRAK4 autoactivation, which primarily activates IRAK1 followed by IRAK2 [194,195] and eventually activates the TNF receptor-associated factor 6 (TRAF6) [196]. Consequently, TRAF6 triggers the MAPK pathway to activate AP-1 and cAMP-response element–binding protein [197], leading to the production of potent proinflammatory cytokines and chemokines that elicit an acute inflammatory response [197]. Targeting this family of proteins will interfere with the host’s innate immune response toward viral infections.
7.9. Transforming Growth Factor-Beta (TGF-β)
TGF-β is one of the highly altered pathways in human cancers affecting cell proliferation, differentiation, apoptosis, and migration [198]. The TGF-β family of cytokines comprises three distinct isoforms, TGF-β1, TGF-β2, and TGF-β3, all of which are dual specificity kinases that bind to the same receptors but with different affinities [199]. TGF-β receptor type 2 (TGFBR2) is a homodimeric receptor that binds to the TGF-β ligand. Upon the ligand binding to the TGFBR2 extracellular domain, a conformational change results in the phosphorylation and activation of TGFBR1, which is a vital propagator of the TGF-β signaling pathway [199]. The Suppressor of Mothers Against Decapentaplegic (SMAD) family of proteins includes secondary effectors of the TGF-signaling pathway [200]. Phosphorylated receptor-specific SMADs associate with SMAD4 and other factors to form a complex that reaches the nucleus and modulates gene expression [201].
Whereas the TGF-β signaling pathway mediates numerous physiological effects, studies have demonstrated that both TGF-β receptors and SMADs crosstalk to other central signaling pathways in the cell, such as MAPK8, MAPK14, PIK3, MAPK1/3 and RAS [200,201]. Some of the downstream mediators of TGF-β signaling are vital cell-cycle checkpoint genes, involving CDKN1A, CDKN1B, and CDKN2B [202,203].
As this family of proteins is involved in gene expression and cell-cycle checkpoints, both of which may be interfered with by viruses for their own replication and transcription, targeting them for antiviral therapy is worth investigating.
7.10. Abelson Murine Leukemia (ABL) Viral Oncogene Homolog 1
The ABL family comprises Abelson murine leukemia viral oncogene homolog 1 (ABL1) and Abelson-Related Gene (ARG or ABL2). The encoded proteins have an SH1 catalytic domain and regulatory SH2 and SH3 domains [204]. ABL1 is ubiquitously expressed in the cytosol and nucleus with essential roles in T-cell receptor signaling, cell adhesion and division, gene transcription, and cell growth [204,205].
In HIV infection, ABL1 was shown to modulate actin to tempt cytoskeletal alterations that are crucial for pore formation, pore expansion, and eventually for HIV-1 invading the target T-cell [204]. In addition, during HIV infection, ABL1 was shown to increase tyrosine phosphorylation of RNApol-II, hence promoting the transcription of viral genetic material in invaded T-cells [205]. Hence, targeting ABL1 proteins may interfere with HIV entry and its gene transcription.
7.11. Bruton’s Tyrosine Kinase (BTK)
BTK is a TEC kinase with a complex role in n several biological processes including B-cell differentiation. Blocking the expression of BTK demonstrated a reduced maturation of B-cells and decreased serum levels of immunoglobulins in mice [206]. In addition, BTK takes part in different signaling pathways that modulate both innate and adaptive immune responses [207]. Several studies have investigated the key role of BTK in viral infections [208]. After the recognition of viral ssRNA in macrophages, TLRs initiate signaling through BTK-dependent activation of NF-κB leading to increased production of various inflammatory cytokines and chemokines, in addition to phagocytosis [208]. BTK would form another target to investigate novel antiviral drugs as well.
8. Kinases Inhibitors in Various Developmental Stages as Antiviral Drugs
In this section we provide an overview of important kinase inhibitors in various developmental stages as antiviral drugs. These inhibitors are listed in Table 4 and are given numbers from 1 to 45.
8.1. Mitogen-Activated Protein Kinases Inhibitors
Biological data showed that U0126 (1), a MEK1/2 inhibitor, exerts an 80% reduction in virus titers in Madin-Darby canine kidney (MDCK) cells at 50 μM and displays low values of a multiplicity of infection; a value of 1 and 0.0025 after 9 and 48 h, respectively [209,210,211]. Moreover, 1 exhibits antiviral activity against mutant ERK. Studies showed that if 1 does not impede viral RNA or protein synthesis, the viral ribonucleoprotein (RNP) sequesters in the nucleus and consequently suppresses viral production [211]; 1 inhibits influenza B virus (IBV) and IAV growth in MDCK cells, avoiding the evolution of resistant strains such as the H1N1 outbreak [210]. Further studies reveal that 1 inhibits astrovirus proliferation in human colorectal adenocarcinoma (Caco-2) cells and suppresses all viral life cycle stages [212]. Recent studies reported that 1 blocks murine coronavirus proliferation [213].
Biological investigation declared that SP600125 (2), a JNK-1 inhibitor, demonstrates a promising effect against Japanese encephalitis virus (JEV) [46,214,215,216]. Studies declared that 2 decreases the inflammatory cytokines generated by JEV-infected microglia (BV-2) and glioma (U251) cell lines [217]. Animal studies reported that 2 reduces the viral titers in infected mice brains and increases the survival rate [46]. Further studies declared that 2 impedes the growth of SARS-CoV in infected Vero E6 (African green monkey kidney epithelial) cells [218].
A kinome analysis experiment declared that SB203580 (3), a p38 inhibitor, and 1 exert an antiviral activity by 45% and 51% at 10 μM, respectively [219,220,221]. Furthermore, 1 exerts more antiviral activity in preinfected cells compared to its inoculation after 2 hours of infection [219]. Biological studies announced that 3 impaired the phosphorylation of heat shock protein 27 (HSP27), cAMP response element-binding protein (CREB), and eukaryotic translation initiation factor 4E (eIF4E) in SARS-CoV-2 infected cells [220].
A repurposing study revealed that the approved drugs selumetinib (4) and trametinib (5), MAPK inhibitors, exert a superior antiproliferative activity (>95%) against MERS-CoV [221,222,223]. Recent studies declared that 4 and 5 induce immune cells, decrease angiotensin-converting enzyme 2 (ACE2) encoding in human cells, and decrease cytokine expression in COVID-19 patients’ blood [224,225,226]. Such findings interrogate that 4 and 5 mitigate SARS-CoV-2 infection and control the body’s immune reactions [224,225,227]. Another repurposing study declared that papaverine (6), an alkaloid, inhibited both IAV and paramyxoviruses with an IC50s range of 2.0-36.4 μM [228,229]. Biological data reported that 6 inhibits MEK and ERK phosphorylation and sequentially impedes the release of viral RNP, while it does not affect viral RNA synthesis [230]. Data propose that 6 exhibits its inhibitory activity at the terminal stage of the viral life cycle [229,230]. Recent studies revealed that 6 suppresses the cytopathogenic effects of SARS-CoV-2 (EC50 = 1.1 μM) [231].
ATR-002 (7), a MEK inhibitor released by Atriva Therapeutics in Tübingen, Germany, shows antiviral activity [232,233,234,235]. Compound 7 is the active metabolite of CI-1040 that was disused due to low plasma concentration, and the development of CI-1040 was ceased despite the fact that it was shown to exert a better MEK inhibitory activity than that of CI-1040 with an IC50 value of 5.3 nM in contrast to 17 nM [236]. Studies claimed that 7 exerts a wide spectrum of antiviral activity in vitro and in vivo mouse models against diverse influenza virus strains of IAV and IBV [232]. In addition, studies reported that 7 exerts a safe, selective, and effective anti-SARS-CoV-2 activity [233,234]. Clinical studies showed that 7 terminates Phase I trials successfully in 70 healthy volunteers (Clinical Trial Identifier: NCT04385420) [31]. Compound 7 is undergoing a Phase II trial to probe its activity against COVID-19 (EU Clinical Trial Register Number: 2020-004206-59) [32].
8.2. Cyclin-Dependent Kinases Inhibitors
Palbociclib (Ibrance®) (8) [237,238], ribociclib (Kisqali®) (Ribotix®) (9) [239,240,241], and abemaciclib (Verzenio®) (10) [242] are FDA-approved CDK4/CDK6 inhibitors that are used therapeutically for breast cancer [37,243]. Biological studies showed that 8 suppresses HIV-1 (EC50 = 0.016 μM) and HSV-1 (EC50 = 0.020 μM) proliferation in vitro [244]. Studies declared that abemaciclib (10) exerts an in vitro inhibitory activity against SARS-CoV-2 with CC50 > 50 mM and IC50 = 6.6 mM [84]. Another study reported that 8 impairs HIV-1 reverse transcription and proliferation as well, as 8 decreases the activation of sterile α motif and HD domain-containing protein-1 (SAMHD1) in CD4+ T-lymphocytes, macrophages [245] and HSV-1 [244]. A recent study declared that CDK4/CDK6 inhibitors (8, 9, and 10) might impede SARS-CoV-2 infection in breast cancer females [241].
Studies declared that (R)-roscovitine (11), a purine-based inhibitor, disrupts DNA synthesis of human cytomegalovirus (HCMV) [82]. A further study reported that 11 exhibits antiproliferative activity against the human Polyomavirus JC virus (JCV) by impairing the transcription of JCV late genes suggesting that CDKs are required for JCV DNA transcription and replication [80]. Flavopiridol (12), a CDK1/CDK2/CDK4 inhibitor, suppresses HIV-1 proliferation by disrupting Tat-transcription [246,247,248]. A high-throughput screening revealed that 12 inhibits multiple IAV strains in human lung adenocarcinoma (A549) cells without cytotoxicity [83]. Recent reports proposed that 11 and 12 could be new therapeutic agents against COVID-19 based on their wide-spectrum antiviral activity and safety window as anticancer agents.
Biological screening studies declared that alsterpaullone (13), CDK1/CDK2 inhibitor, a purine analogue, exerts potent activity against HIV-1 [80,249]. Studies reported that 13 exerts a selective and dose-dependent antiproliferative activity in an infected chronic HIV-1 infection model (ACH2), acute HIV-1 infection model (OM10.1), latent HIV-1 infected T-lymphocytic model (J1.1) cells [80]. Studies reported that 13 exerts 100% inhibition against CDK2 in an infected cell at 0.5 mΜ [80]. Also, studies revealed that CDK2 expression and protein levels are suppressed in infected HIV-1 cells following 13 inoculations. And, studies reported that 11 and 13 demonstrate a synergism effect in infected human peripheral blood mononuclear cells confirming that targeting numerous CDKs might be promising to eradicate HIV-1 [80]. Studies reported that 13 might exert anti-SARS-CoV-2 activity [81].
A screening study disclosed FIT-039 (14) as a CDK9 competitive inhibitor (IC50 = 5.8 μM) [250]. Data showed that 14 exerts dose-dependent inhibitory activity against HSV-1 in infected cells [250]. Furthermore, 14 attenuates the phosphorylation of RNApol-II CTD in HSV-1 infected human embryonic kidney (HEK293) cells, hypothesizing that 14 might impede viral transcription. The biological investigation reported that 14 ameliorates skin injury and survival rates in mice models with HSV-1 infected skin. Data showed that 14 exerts antiproliferative activity against human adenovirus (HAdV) type 5, HSV-2, HBV, and HCMV [83]. Studies announced that dinaciclib (15), a CDK1/CDK2/CDK5/CDK9 inhibitor [251], exerts antiviral activity against SARS-CoV-2 [252]. Another investigation declared that a combination of 12 and 15 induces a synergistic effect [83]. Biological studies revealed that PHA-690509 (16), a CDK2 inhibitor, prevents Zika virus (ZIKV) infection in human glioblastoma (SNB-19) cells in a dose-dependent manner (IC50 = 1.72 μM) by inhibiting RNA replication [81].
8.3. Receptor Tyrosine Kinase Inhibitors
Studies reported that genistein (17), RTK inhibitor, and gefitinib (18), narrow spectrum EGFR inhibitor, impede IAV entry in A549 cells [253]. Additional studies showed that 17 inhibits the proliferation of HIV-1 [254], HSV-1 [255], arenavirus [147], and SARS-CoV [256,257]. Computational studies disclosed that 17 might be a possible SARS-CoV-2 inhibitor [258]. Biological studies showed that 18 impede SARS-CoV-2 proliferation in Caco-2 and Vero E6 cells [259]. Further studies declared that AG1478 (19), selective EGFR, hinders transmissible gastroenteritis virus (TGEV) uptake by porcine intestinal columnar epithelial cells (IPEC) [147]. However, studies showed that 19 exhibits adverse effects in rhinovirus and IAV-infected human bronchial epithelial (BEAS-2b) cells [147]. A recent hypothesis stated that EGFR inhibitors exemplified by gefitinib (18) and erlotinib (20), as well as platelet-derived growth factor receptor (PDGFR) inhibitors such as sorafenib (21), impede the development of lung fibrosis [112,260]. Though, the advantageous effect of such kinase inhibitors is not related to antiviral activity [149]. Studies revealed that 21 inhibits HBV gene expression by downregulating the JNK cascade, which forms FXR; a transcription protein that incites HBV growth and gene expression [261]. Further studies showed that 21 suppresses the proliferation of SARS-CoV-2 and DNA viruses in vitro [262,263]. Nintedanib (22), a fibroblast growth factor receptor (FGFR) inhibitor, reaches clinical trials as a treatment for lung fibrosis for moderate symptoms of COVID-19 [70].
Studies disclosed that R428 (23) impairs the kinase activity of the AXL receptor, a member of the RTKs family, and consequently inhibits viral entry and enhances IFN-1 signaling transduction [54]. Recent studies reported that suppressing the AXL receptor attenuates the viral infection in human lung adenocarcinoma (H1299) and lung epithelial cells [264]. Studies reported that AG879 (24), tropomyosin receptor kinase-A (TRK-A) and EGFR/HER2 inhibitor, and tyrphostin A9 (25), PDGFR inhibitor, block the replication of IAV [110]. Another study declared that 25 impedes TGEV replication in PK-15 or ST cells [55].
8.4. Numb-Associated Kinase Inhibitors
The human Numb-associated kinase family of Ser/Thr kinases harbors adaptor-associated kinase 1 (AAK1), BMP-2 inducible kinase (BMP2K/BIKE), GAK, and Ser/Thr kinase 16 (STK16) [265]. Biological data recorded that erlotinib (20) and sunitinib (26), AAK1 and GAK inhibitors, exert a wide-spectrum antiviral activity against RNA viruses descending from Arenaviridae, Flaviviridae, Filoviridae, Paramyxoviridae, and Togaviridae [108,266]. Another study revealed that 26 attenuates SARS-CoV-2, SARS-CoV, and MERS-CoV infection by blocking the adaptor-related protein complex 2 subunit mu 1 (AP2M1) phosphorylation [267]. Biological studies suggested that baricitinib (27), an effective AAK1 and GAK inhibitor, might be a potent therapy for COVID-19 [265,268,269]. Clinical studies reported that 27 reduces ICU admission and ameliorates signs and symptoms of COVID-19 patients [270]. Another study showed that 27 blocks of Janus kinase (JNK1/2) anticipate potential risks to take place [271]. Therefore, treatment with 27 is recommended for short intervals (7–14 days) to bypass opportunistic viral infections [73].
8.5. Src Kinases Inhibitors
Studies reported that saracatinib (29) exerts antiproliferative activity against MERS-CoV (EC50 = 2.9 μM and CC50 = 50 μM), human coronaviruses (HCoV-229E, EC50 = 2.4 μM) and (HCoV-OC43, EC50 = 5.1 μM) [204]. Further studies revealed a strong correlation between Src kinase suppression and Dengue virus (DENV) inhibitory profile [108]. Studies reported that 28 and 29 decreased DENV infection in Huh7.5.1, clone C6/36 (derived from Aedes albopictus mosquito), and Vero (derived from an African green monkey kidney epithelial) cells in a dose-dependent manner starting from 0.05–5 μM. Recent studies showed that 28 and 29 suppressed RNA replication of DENV through Fyn kinase [272]. Studies declared that bosutinib (30), an Src kinase inhibitor, inhibits SARS-CoV-2 entry with (EC50 = 2.45 µM) [273,274,275]. Studies reported that ponatinib (31) suppresses cytokines release responding to the SARS-CoV-2 N-terminal domain. Also, studies showed that 31 inhibits in vitro cytokine release in myeloid cells and in vivo in lung inflammation mouse models [276]. Biological investigations disclosed that vandetanib (32) inhibits SARS-CoV-2 in A549 cells expressing ACE2 (A549-hACE2) with an IC50 value of 0.79 μM as well as it suppresses HCoV-229E proliferation [277]. In addition, studies proposed that 32 impede COVID-19 cytokine storm in mice models [277]. The in vivo findings revealed that 32 decreases IL-6 and 10, TNF-α, and inflammatory infiltrate in infected mice lungs without decreasing viral titers [277].
Studies reported that dasatinib (28), an Src kinase inhibitor, impedes HIV-1 infection through the entry process at the membrane hemifusion stage and targets the viral restriction factor SAMHD1 [278]. Biological studies showed that inoculation of glioblastoma (U87/CD4/CCR5) cells with 300 nM of 28 reduces cellular fusion with HIV-1 by 93% [204]. Studies showed that a combination of 28 with sofosbuvir, a viral entry inhibitor, exhibits a synergistic activity against HCV. Such synergism effect potentiates HCV antiviral activity and improves IC50 values by 210-fold in human hepatoma (Huh7.5.1) cells [110]. Studies demonstrated that 28 exhibit antiviral activity against MERS-CoV and SARS-CoV [279,280].
8.6. Phosphatidylinositol-3-Phosphate-5 Kinase Inhibitors
Studies declared that apilimod (33), YM-201636 (34), and vacuolin-1 (35), phosphatidylinositol-3 phosphate-5 kinase (Pikfyve) inhibitors, inhibit filoviruses and Ebola virus (EBOV) [281], disturb the endosomal trafficking, and consequently block SARS-CoV-2 entry [282]. Further studies showed that 33 exerts antiviral activity against the SARS-CoV-2 and Omicron variants [283]. Compound 33 has reached Phase II clinical trials (NCT04446377) to probe its effect on the progress of COVID-19 [108].
8.7. G-Protein Coupled Receptor Kinase Inhibitors
Studies showed that methyl 5-[2-(5-nitro-2-furyl) vinyl]-2-furoate (36), G-protein coupled receptor kinase (GRK) inhibitor, blocks IAV entry and replication [47].
8.8. Abelson Tyrosine Kinase Inhibitors
Drug repurposing studies identified imatinib (37) and 28, ABL inhibitors, as anti- MERS-CoV and SARS-CoV agents, whereas nilotinib (38) exerts anti-SARS-CoV activity [280]. Recent studies showed that 35 inhibits SARS-CoV in Vero E6 (EC50 = 9.82 μM) [284]. Furthermore, 37 has reached Phase III clinical trials for COVID-19 (NCT04394416) [114]. Another study displayed that GNF-2 (39), an ABL inhibitor (IC50 = 138 nM), exerts anti-DENV activity by inhibiting viral entry and proliferation [285]. Studies confirmed that 39 targets both ABL (intracellular) and DENV E protein (extracellular). Data showed that 39 exerts efficient inhibitory activity against DENV E protein (IC50 = 5−25 μM) [285]. Further studies demonstrated that 37, 39, and GNF-5 (40) impede the fusion of the S protein of MERS-CoV and SARS-CoV and consequently block coronavirus entry [286,287].
8.9. Calcium/Calmodulin-Dependent Protein Kinase II Inhibitors
8.10. Ataxia Telangiectasia and Rad3-Related Kinase Inhibitors
Studies showed that berzosertib (42), ataxia telangiectasia, and rad3-related kinase (ATR) inhibitors exhibit antiviral activity in cells overexpressing ACE2, such as human embryonic kidney (HEK293T) and Vero-E6 cells [291]. ATR inhibitors causing DNA damage are suggested as a new therapeutic agent for SARS-CoV-2 [292]. Another study declared that 42 exerts anti-SARS-CoV-2 and MERS-CoV activity in an array of cell lines [292].
8.11. Adenosine 5′-Monophosphate-Activated Protein Kinase Inhibitors
Studies revealed that dorsomorphin (43), an adenosine 5′-monophosphate-activated protein kinase (AMPK) inhibitor, impairs EBOV proliferation in a dose-dependent manner in vitro in Vero cells and mitigates human macrophages infection that is EBOV targets in vivo [293,294,295]. Studies spotlighted the significance of 43 as anti-SARS-CoV-2[296].
8.12. Phosphatidylinositol 3-Kinase/Akt/mTOR Pathway Inhibitors
8.13. Sphingosine Kinase Inhibitors
Studies displayed that sphingosine kinase (SphK) inhibitors decrease SARS-CoV-2 replication and viral titer [300]. Studies showed that opaganib (45), SphK2 inhibitor (Ki = 9.8 μM) [301], mitigates inflammatory signaling and decreases viral growth in vitro in human bronchial tissue [302]. Compound 45 has reached Phase II/III clinical studies for COVID-19 pneumonia patients (NCT04467840) treatment [119].
9. Repurposing Old Kinase Inhibitors as Antivirals
Currently, most approved kinase inhibitors are clinically used for the treatment of cancer and inflammatory processes [38,303,304]. However, only 50 of the 500 known protein kinases encoded by the human genome have been targeted by drugs for cancer treatments [305,306,307]. The available kinase inhibitors provide a unique opportunity to expand therapeutic avenues beyond their current uses. Viral infections stand as a global issue challenging the health system and demanding successful therapeutic strategies to tackle this issue. Viruses are dependent on several host cellular proteins, and phosphorylation events appear to have a crucial role during viral progression at distinct stages of infection [221,308,309,310].
Repurposing the existing kinase inhibitors to combat viral infections may provide a better understanding of the kinases’ roles during a viral infection. In addition, since clinical data on toxicity, pharmacokinetics, and pharmacodynamics are already available for the approved inhibitors, repurposing them for a new clinical application would shorten the clinical pathway and limit the cost of their release as approved drugs.
10. Pros and Cons of Targeting Host Proteins for Antiviral Drug Discovery
Targeting host proteins via the use of HTA drugs is considered a new approach to the treatment of viral infections. In contrast to DAA drugs, HTA drugs have been shown to have broad spectrum of activity and low susceptibility to viral resistance [5,311]. This may be justified as host-cellular factors and/or mechanisms are used by viruses for their replication and transcription, as discussed earlier in this review, irrespective of their type, hence increasing the antiviral spectrum. In addition, viral replication errors, especially RNA viruses, contribute to resistance and negatively affect DAAs’ activity [5]. As host proteins are not subjected to viral genetic control and their resistance-contributing replication errors, along with low host genetic variability compared to viruses, HTA drugs are less likely to be ineffective against viruses [5,311].
On the other hand, HTA drugs have some disadvantages. HTA drugs target host cell cellular pathways required for survival, thus contributing to adverse effects and cytotoxicity [312,313]. In phase II clinical trials, alisporivir has been shown to cause hyperbilirubinemia and hypertriglyceridemia [312,314,315]. The use of HTA drugs may also cause the viruses to use alternate hosts or modify their affinity towards them [311]. Moreover, genetic polymorphism among patients may contribute to the reduced efficacy of HTA drugs to block their targets [314]. For example, suboptimal response, using alisoprivir against HCV, was observed in 10–15% of patients in the study [311]. Furthermore, poor correlation between in vitro, in vivo, or clinical trials responses to HTA drugs were observed, such as the use of VX-497 and statins for the treatment of HCV infection [316,317,318].
In relation to kinases, an in vitro study has investigated the use of specific kinase inhibitors targeting MEK and Src kinases to evaluate their activity against the proliferation of flavivirus infections inside BHK21 and Vero cells [221]. BHK21 and Vero cells are mammalian cells usually used to study the growth of viruses in laboratories [308,309]. The MEK inhibitors (trametinib and selumetinib) and Src inhibitors (saracatinib and bosutinib), which are being designed to treat cancers, showed antiviral activity against several flaviviruses (Zika virus, dengue virus, and yellow fever virus) [221]. However, the most effective and safest among them was trametinib. Safety was evaluated via the calculation of the selectivity index (SI), in which a ratio between the 50% cytotoxic concentration (inhibitors against cells only) and the 50% effective concentration (antiviral activity) is calculated. Trametinib had the highest SI compared to the other kinase inhibitorsv [221]. Hence, there is a risk of cytotoxicity when repurposing kinase inhibitors to target viral proliferation.
RV replication depends on phosphatidylinositol 4-kinase III beta (PI4KIIIβ) [310]. In in vitro and in vivo studies, the replication of RV has been prevented by the use of aminothiazole compounds, Compound 1 and Compound 2, which are PI4KIIIβ inhibitors [310]. However, the in vivo study has shown several adverse effects on mice, including muscle weakness and difficulty in breathing [310]. Although it was concluded that these adverse effects could possibly be species-specific and not relevant to humans, further in vivo studies are required to check the mechanism of toxicities and their relevance to humans [310].
Kinase inhibitors were also recently being investigated clinically in patients with COVID-19, as extensively summarized by Malekinejad et al. [319]. Clinical trials have studied, with some still ongoing, the use of JAK/STAT and BTK inhibitors for patients with COVID-19. Although a number of JAK/STAT kinase inhibitors, such as NCT05187793 and NCT04390061, resulted in clinical recovery and prevention of severe respiratory failure, respectively, others have shown adverse outcomes [319]. Among the adverse outcomes resulted due to the use of JAK/STAT and BTK inhibitors include hospitalization, the need for mechanical ventilation, extracorporeal membrane oxygenation (ECMO), respiratory failure, the need for supplemental oxygen, renal failure, disease progression, ICU admission, and death [319].
HTA drugs that target kinases, in particular, may halt viral life cycles and be curative if we are successful in detecting viral infections early on. Although most anticancer kinase inhibitors are not curative as tumors find escape routes, this may or may not be the case if they were to be used for the treatment of viral infections. Resistance to kinase inhibitors in cancer patients could be either primary (innate) or acquired. Innate resistance is caused by tumors that harbor specific genetic mutations, which cause cancer cells to become refractory to target inhibition. Acquired resistance, on the other hand, occurs due to complex, diverse mechanisms, including secondary target mutations that result in the acquisition of ‘bypass’ signaling pathways, the alteration of the cancer microenvironment, and increased expression of drug transporters, such as multidrug resistance proteins or brain cancer resistance proteins. These mechanisms, if they occur in infected host cells, may prevent antiviral kinase-inhibiting drugs from reaching clinically effective concentrations inside infected cells.
Targeted therapies that involve the use of enzyme inhibitors, such as kinase inhibitors, can alter normal physiological cell functions, thus altering normal cell behavioral phenotype via interacting with other pathways that are not intended to act upon [320,321,322]. These abnormal effects could arise either directly due to off-target interactions with additional proteins within the targeted pathway [323,324] or indirectly due to crosstalk between the targeted pathway and other pathways regulating a behavioral response [325]. In addition, off-target effects could arise due to retroactivity, in which a downstream disruption in a signaling pathway produces an upstream effect even though there is no association of negative feedback inhibition [326]. Although retroactivity does occur naturally in covalently modified cascades, the movement of signals within signaling pathways is usually in a downstream manner [327]. Kinase inhibitors have been shown to produce off-target effects via retroactivity [327]. Cell behaviors and off-target effects in the presence of kinase inhibitors or other drugs could be predicted computationally using a partial least square regression framework based on the signals of several key signaling pathways [328,329,330].
A summary of the pros and cons of targeting viral proteins or host proteins is presented in Table 5.
11. Drug Design Considerations for Kinase-Targeted Antivirals
11.1. Binding Sites
Kinase inhibitors can be classified into six main groups according to their binding sites (Table 6): (1) inhibitors that bind to the ATP pocket in the active conformation of a kinase and are known as type I inhibitors; (2) inhibitors that bind adjacent to the ATP pocket (adenine binding residues) of the unphosphorylated inactive conformation of kinases and are knowns as type II inhibitors; (3) non-ATP competitive inhibitors that bind within the cleft between the small and large lobes close to the ATP binding pocket and are known as type III inhibitors; (4) allosteric inhibitors that bind away from the ATP cleft and are knowns as type IV inhibitors; (5) agents that span two distinct regions of the protein kinase domain and are known as type V inhibitors; and (6) agents that form covalent bonds with their target kinases and are known as type VI inhibitors.
11.2. Physicochemical Properties
One feature of kinase inhibitors that have transformed clinical cancer care is their ability to penetrate the blood–brain barrier [305]. This could be advantageous for antivirals since the drug needs to overcome drug delivery barriers to reach its intracellular or intranuclear targets.
11.3. Kinase Inhibitor Combinations
Designing drug combinations of kinase inhibitors, as well as combinations of kinase inhibitors with other therapeutic modalities, holds the promise of identifying effective antiviral drugs by targeting signal transduction pathways at multiple points, which could impede the virus life cycle faster, in addition to better modulation of the immune response and inflammatory pathways that are turned on or off in response to viral infections.
12. Conclusions
HTAs have vast potential to treat or prevent viral infections in addition to combating emerging novel viruses. They are conceivable to develop universal antivirals with an increased antiviral spectrum and reduced resistance. Certainly, viruses exploit host proteins to enter host cells, replicate their genomes, synthesize their own viral proteins, and produce a progeny of infectious viral particles. This review provided a summary of host proteins involved in the life cycle of viruses and provided an important update on the antiviral drug development pipeline with a special focus on kinase-targeting antivirals detailing their role in signal transduction pathways and providing drug discovery intelligence on drugs at different developmental stages. We also discussed repurposing approved kinase inhibitors, which have been clinically used for the treatment of cancers and inflammation, to combat viral diseases. However, more progress is needed to ensure that R&D efforts continue to identify novel kinase ligands or repurpose already existing kinase-targeting drugs to treat or prevent existing and emerging viral diseases. Indeed, the potential for developing novel antiviral kinase inhibitors is massive and will continue to be a major growth area for modulating infectious and auto-immune diseases in the future.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/v15020568/s1, Table S1: Enrichment results using 51 higher validity CDDI kinase biomarkers for viral diseases.
Author Contributions
Conceptualization, R.H.; software, R.H.; formal analysis, R.H.; investigation, R.H., D.A.S., O.H.A. and S.B.; resources, R.H., D.A.S., O.H.A. and S.B.; biomarker data curation, R.H.; writing—original draft preparation, R.H., D.A.S., O.H.A., R.K. and S.B.; writing—review and editing, R.H., D.A.S., O.H.A. and S.B.; visualization, R.H.; supervision, R.H.; project administration, R.H.; funding acquisition, R.H. and D.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
R.H. and D.A.S. acknowledge support from the Deanship of Scientific Research at Al-Zaytoonah University of Jordan (Grant number 2020-2019/17/03).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data supporting the reported results can be requested by contacting the corresponding author directly.
Acknowledgments
R.H. and D.A.S. acknowledge funding from the Deanship of Scientific Research at Al-Zaytoonah University of Jordan (Grant number 2020-2019/17/03).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| (+) | positive-sense |
| (−) | negative-sense |
| APC | Antigen-presenting cell |
| BC | Baltimore Class |
| CDDI | Cortellis Drug Discovery Intelligence |
| CDKis | CDK inhibitors |
| CDKs | Cyclin-dependent kinases |
| GAK | Cyclin G associated kinase (GAK) |
| DAA | Direct acting antiviral |
| ds | double-stranded |
| FDA | Food and Drug Administration |
| FDR | False discovery rate |
| HAND | HIV-1-associated neurocognitive disorders |
| HTA | Host targeted antiviral |
| ICTV | International Committee on Taxonomy of Viruses |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MAPK | Mitogen-activated protein kinase |
| MLKL | Mixed lineage kinase-like protein |
| PKC | Protein kinase C |
| PKR | Protein kinase RNA-activated |
| PRR | Pattern recognition receptors |
| R&D | Research and development |
| RdRp | RNA-dependent RNA polymerase |
| RIG-1 | Retinoic acid-inducible gene 1 |
| RIPK | Receptor interacting protein kinase |
| RLR | Retinoic acid-inducible gene 1(RIG-I)-like receptor |
| RT | reverse transcribing |
| RTK | Receptor tyrosine kinase |
| RTz | Reverse transcriptase enzyme |
| ss | single-stranded |
| TLR | Toll-like receptor |
| UAD | Under active development |
References
- Li, G.; De Clercq, E. Overview of Antiviral Drug Discovery and Development: ViralVersusHost Targets. In Antiviral Discovery for Highly Pathogenic Emerging Viruses; The Royal Society of Chemistry: London, UK, 2021; pp. 1–27. [Google Scholar]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. An Updated Review on SARS-CoV-2 Main Proteinase (M(Pro)): Protein Structure and Small-Molecule Inhibitors. Curr. Top. Med. Chem. 2021, 21, 442–460. [Google Scholar] [CrossRef]
- Mahmoud, I.S.; Jarrar, Y.B. Targeting the intestinal TMPRSS2 protease to prevent SARS-CoV-2 entry into enterocytes-prospects and challenges. Mol. Biol. Rep. 2021, 48, 4667–4675. [Google Scholar] [CrossRef]
- Strasfeld, L.; Chou, S. Antiviral drug resistance: Mechanisms and clinical implications. Infect. Dis. Clin. N. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Li, Z. Medicinal chemistry strategies toward host targeting antiviral agents. Med. Res. Rev. 2020, 40, 1519–1557. [Google Scholar] [CrossRef]
- Ikhmais, B.; Hammad, A.M.; Al-Qerem, W.; Abusara, O.H.; Ling, J. Conducting COVID-19-Related Research in Jordan: Are We Ready? Disaster Med. Public Health Prep. 2022, 16, 967–974. [Google Scholar] [CrossRef]
- Schang, L.M.; St Vincent, M.R.; Lacasse, J.J. Five years of progress on cyclin-dependent kinases and other cellular proteins as potential targets for antiviral drugs. Antivir. Chem. Chemother. 2006, 17, 293–320. [Google Scholar] [CrossRef]
- Richman, D.D.; Nathanson, N. Antiviral Therapy. In Viral Pathogenesis; Elsevier: Amesterdam, The Netherlands, 2016; pp. 271–287. [Google Scholar]
- Richman, D.D. Antiviral drug resistance. Antiviral Res. 2006, 71, 117–121. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. An Updated Review on Betacoronavirus Viral Entry Inhibitors: Learning from Past Discoveries to Advance COVID-19 Drug Discovery. Curr. Top. Med. Chem. 2021, 21, 571–596. [Google Scholar] [CrossRef]
- Prasad, B.V.; Schmid, M.F. Principles of virus structural organization. Adv. Exp. Med. Biol. 2012, 726, 17–47. [Google Scholar] [CrossRef]
- Khirfan, F.; Jarrar, Y.; Al-Qirim, T.; Goh, K.W.; Jarrar, Q.; Ardianto, C.; Awad, M.; Al-Ameer, H.J.; Al-Awaida, W.; Moshawih, S.; et al. Analgesics Induce Alterations in the Expression of SARS-CoV-2 Entry and Arachidonic-Acid-Metabolizing Genes in the Mouse Lungs. Pharmaceuticals 2022, 15, 696. [Google Scholar] [CrossRef]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of virology. Handb. Clin. Neurol. 2014, 123, 45–66. [Google Scholar] [CrossRef]
- Liu, Y.; Dyall-Smith, M.; Oksanen, H.M. ICTV Virus Taxonomy Profile: Pleolipoviridae 2022. J. Gen. Virol. 2022, 103. [Google Scholar] [CrossRef]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2011; Volume 9. [Google Scholar]
- ICTV. About Virus Taxonomic Classification. Available online: https://ictv.global/taxonomy/about (accessed on 2 December 2022).
- ICTV. Current ICTV Taxonomy Release. Available online: https://ictv.global/taxonomy (accessed on 2 December 2022).
- ICTV. ICTV Report Chapters by Genome. Available online: https://ictv.global/report/genome (accessed on 2 December 2022).
- Baltimore, D. Expression of animal virus genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef]
- Baltimore, D. Viral genetic systems. Trans. N. Y. Acad. Sci. 1971, 33, 327–332. [Google Scholar] [CrossRef]
- Baltimore, D. The strategy of RNA viruses. Harvey Lect. 1974, 70, 57–74. [Google Scholar]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e0005321. [Google Scholar] [CrossRef]
- Bardaweel, S.K.; Hajjo, R.; Sabbah, D.A. Sitagliptin: A potential drug for the treatment of COVID-19? Acta Pharm. 2021, 71, 175–184. [Google Scholar] [CrossRef]
- Plemper, R.K. Cell entry of enveloped viruses. Curr. Opin. Virol. 2011, 1, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Melikyan, G.B.; Barnard, R.J.; Abrahamyan, L.G.; Mothes, W.; Young, J.A. Imaging individual retroviral fusion events: From hemifusion to pore formation and growth. Proc. Natl. Acad. Sci. USA 2005, 102, 8728–8733. [Google Scholar] [CrossRef] [Green Version]
- Cosset, F.L.; Lavillette, D. Cell entry of enveloped viruses. Adv. Genet. 2011, 73, 121–183. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol. Biol. Cell 2000, 11, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Chernomordik, L.V.; Frolov, V.A.; Leikina, E.; Bronk, P.; Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: Restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol. 1998, 140, 1369–1382. [Google Scholar] [CrossRef]
- Borrego-Diaz, E.; Peeples, M.E.; Markosyan, R.M.; Melikyan, G.B.; Cohen, F.S. Completion of trimeric hairpin formation of influenza virus hemagglutinin promotes fusion pore opening and enlargement. Virology 2003, 316, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Moyer, C.L.; Nemerow, G.R. Viral weapons of membrane destruction: Variable modes of membrane penetration by non-enveloped viruses. Curr. Opin. Virol. 2011, 1, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, M.; Greber, U.F. Uncoating of non-enveloped viruses. Curr. Opin. Virol. 2013, 3, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Kilcher, S.; Mercer, J. DNA virus uncoating. Virology 2015, 479–480, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Galibert, F.; Chen, T.N.; Mandart, E. Nucleotide sequence of a cloned woodchuck hepatitis virus genome: Comparison with the hepatitis B virus sequence. J. Virol. 1982, 41, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef]
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. Viruses 2018, 55–82. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. Rev. 2014, 78, 278–303. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Narayanan, S.; Yang, D.-H. Chapter 9—CDK Inhibitors as Sensitizing Agents for Cancer Chemotherapy. In Protein Kinase Inhibitors as Sensitizing Agents for Chemotherapy; Chen, Z.-S., Yang, D.-H., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 4, pp. 125–149. [Google Scholar]
- Hajjo, R.; Sabbah, D.A.; Bardaweel, S.K. Chemocentric Informatics Analysis: Dexamethasone Versus Combination Therapy for COVID-19. ACS Omega 2020, 5, 29765–29779. [Google Scholar] [CrossRef]
- Hajjo, R.; Sabbah, D.A.; Bardaweel, S.K.; Tropsha, A. Shedding the Light on Post-Vaccine Myocarditis and Pericarditis in COVID-19 and Non-COVID-19 Vaccine Recipients. Vaccines 2021, 9, 1186. [Google Scholar] [CrossRef]
- Schwartz, P.A.; Murray, B.W. Protein kinase biochemistry and drug discovery. Bioorg. Chem. 2011, 39, 192–210. [Google Scholar] [CrossRef]
- García-Cárceles, J.; Caballero, E.; Gil, C.; Martínez, A. Kinase Inhibitors as Underexplored Antiviral Agents. J. Med. Chem. 2022, 65, 935–954. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases--the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G. Quantitative, wide-spectrum kinase profiling in live cells for assessing the effect of cellular ATP on target engagement. Cell Chem. Biol. 2018, 25, 206–214.e211. [Google Scholar] [CrossRef] [Green Version]
- Lahiry, P.; Torkamani, A.; Schork, N.J.; Hegele, R.A. Kinase mutations in human disease: Interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, H.; He, W.; Zhu, B.; Zhou, D.; Chen, Z.; Ashraf, U.; Wei, Y.; Liu, Z.; Fu, Z.F.; et al. Quantitative phosphoproteomic analysis identifies the critical role of JNK1 in neuroinflammation induced by Japanese encephalitis virus. Sci. Signal. 2016, 9, ra98. [Google Scholar] [CrossRef]
- Yángüez, E.; Hunziker, A.; Dobay, M.P.; Yildiz, S.; Schading, S.; Elshina, E.; Karakus, U.; Gehrig, P.; Grossmann, J.; Dijkman, R.; et al. Phosphoproteomic-based kinase profiling early in influenza virus infection identifies GRK2 as antiviral drug target. Nat. Commun. 2018, 9, 3679. [Google Scholar] [CrossRef] [Green Version]
- Schang, L.M. The cell cycle, cyclin-dependent kinases, and viral infections: New horizons and unexpected connections. Prog. Cell Cycle Res. 2003, 5, 103–124. [Google Scholar]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. 2014, 1170, 165–227. [Google Scholar] [CrossRef]
- Gutierrez-Chamorro, L.; Felip, E.; Ezeonwumelu, I.J.; Margelí, M.; Ballana, E. Cyclin-dependent Kinases as Emerging Targets for Developing Novel Antiviral Therapeutics. Trends Microbiol. 2021, 29, 836–848. [Google Scholar] [CrossRef]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 2001, re2. [Google Scholar] [CrossRef]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interferon Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Dong, W.; Xie, W.; Liu, Y.; Sui, B.; Zhang, H.; Liu, L.; Tan, Y.; Tong, X.; Fu, Z.F.; Yin, P.; et al. Receptor tyrosine kinase inhibitors block proliferation of TGEV mainly through p38 mitogen-activated protein kinase pathways. Antivir. Res. 2020, 173, 104651. [Google Scholar] [CrossRef]
- Kumar, R.; Khandelwal, N.; Thachamvally, R.; Tripathi, B.N.; Barua, S.; Kashyap, S.K.; Maherchandani, S.; Kumar, N. Role of MAPK/MNK1 signaling in virus replication. Virus Res. 2018, 253, 48–61. [Google Scholar] [CrossRef]
- Katagiri, K.; Hattori, M.; Minato, N.; Kinashi, T. Rap1 functions as a key regulator of T-cell and antigen-presenting cell interactions and modulates T-cell responses. Mol. Cell Biol. 2002, 22, 1001–1015. [Google Scholar] [CrossRef] [Green Version]
- Jaśkiewicz, A.; Pająk, B.; Orzechowski, A. The Many Faces of Rap1 GTPase. Int. J. Mol. Sci. 2018, 19, 2848. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Mita, M.M. Activated ras signaling pathways and reovirus oncolysis: An update on the mechanism of preferential reovirus replication in cancer cells. Front. Oncol. 2014, 4, 167. [Google Scholar] [CrossRef] [Green Version]
- Kleinehr, J.; Wilden, J.J.; Boergeling, Y.; Ludwig, S.; Hrincius, E.R. Metabolic Modifications by Common Respiratory Viruses and Their Potential as New Antiviral Targets. Viruses 2021, 13, 2068. [Google Scholar] [CrossRef]
- Appelberg, S.; Gupta, S.; Svensson Akusjärvi, S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M.; et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K.; Zhong, H.A. Phosphatidylinositol 3-kinase (PI3K) inhibitors: A recent update on inhibitor design and clinical trials (2016–2020). Expert Opin. Ther. Pat. 2021, 31, 877–892. [Google Scholar] [CrossRef]
- Garbe, C.; Zibert, J. Ingenol mebutate gel, 0.015% repeat use for Multiple Actinic Keratoses on face and scalp: A review of the phase 3 clinical study rationale and design. J. Dtsch. Dermatol. Ges. 2013, 131. [Google Scholar]
- Solares, A.M.; Santana, A.; Baladrón, I.; Valenzuela, C.; González, C.A.; Díaz, A.; Castillo, D.; Ramos, T.; Gómez, R.; Alonso, D.F. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 2009, 9, 146. [Google Scholar] [CrossRef] [Green Version]
- Soriano-García, J.; López-Díaz, A.; Solares-Asteasuainzarra, M.; Baladrón-Castrillo, I.; Batista-Albuerne, N.; García-García, I.; González-Méndez, L.; Perera-Negrín, Y.; Valenzuela-Silva, C.; Pedro, A. Pharmacological and safety evaluation of CIGB-300, a casein kinase 2 inhibitor peptide, administered intralesionally to patients with cervical cancer stage IB2/II. J. Cancer Res. Ther. 2013, 1, 163–173. [Google Scholar]
- Lin, C.-Y.; Chu, J.C.-H.-C. Management of Toxicities of Targeted Therapies. In IASLC Thoracic Oncology, 2nd ed.; Pass, H.I., Ball, D., Scagliotti, G.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 490–500.e493. [Google Scholar]
- ClinicalTrials.gov, Identifier: NCT04480957. Ascending Dose Study of Investigational SARS-CoV-2 Vaccine ARCT-021 in Healthy Adult Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT04480957 (accessed on 21 November 2022).
- Khanna, N.; Dalby, R.; Tan, M.; Arnold, S.; Stern, J.; Frazer, N. Phase I/II clinical safety studies of terameprocol vaginal ointment. Gynecol. Oncol. 2007, 107, 554–562. [Google Scholar] [CrossRef]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A comprehensive review of the global efforts on COVID-19 vaccine development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef]
- Dambach, D.M. Potential adverse effects associated with inhibition of p38α/β MAP kinases. Curr. Top. Med. Chem. 2005, 5, 929–939. [Google Scholar] [CrossRef]
- Favalli, E.G.; Biggioggero, M.; Maioli, G.; Caporali, R. Baricitinib for COVID-19: A suitable treatment? Lancet Infect. Dis. 2020, 20, 1012–1013. [Google Scholar] [CrossRef]
- Pavletich, N.P. Mechanisms of cyclin-dependent kinase regulation: Structures of Cdks, their cyclin activators, and Cip and INK4 inhibitors. J. Mol. Biol. 1999, 287, 821–828. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey, P.D.; Tong, L.; Pavletich, N.P. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev. 2000, 14, 3115–3125. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking Bad: How Viruses Subvert the Cell Cycle. Front. Cell Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [Green Version]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to Peptide inhibitors. Cancers 2015, 7, 179. [Google Scholar] [CrossRef] [Green Version]
- Guendel, I.; Agbottah, E.T.; Kehn-Hall, K.; Kashanchi, F. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res. Ther. 2010, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Perwitasari, O.; Yan, X.; O’Donnell, J.; Johnson, S.; Tripp, R.A. Repurposing Kinase Inhibitors as Antiviral Agents to Control Influenza A Virus Replication. Assay. Drug Dev. Technol. 2015, 13, 638–649. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Okuyama-Dobashi, K.; Murakami, S.; Chen, W.; Okamoto, T.; Ueda, K.; Hosoya, T.; Matsuura, Y.; Ryo, A.; Tanaka, Y.; et al. Inhibitory effect of CDK9 inhibitor FIT-039 on hepatitis B virus propagation. Antiviral Res. 2016, 133, 156–164. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob. Agents Chemother. 2020, 64, e00819-20. [Google Scholar] [CrossRef]
- Gupta, A.; Zhou, H.X. Profiling SARS-CoV-2 Main Protease (M(PRO)) Binding to Repurposed Drugs Using Molecular Dynamics Simulations in Classical and Neural Network-Trained Force Fields. ACS Comb. Sci. 2020, 22, 826–832. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Eby, H.; Henkel, N.D.; Creeden, J.; Imami, A.; Asah, S.; Zhang, X.; Wu, X.; Alnafisah, R.; Taylor, R.T.; et al. Identification of new drug treatments to combat COVID19: A signature-based approach using iLINCS. Res. Sq. 2020, rs.3, rs-25643. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.-M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Bannwarth, S.; Gatignol, A. HIV-1 TAR RNA: The target of molecular interactions between the virus and its host. Curr. HIV Res. 2005, 3, 61–71. [Google Scholar] [CrossRef]
- Price, D.H. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol. Cell Biol. 2000, 20, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Hajjo, R.; Sabbah, D.A.; Tropsha, A. Analyzing the Systems Biology Effects of COVID-19 mRNA Vaccines to Assess Their Safety and Putative Side Effects. Pathogens 2022, 11, 743. [Google Scholar] [CrossRef]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Rose, B.A.; Force, T.; Wang, Y. Mitogen-activated protein kinase signaling in the heart: Angels versus demons in a heart-breaking tale. Physiol. Rev. 2010, 90, 1507–1546. [Google Scholar] [CrossRef] [Green Version]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Ceballos-Olvera, I.; Chávez-Salinas, S.; Medina, F.; Ludert, J.E.; del Angel, R.M. JNK phosphorylation, induced during dengue virus infection, is important for viral infection and requires the presence of cholesterol. Virology 2010, 396, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Hou, X.; Li, X.; Peng, H.; Shi, M.; Jiang, Q.; Liu, X.; Ji, Y.; Yao, Y.; He, C.; et al. Differential gene expressions of the MAPK signaling pathway in enterovirus 71-infected rhabdomyosarcoma cells. Braz. J. Infect. Dis. 2013, 17, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [Green Version]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Gaur, P.; Munjhal, A.; Lal, S.K. Influenza virus and cell signaling pathways. Med. Sci. Monit. 2011, 17, Ra148–Ra154. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Pleschka, S.; Planz, O.; Wolff, T. Ringing the alarm bells: Signalling and apoptosis in influenza virus infected cells. Cell Microbiol. 2006, 8, 375–386. [Google Scholar] [CrossRef]
- Rodríguez, M.E.; Brunetti, J.E.; Wachsman, M.B.; Scolaro, L.A.; Castilla, V. Raf/MEK/ERK pathway activation is required for Junín virus replication. J. Gen. Virol. 2014, 95, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.A.; Silva, P.N.; Pereira, A.C.; De Sousa, L.P.; Ferreira, P.C.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Identifier: NCT00404248. Tetra-O-Methyl Nordihydroguaiaretic Acid in Treating Patients with Recurrent High-Grade Glioma. Available online: https://clinicaltrials.gov/ct2/show/NCT00404248 (accessed on 2 February 2023).
- Le Sommer, C.; Barrows, N.J.; Bradrick, S.S.; Pearson, J.L.; Garcia-Blanco, M.A. G protein-coupled receptor kinase 2 promotes flaviviridae entry and replication. PLoS Negl. Trop. Dis. 2012, 6, e1820. [Google Scholar] [CrossRef] [Green Version]
- Hemonnot, B.; Cartier, C.; Gay, B.; Rebuffat, S.; Bardy, M.; Devaux, C.; Boyer, V.; Briant, L. The host cell MAP kinase ERK-2 regulates viral assembly and release by phosphorylating the p6gag protein of HIV-1. J. Biol. Chem. 2004, 279, 32426–32434. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.J.; Yang, P.L. c-Src protein kinase inhibitors block assembly and maturation of dengue virus. Proc. Natl. Acad. Sci. USA 2007, 104, 3520–3525. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, A.J.; Medigeshi, G.R.; Meyers, H.L.; DeFilippis, V.; Früh, K.; Briese, T.; Lipkin, W.I.; Nelson, J.A. The Src family kinase c-Yes is required for maturation of West Nile virus particles. J. Virol. 2005, 79, 11943–11951. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Liang, Y.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication. J. Virol. 2011, 85, 2818–2827. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Z.; Bradel-Tretheway, B.; Sumagin, S.; Bidlack, J.M.; Dewhurst, S. The human herpesvirus 6 G protein-coupled receptor homolog U51 positively regulates virus replication and enhances cell-cell fusion in vitro. J. Virol. 2005, 79, 11914–11924. [Google Scholar] [CrossRef] [Green Version]
- Purcaru, O.S.; Artene, S.A.; Barcan, E.; Silosi, C.A.; Stanciu, I.; Danoiu, S.; Tudorache, S.; Tataranu, L.G.; Dricu, A. The Interference between SARS-CoV-2 and Tyrosine Kinase Receptor Signaling in Cancer. Int. J. Mol. Sci. 2021, 22, 4830. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- GeneCards-The Human Gene Database. KRR1 Gene—KRR1 Small Subunit Processome Component Homolog. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=KRR1 (accessed on 8 December 2022).
- National Library of Medicine—National Center for Biotechnology Information. KRR1 Small Subunit Processome Component Homolog [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/11103 (accessed on 9 December 2022).
- Battiste, J.L.; Mao, H.; Rao, N.S.; Tan, R.; Muhandiram, D.R.; Kay, L.E.; Frankel, A.D.; Williamson, J.R. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science 1996, 273, 1547–1551. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.; Chen, L.; Buettner, J.A.; Hudson, D.; Frankel, A.D. RNA recognition by an isolated alpha helix. Cell 1993, 73, 1031–1040. [Google Scholar] [CrossRef]
- Malim, M.H.; Hauber, J.; Le, S.Y.; Maizel, J.V.; Cullen, B.R. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 1989, 338, 254–257. [Google Scholar] [CrossRef]
- Heaphy, S.; Dingwall, C.; Ernberg, I.; Gait, M.J.; Green, S.M.; Karn, J.; Lowe, A.D.; Singh, M.; Skinner, M.A. HIV-1 regulator of virion expression (Rev) protein binds to an RNA stem-loop structure located within the Rev response element region. Cell 1990, 60, 685–693. [Google Scholar] [CrossRef]
- Daelemans, D.; Costes, S.V.; Cho, E.H.; Erwin-Cohen, R.A.; Lockett, S.; Pavlakis, G.N. In vivo HIV-1 Rev multimerization in the nucleolus and cytoplasm identified by fluorescence resonance energy transfer. J. Biol. Chem. 2004, 279, 50167–50175. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Byrn, R.; Groopman, J.; Baltimore, D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: Evidence for differential gene expression. J. Virol. 1989, 63, 3708–3713. [Google Scholar] [CrossRef] [Green Version]
- Klotman, M.E.; Kim, S.; Buchbinder, A.; DeRossi, A.; Baltimore, D.; Wong-Staal, F. Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 5011–5015. [Google Scholar] [CrossRef] [Green Version]
- Arizala, J.A.C.; Takahashi, M.; Burnett, J.C.; Ouellet, D.L.; Li, H.; Rossi, J.J. Nucleolar Localization of HIV-1 Rev Is Required, Yet Insufficient for Production of Infectious Viral Particles. AIDS Res. Hum. Retrovir. 2018, 34, 961–981. [Google Scholar] [CrossRef] [Green Version]
- Gou, H.; Bian, Z.; Cai, R.; Chu, P.; Song, S.; Li, Y.; Jiang, Z.; Zhang, K.; Yang, D.; Li, C. RIPK3-Dependent Necroptosis Limits PRV Replication in PK-15 Cells. Front. Microbiol. 2021, 12, 664353. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Nailwal, H.; Chan, F.K. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019, 26, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Wu, X.N.; Yang, Z.H.; Wang, X.K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Viral modulation of programmed necrosis. Curr. Opin. Virol. 2013, 3, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Schock, S.N.; Chandra, N.V.; Sun, Y.; Irie, T.; Kitagawa, Y.; Gotoh, B.; Coscoy, L.; Winoto, A. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ. 2017, 24, 615–625. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef]
- Chang, W.; Lim, J.G.; Hellström, I.; Gentry, L.E. Characterization of vaccinia virus growth factor biosynthetic pathway with an antipeptide antiserum. J. Virol. 1988, 62, 1080–1083. [Google Scholar] [CrossRef] [Green Version]
- King, C.S.; Cooper, J.A.; Moss, B.; Twardzik, D.R. Vaccinia virus growth factor stimulates tyrosine protein kinase activity of A431 cell epidermal growth factor receptors. Mol. Cell Biol. 1986, 6, 332–336. [Google Scholar] [CrossRef]
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304. [Google Scholar] [CrossRef] [Green Version]
- Stroobant, P.; Rice, A.P.; Gullick, W.J.; Cheng, D.J.; Kerr, I.M.; Waterfield, M.D. Purification and characterization of vaccinia virus growth factor. Cell 1985, 42, 383–393. [Google Scholar] [CrossRef]
- Sanderson, C.M.; Way, M.; Smith, G.L. Virus-induced cell motility. J. Virol. 1998, 72, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Beerli, C.; Yakimovich, A.; Kilcher, S.; Reynoso, G.V.; Fläschner, G.; Müller, D.J.; Hickman, H.D.; Mercer, J. Vaccinia virus hijacks EGFR signalling to enhance virus spread through rapid and directed infected cell motility. Nat. Microbiol. 2019, 4, 216–225. [Google Scholar] [CrossRef]
- Langhammer, S.; Koban, R.; Yue, C.; Ellerbrok, H. Inhibition of poxvirus spreading by the anti-tumor drug Gefitinib (Iressa). Antiviral Res. 2011, 89, 64–70. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [Green Version]
- Ueki, I.F.; Min-Oo, G.; Kalinowski, A.; Ballon-Landa, E.; Lanier, L.L.; Nadel, J.A.; Koff, J.L. Respiratory virus–induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J. Exp. Med. 2013, 210, 1929–1936. [Google Scholar] [CrossRef]
- Wang, W.; Wu, J.; Zhang, X.; Hao, C.; Zhao, X.; Jiao, G.; Shan, X.; Tai, W.; Yu, G. Inhibition of Influenza A Virus Infection by Fucoidan Targeting Viral Neuraminidase and Cellular EGFR Pathway. Sci. Rep. 2017, 7, 40760. [Google Scholar] [CrossRef] [Green Version]
- Currier, M.G.; Lee, S.; Stobart, C.C.; Hotard, A.L.; Villenave, R.; Meng, J.; Pretto, C.D.; Shields, M.D.; Nguyen, M.T.; Todd, S.O.; et al. EGFR Interacts with the Fusion Protein of Respiratory Syncytial Virus Strain 2-20 and Mediates Infection and Mucin Expression. PLoS Pathog. 2016, 12, e1005622. [Google Scholar] [CrossRef] [Green Version]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef]
- Chua, B.A.; Ngo, J.A.; Situ, K.; Morizono, K. Roles of phosphatidylserine exposed on the viral envelope and cell membrane in HIV-1 replication. Cell Commun. Signal. 2019, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Richard, A.S.; Jackson, C.B.; Ojha, A.; Choe, H. Phosphatidylethanolamine and Phosphatidylserine Synergize To Enhance GAS6/AXL-Mediated Virus Infection and Efferocytosis. J. Virol. 2020, 95, e02079-20. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and non-canonical aspects of JAK–STAT signaling: Lessons from interferons for cytokine responses. Front. immunol. 2017, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [Green Version]
- Bazan, J.F. Shared architecture of hormone binding domains in type I and II interferon receptors. Cell 1990, 61, 753–754. [Google Scholar] [CrossRef]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.-Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [Green Version]
- Harrison, A.R.; Moseley, G.W. The dynamic interface of viruses with STATs. J. Virol. 2020, 94, e00856-20. [Google Scholar] [CrossRef]
- Nguyen, N.V.; Tran, J.T.; Sanchez, D.J. HIV blocks Type I IFN signaling through disruption of STAT1 phosphorylation. Innate Immun. 2018, 24, 490–500. [Google Scholar] [CrossRef] [Green Version]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- De Armas, L.R.; Gavegnano, C.; Pallikkuth, S.; Rinaldi, S.; Pan, L.; Battivelli, E.; Verdin, E.; Younis, R.T.; Pahwa, R.; Williams, S.L. The Effect of JAK1/2 Inhibitors on HIV Reservoir Using Primary Lymphoid Cell Model of HIV Latency. Front. Immunol. 2021, 12, 720697. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Zagórska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Nassar, M.; Tabib, Y.; Capucha, T.; Mizraji, G.; Nir, T.; Pevsner-Fischer, M.; Zilberman-Schapira, G.; Heyman, O.; Nussbaum, G.; Bercovier, H. GAS6 is a key homeostatic immunological regulator of host–commensal interactions in the oral mucosa. Proc. Natl. Acad. Sci. USA 2017, 114, E337–E346. [Google Scholar] [CrossRef] [Green Version]
- Adomati, T.; Cham, L.B.; Hamdan, T.A.; Bhat, H.; Duhan, V.; Li, F.; Ali, M.; Lang, E.; Huang, A.; Naser, E. Dead cells induce innate anergy via mertk after acute viral infection. Cell Rep. 2020, 30, 3671–3681.e3675. [Google Scholar] [CrossRef]
- Zheng, G.; Li, L.-F.; Zhang, Y.; Qu, L.; Wang, W.; Li, M.; Yu, S.; Zhou, M.; Luo, Y.; Sun, Y. MERTK is a host factor that promotes classical swine fever virus entry and antagonizes innate immune response in PK-15 cells. Emerg. Microbes Infect. 2020, 9, 571–581. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Neveu, G.; Barouch-Bentov, R.; Ziv-Av, A.; Gerber, D.; Jacob, Y.; Einav, S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 2012, 8, e1002845. [Google Scholar] [CrossRef] [Green Version]
- Kovackova, S.; Chang, L.; Bekerman, E.; Neveu, G.; Barouch-Bentov, R.; Chaikuad, A.; Heroven, C.; Šála, M.; De Jonghe, S.; Knapp, S.; et al. Selective Inhibitors of Cyclin G Associated Kinase (GAK) as Anti-Hepatitis C Agents. J. Med. Chem. 2015, 58, 3393–3410. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [Green Version]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204 (Suppl. 3), S957–S967. [Google Scholar] [CrossRef] [Green Version]
- Sikka, V.; Chattu, V.K.; Popli, R.K.; Galwankar, S.C.; Kelkar, D.; Sawicki, S.G.; Stawicki, S.P.; Papadimos, T.J. The Emergence of Zika Virus as a Global Health Security Threat: A Review and a Consensus Statement of the INDUSEM Joint working Group (JWG). J. Glob. Infect. Dis. 2016, 8, 3–15. [Google Scholar] [CrossRef]
- Sloan, R.D.; Kuhl, B.D.; Mesplède, T.; Münch, J.; Donahue, D.A.; Wainberg, M.A. Productive entry of HIV-1 during cell-to-cell transmission via dynamin-dependent endocytosis. J. Virol. 2013, 87, 8110–8123. [Google Scholar] [CrossRef] [Green Version]
- Wouters, R.; Pu, S.Y.; Froeyen, M.; Lescrinier, E.; Einav, S.; Herdewijn, P.; De Jonghe, S. Cyclin G-associated kinase (GAK) affinity and antiviral activity studies of a series of 3-C-substituted isothiazolo [4,3-b]pyridines. Eur. J. Med. Chem. 2019, 163, 256–265. [Google Scholar] [CrossRef]
- Pu, S.Y.; Wouters, R.; Schor, S.; Rozenski, J.; Barouch-Bentov, R.; Prugar, L.I.; O’Brien, C.M.; Brannan, J.M.; Dye, J.M.; Herdewijn, P.; et al. Optimization of Isothiazolo [4,3-b]pyridine-Based Inhibitors of Cyclin G Associated Kinase (GAK) with Broad-Spectrum Antiviral Activity. J. Med. Chem. 2018, 61, 6178–6192. [Google Scholar] [CrossRef]
- Martinez-Gualda, B.; Pu, S.Y.; Froeyen, M.; Herdewijn, P.; Einav, S.; De Jonghe, S. Structure-activity relationship study of the pyridine moiety of isothiazolo [4,3-b]pyridines as antiviral agents targeting cyclin G-associated kinase. Bioorg. Med. Chem. 2020, 28, 115188. [Google Scholar] [CrossRef]
- Li, J.; Kovackova, S.; Pu, S.; Rozenski, J.; De Jonghe, S.; Einav, S.; Herdewijn, P. Isothiazolo [4,3-b]pyridines as inhibitors of cyclin G associated kinase: Synthesis, structure-activity relationship studies and antiviral activity. Medchemcomm 2015, 6, 1666–1672. [Google Scholar] [CrossRef]
- Melrose, H.; Lincoln, S.; Tyndall, G.; Dickson, D.; Farrer, M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience 2006, 139, 791–794. [Google Scholar] [CrossRef]
- Seol, W. Biochemical and molecular features of LRRK2 and its pathophysiological roles in Parkinson’s disease. BMB Rep. 2010, 43, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, P.N.; Chen, S.G.; Wilson-Delfosse, A.L. Leucine-rich repeat kinase 2 (LRRK2): A key player in the pathogenesis of Parkinson’s disease. J. Neurosci. Res. 2009, 87, 1283–1295. [Google Scholar] [CrossRef] [Green Version]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Cross, H.M.; Combrinck, M.I.; Joska, J.A. HIV-associated neurocognitive disorders: Antiretroviral regimen, central nervous system penetration effectiveness, and cognitive outcomes. S. Afr. Med. J. 2013, 103, 758–762. [Google Scholar] [CrossRef] [Green Version]
- Hopcroft, L.; Bester, L.; Clement, D.; Quigley, A.; Sachdeva, M.; Rourke, S.B.; Nixon, S.A. “My body’s a 50 year-old but my brain is definitely an 85 year-old”: Exploring the experiences of men ageing with HIV-associated neurocognitive challenges. J. Int. AIDS Soc. 2013, 16, 18506. [Google Scholar] [CrossRef]
- Kure, K.; Lyman, W.D.; Weidenheim, K.M.; Dickson, D.W. Cellular localization of an HIV-1 antigen in subacute AIDS encephalitis using an improved double-labeling immunohistochemical method. Am. J. Pathol. 1990, 136, 1085–1092. [Google Scholar]
- Anderson, E.; Zink, W.; Xiong, H.; Gendelman, H.E. HIV-1-associated dementia: A metabolic encephalopathy perpetrated by virus-infected and immune-competent mononuclear phagocytes. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. 2), S43–S54. [Google Scholar] [CrossRef]
- Puccini, J.M.; Marker, D.F.; Fitzgerald, T.; Barbieri, J.; Kim, C.S.; Miller-Rhodes, P.; Lu, S.M.; Dewhurst, S.; Gelbard, H.A. Leucine-rich repeat kinase 2 modulates neuroinflammation and neurotoxicity in models of human immunodeficiency virus 1-associated neurocognitive disorders. J. Neurosci. 2015, 35, 5271–5283. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Dzamko, N.; Prescott, A.; Davies, P.; Liu, Q.; Yang, Q.; Lee, J.D.; Patricelli, M.P.; Nomanbhoy, T.K.; Alessi, D.R.; et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011, 7, 203–205. [Google Scholar] [CrossRef] [Green Version]
- Marker, D.F.; Puccini, J.M.; Mockus, T.E.; Barbieri, J.; Lu, S.M.; Gelbard, H.A. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J. Neuroinflamm. 2012, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- De Nardo, D. Toll-like receptors: Activation, signalling and transcriptional modulation. Cytokine 2015, 74, 181–189. [Google Scholar] [CrossRef]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88–IRAK4–IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Qiao, Q.; Ferrao, R.; Shen, C.; Hatcher, J.M.; Buhrlage, S.J.; Gray, N.S.; Wu, H. Crystal structure of human IRAK1. Proc. Natl. Acad. Sci. USA 2017, 114, 13507–13512. [Google Scholar] [CrossRef] [Green Version]
- Kawagoe, T.; Sato, S.; Matsushita, K.; Kato, H.; Matsui, K.; Kumagai, Y.; Saitoh, T.; Kawai, T.; Takeuchi, O.; Akira, S. Sequential control of Toll-like receptor–dependent responses by IRAK1 and IRAK2. Nat. Immunol. 2008, 9, 684–691. [Google Scholar] [CrossRef]
- Ferrao, R.; Zhou, H.; Shan, Y.; Liu, Q.; Li, Q.; Shaw, D.E.; Li, X.; Wu, H. IRAK4 dimerization and trans-autophosphorylation are induced by Myddosome assembly. Mol. Cell 2014, 55, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Ye, H.; Arron, J.R.; Lamothe, B.; Cirilli, M.; Kobayashi, T.; Shevde, N.K.; Segal, D.; Dzivenu, O.K.; Vologodskaia, M.; Yim, M. Distinct molecular mechanism for initiating TRAF6 signalling. Nature 2002, 418, 443–447. [Google Scholar] [CrossRef]
- Qian, Y.; Commane, M.; Ninomiya-Tsuji, J.; Matsumoto, K.; Li, X. IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFκB. J. Biol. Chem. 2001, 276, 41661–41667. [Google Scholar] [CrossRef] [Green Version]
- Akhurst, R.J. TGFβ signaling in health and disease. Nat. Genet. 2004, 36, 790–792. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Gal, A.; Sjöblom, T.; Fedorova, L.; Imreh, S.; Beug, H.; Moustakas, A. Sustained TGFβ exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008, 27, 1218–1230. [Google Scholar] [CrossRef] [Green Version]
- Dillon, R.; White, D.; Muller, W. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.-F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [Green Version]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massagué, J. Transforming growth factor β signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef] [Green Version]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef] [Green Version]
- Bermejo, M.; López-Huertas, M.R.; García-Pérez, J.; Climent, N.; Descours, B.; Ambrosioni, J.; Mateos, E.; Rodríguez-Mora, S.; Rus-Bercial, L.; Benkirane, M. Dasatinib inhibits HIV-1 replication through the interference of SAMHD1 phosphorylation in CD4+ T cells. Biochem. Pharmacol. 2016, 106, 30–45. [Google Scholar] [CrossRef]
- Khan, W.N.; Alt, F.W.; Gerstein, R.M.; Malynn, B.A.; Larsson, I.; Rathbun, G.; Davidson, L.; Müller, S.; Kantor, A.B.; Herzenberg, L.A. Defective B cell development and function in Btk-deficient mice. Immunity 1995, 3, 283–299. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Zhou, C.; Guo, H.; Zheng, M. Effects of BTK signalling in pathogenic microorganism infections. J. Cell Mol. Med. 2019, 23, 6522–6529. [Google Scholar] [CrossRef] [Green Version]
- Page, T.H.; Urbaniak, A.M.; Santo, A.I.E.; Danks, L.; Smallie, T.; Williams, L.M.; Horwood, N.J. Bruton’s tyrosine kinase regulates TLR7/8-induced TNF transcription via nuclear factor-κB recruitment. Biochem. Biophys. Res. Commun. 2018, 499, 260–266. [Google Scholar] [CrossRef]
- Droebner, K.; Pleschka, S.; Ludwig, S.; Planz, O. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. 2011, 92, 195–203. [Google Scholar] [CrossRef]
- Battcock, S.M.; Collier, T.W.; Zu, D.; Hirasawa, K. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J. Virol. 2006, 80, 4422–4430. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, S.; Wolff, T.; Ehrhardt, C.; Wurzer, W.J.; Reinhardt, J.; Planz, O.; Pleschka, S. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Moser, L.A.; Schultz-Cherry, S. Suppression of astrovirus replication by an ERK1/2 inhibitor. J. Virol. 2008, 82, 7475–7482. [Google Scholar] [CrossRef] [Green Version]
- Ghasemnejad-Berenji, M.; Pashapour, S. SARS-CoV-2 and the possible role of Raf/MEK/ERK pathway in viral survival: Is this a potential therapeutic strategy for COVID-19? Pharmacology 2021, 106, 119–122. [Google Scholar] [CrossRef]
- Tang, Y.; Yang, G.; Li, Y.; Wang, M.; Li, G.; Hu, Y. Protective effects of SP600125 on mice infected with H1N1 influenza A virus. Arch. Virol. 2021, 166, 2151–2158. [Google Scholar] [CrossRef]
- Pereira, A.C.; Soares-Martins, J.A.; Leite, F.G.; Da Cruz, A.F.; Torres, A.A.; Souto-Padrón, T.; Kroon, E.G.; Ferreira, P.C.; Bonjardim, C.A. SP600125 inhibits Orthopoxviruses replication in a JNK1/2 -independent manner: Implication as a potential antipoxviral. Antiviral Res. 2012, 93, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Sreekanth, G.P.; Chuncharunee, A.; Cheunsuchon, B.; Noisakran, S.; Yenchitsomanus, P.T.; Limjindaporn, T. JNK1/2 inhibitor reduces dengue virus-induced liver injury. Antiviral Res. 2017, 141, 7–18. [Google Scholar] [CrossRef]
- He, Z.; Chen, X.; Fu, M.; Tang, J.; Li, X.; Cao, H.; Wang, Y.; Zheng, S.J. Inhibition of fowl adenovirus serotype 4 replication in Leghorn male hepatoma cells by SP600125 via blocking JNK MAPK pathway. Vet. Microbiol. 2019, 228, 45–52. [Google Scholar] [CrossRef]
- Ye, Z.; Wong, C.; Li, P.; Xie, Y. The role of SARS-CoV protein, ORF-6, in the induction of host cell death. Hong Kong Med. J. 2010, 16, 22. [Google Scholar]
- Zhang, R.; Hristovski, D.; Schutte, D.; Kastrin, A.; Fiszman, M.; Kilicoglu, H. Drug repurposing for COVID-19 via knowledge graph completion. J. Biomed. Inform. 2021, 115, 103696. [Google Scholar] [CrossRef]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef]
- Valencia, H.J.; de Aguiar, M.; Costa, M.A.; Mendonça, D.C.; Reis, E.V.; Arias, N.E.C.; Drumond, B.P.; Bonjardim, C.A. Evaluation of kinase inhibitors as potential therapeutics for flavivirus infections. Arch. Virol. 2021, 166, 1433–1438. [Google Scholar] [CrossRef]
- Haasbach, E.; Hartmayer, C.; Planz, O. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro. Antiviral Res. 2013, 98, 319–324. [Google Scholar] [CrossRef]
- Schräder, T.; Dudek, S.E.; Schreiber, A.; Ehrhardt, C.; Planz, O.; Ludwig, S. The clinically approved MEK inhibitor Trametinib efficiently blocks influenza A virus propagation and cytokine expression. Antiviral Res. 2018, 157, 80–92. [Google Scholar] [CrossRef]
- Zhou, L.; Huntington, K.; Zhang, S.; Carlsen, L.; So, E.Y.; Parker, C.; Sahin, I.; Safran, H.; Kamle, S.; Lee, C.M.; et al. MEK inhibitors reduce cellular expression of ACE2, pERK, pRb while stimulating NK-mediated cytotoxicity and attenuating inflammatory cytokines relevant to SARS-CoV-2 infection. Oncotarget 2020, 11, 4201–4223. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Imami, A.; Eby, H.; Henkel, N.D.; Creeden, J.F.; Asah, S.; Zhang, X.; Wu, X.; Alnafisah, R.; Taylor, R.T.; et al. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci. Rep. 2021, 11, 4495. [Google Scholar] [CrossRef]
- Sendama, W. L1000 connectivity map interrogation identifies candidate drugs for repurposing as SARS-CoV-2 antiviral therapies. Comput. Struct. Biotechnol. J. 2020, 18, 3947–3949. [Google Scholar] [CrossRef]
- Shen, Z.; Halberg, A.; Fong, J.Y.; Guo, J.; Song, G.; Louie, B.; Luedtke, G.R.; Visuthikraisee, V.; Protter, A.A.; Koh, X.; et al. Elucidating host cell response pathways and repurposing therapeutics for SARS-CoV-2 and other coronaviruses. Sci. Rep. 2022, 12, 18811. [Google Scholar] [CrossRef]
- Aggarwal, M.; Leser, G.P.; Lamb, R.A. Repurposing Papaverine as an Antiviral Agent against Influenza Viruses and Paramyxoviruses. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, B.L.; de Oliveira, N.C.; Ritter, M.R.; Tonin, F.S.; Melo, E.B.; Sanches, A.C.C.; Fernandez-Llimos, F.; Petruco, M.V.; de Mello, J.C.P.; Chierrito, D.; et al. The naturally-derived alkaloids as a potential treatment for COVID-19: A scoping review. Phytother. Res. 2022, 36, 2686–2709. [Google Scholar] [CrossRef]
- Ellinger, B.; Bojkova, D.; Zaliani, A.; Cinatl, J.; Claussen, C.; Westhaus, S.; Keminer, O.; Reinshagen, J.; Kuzikov, M.; Wolf, M.; et al. A SARS-CoV-2 cytopathicity dataset generated by high-content screening of a large drug repurposing collection. Sci. Data 2021, 8, 70. [Google Scholar] [CrossRef]
- Valipour, M.; Irannejad, H.; Emami, S. Papaverine, a promising therapeutic agent for the treatment of COVID-19 patients with underlying cardiovascular diseases (CVDs). Drug Dev. Res. 2022, 83, 1246–1250. [Google Scholar] [CrossRef]
- Laure, M.; Hamza, H.; Koch-Heier, J.; Quernheim, M.; Müller, C.; Schreiber, A.; Müller, G.; Pleschka, S.; Ludwig, S.; Planz, O. Antiviral efficacy against influenza virus and pharmacokinetic analysis of a novel MEK-inhibitor, ATR-002, in cell culture and in the mouse model. Antivir. Res. 2020, 178, 104806. [Google Scholar] [CrossRef]
- Schreiber, A.; Ambrosy, B.; Planz, O.; Schloer, S.; Rescher, U.; Ludwig, S. The MEK1/2 Inhibitor ATR-002 (Zapnometinib) Synergistically Potentiates the Antiviral Effect of Direct-Acting Anti-SARS-CoV-2 Drugs. Pharmaceutics 2022, 14, 1776. [Google Scholar] [CrossRef]
- Schreiber, A.; Viemann, D.; Schöning, J.; Schloer, S.; Mecate Zambrano, A.; Brunotte, L.; Faist, A.; Schöfbänker, M.; Hrincius, E.; Hoffmann, H.; et al. The MEK1/2-inhibitor ATR-002 efficiently blocks SARS-CoV-2 propagation and alleviates pro-inflammatory cytokine/chemokine responses. Cell Mol. Life Sci. 2022, 79, 65. [Google Scholar] [CrossRef]
- Hamza, H.; Shehata, M.M.; Mostafa, A.; Pleschka, S.; Planz, O. Improved in vitro Efficacy of Baloxavir Marboxil Against Influenza A Virus Infection by Combination Treatment With the MEK Inhibitor ATR-002. Front. Microbiol. 2021, 12, 611958. [Google Scholar] [CrossRef]
- Bruchhagen, C.; Jarick, M.; Mewis, C.; Hertlein, T.; Niemann, S.; Ohlsen, K.; Peters, G.; Planz, O.; Ludwig, S.; Ehrhardt, C. Metabolic conversion of CI-1040 turns a cellular MEK-inhibitor into an antibacterial compound. Sci. Rep. 2018, 8, 9114. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Tiedt, R.; Loo, A.; Horn, T.; Delach, S.; Kovats, S.; Haas, K.; Engstler, B.S.; Cao, A.; Pinzon-Ortiz, M.; et al. Correction: The potent and selective cyclin-dependent kinases 4 and 6 inhibitor ribociclib (LEE011) is a versatile combination partner in preclinical cancer models. Oncotarget 2020, 11, 1289. [Google Scholar] [CrossRef]
- Barba, M.; Krasniqi, E.; Pizzuti, L.; Mazzotta, M.; Marinelli, D.; Giuliano, G.; Di Liso, F.S.; Cappuzzo, F.; Landi, L.; Tomao, S.; et al. COVID-19 risk in breast cancer patients receiving CDK4/6 inhibitors: Literature data and a monocentric experience. Breast J. 2021, 27, 359–362. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [CrossRef]
- Badia, R.; Angulo, G.; Riveira-Muñoz, E.; Pujantell, M.; Puig, T.; Ramirez, C.; Torres-Torronteras, J.; Martí, R.; Pauls, E.; Clotet, B.; et al. Inhibition of herpes simplex virus type 1 by the CDK6 inhibitor PD-0332991 (palbociclib) through the control of SAMHD1. J. Antimicrob. Chemother. 2016, 71, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Pauls, E.; Badia, R.; Torres-Torronteras, J.; Ruiz, A.; Permanyer, M.; Riveira-Muñoz, E.; Clotet, B.; Marti, R.; Ballana, E.; Esté, J.A. Palbociclib, a selective inhibitor of cyclin-dependent kinase4/6, blocks HIV-1 reverse transcription through the control of sterile α motif and HD domain-containing protein-1 (SAMHD1) activity. AIDS 2014, 28, 2213–2222. [Google Scholar] [CrossRef]
- Chao, S.H.; Fujinaga, K.; Marion, J.E.; Taube, R.; Sausville, E.A.; Senderowicz, A.M.; Peterlin, B.M.; Price, D.H. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J. Biol. Chem. 2000, 275, 28345–28348. [Google Scholar] [CrossRef] [Green Version]
- Biglione, S.; Byers, S.A.; Price, J.P.; Nguyen, V.T.; Bensaude, O.; Price, D.H.; Maury, W. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex. Retrovirology 2007, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Ghosh, A.; Nathans, R.S.; Sharova, N.; O’Brien, S.; Cao, H.; Stevenson, M.; Rana, T.M. Identification of flavopiridol analogues that selectively inhibit positive transcription elongation factor (P-TEFb) and block HIV-1 replication. Chembiochem 2009, 10, 2072–2080. [Google Scholar] [CrossRef]
- Watanabe, T.; Sato, Y.; Masud, H.; Takayama, M.; Matsuda, H.; Hara, Y.; Yanagi, Y.; Yoshida, M.; Goshima, F.; Murata, T.; et al. Antitumor activity of cyclin-dependent kinase inhibitor alsterpaullone in Epstein-Barr virus-associated lymphoproliferative disorders. Cancer Sci. 2020, 111, 279–287. [Google Scholar] [CrossRef]
- Yamamoto, M.; Onogi, H.; Kii, I.; Yoshida, S.; Iida, K.; Sakai, H.; Abe, M.; Tsubota, T.; Ito, N.; Hosoya, T.; et al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses. J. Clin. Invest. 2014, 124, 3479–3488. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e619. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef] [Green Version]
- Stantchev, T.S.; Markovic, I.; Telford, W.G.; Clouse, K.A.; Broder, C.C. The tyrosine kinase inhibitor genistein blocks HIV-1 infection in primary human macrophages. Virus Res. 2007, 123, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Yura, Y.; Yoshida, H.; Sato, M. Inhibition of herpes simplex virus replication by genistein, an inhibitor of protein-tyrosine kinase. Arch. Virol. 1993, 132, 451–461. [Google Scholar] [CrossRef]
- Andres, A.; Donovan, S.M.; Kuhlenschmidt, M.S. Soy isoflavones and virus infections. J. Nutr. Biochem. 2009, 20, 563–569. [Google Scholar] [CrossRef]
- Marmitt, D.J.; Goettert, M.I.; Rempel, C. Compounds of plants with activity against SARS-CoV-2 targets. Expert Rev. Clin. Pharmacol. 2021, 14, 623–633. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, L.; Gao, J.; Zhang, B.; Liu, X.; Yang, N.; Liu, X.; Liu, X.; Cheng, Y. Discovery of genistein derivatives as potential SARS-CoV-2 main protease inhibitors by virtual screening, molecular dynamics simulations and ADMET analysis. Front. Pharmacol. 2022, 13, 961154. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Liang, R.; Wang, S.J.; Ye, Z.W.; Wang, T.Y.; Chen, M.; Liu, J.; Na, L.; Yang, Y.L.; Yang, Y.B.; et al. SARS-CoV-2 hijacks macropinocytosis to facilitate its entry and promote viral spike-mediated cell-to-cell fusion. J. Biol. Chem. 2022, 298, 102511. [Google Scholar] [CrossRef]
- Venkataraman, T.; Frieman, M.B. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antiviral Res. 2017, 143, 142–150. [Google Scholar] [CrossRef]
- Cho, H.K.; Kim, J.R.; Kim, S.Y.; Kyaw, Y.Y.; Win, A.A.; Cheong, J.H. Sorafenib suppresses hepatitis B virus gene expression via inhibiting JNK pathway. Hepatoma Res. 2015, 1, 97–103. [Google Scholar]
- Meyer, B.; Chiaravalli, J.; Gellenoncourt, S.; Brownridge, P.; Bryne, D.P.; Daly, L.A.; Grauslys, A.; Walter, M.; Agou, F.; Chakrabarti, L.A.; et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [Google Scholar] [CrossRef]
- Sun, L.; Li, P.; Ju, X.; Rao, J.; Huang, W.; Ren, L.; Zhang, S.; Xiong, T.; Xu, K.; Zhou, X.; et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 2021, 184, 1865–1883.e1820. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef]
- Sorrell, F.J.; Szklarz, M.; Abdul Azeez, K.R.; Elkins, J.M.; Knapp, S. Family-wide Structural Analysis of Human Numb-Associated Protein Kinases. Structure 2016, 24, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.S.; Jung, E.; Kim, M.; Baric, R.S.; Go, Y.Y. Saracatinib Inhibits Middle East Respiratory Syndrome-Coronavirus Replication In Vitro. Viruses 2018, 10, 283. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.G.; Tang, D.J.; Hua, Z.; Wang, Z.; An, J. Sunitinib reduces the infection of SARS-CoV, MERS-CoV and SARS-CoV-2 partially by inhibiting AP2M1 phosphorylation. Cell Discov. 2020, 6, 71. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.N.; Pino, M.; Boddapati, A.K.; Viox, E.G.; Starke, C.E.; Upadhyay, A.A.; Gumber, S.; Nekorchuk, M.; Busman-Sahay, K.; Strongin, Z.; et al. Baricitinib treatment resolves lower-airway macrophage inflammation and neutrophil recruitment in SARS-CoV-2-infected rhesus macaques. Cell 2021, 184, 460–475.e421. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Matarrese, D.; Nicastri, E.; Stobbione, P.; Goletti, D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J. Infect. 2020, 81, 318–356. [Google Scholar] [CrossRef]
- Praveen, D.; Puvvada, R.C.; M, V.A. Janus kinase inhibitor baricitinib is not an ideal option for management of COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105967. [Google Scholar] [CrossRef]
- de Wispelaere, M.; LaCroix, A.J.; Yang, P.L. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J. Virol. 2013, 87, 7367–7381. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Pei, R.-J.; Li, H.; Ma, X.-N.; Zhou, Y.; Zhu, F.-H.; He, P.-L.; Tang, W.; Zhang, Y.-C.; Xiong, J.; et al. Identification of SARS-CoV-2 entry inhibitors among already approved drugs. Acta Pharmacol. Sin. 2021, 42, 1347–1353. [Google Scholar] [CrossRef]
- Galimberti, S.; Baldini, C.; Baratè, C.; Ricci, F.; Balducci, S.; Grassi, S.; Ferro, F.; Buda, G.; Benedetti, E.; Fazzi, R.; et al. The CoV-2 outbreak: How hematologists could help to fight Covid-19. Pharmacol. Res. 2020, 157, 104866. [Google Scholar] [CrossRef]
- Sencanski, M.; Perovic, V.; Milicevic, J.; Todorovic, T.; Prodanovic, R.; Veljkovic, V.; Paessler, S.; Glisic, S. Identification of SARS-CoV-2 Papain-like Protease (PLpro) Inhibitors Using Combined Computational Approach. ChemistryOpen 2022, 11, e202100248. [Google Scholar] [CrossRef]
- Chan, M.; Vijay, S.; McNevin, J.; McElrath, M.J.; Holland, E.C.; Gujral, T.S. Machine learning identifies molecular regulators and therapeutics for targeting SARS-CoV2-induced cytokine release. Mol. Syst. Biol. 2021, 17, e10426. [Google Scholar] [CrossRef]
- Puhl, A.C.; Gomes, G.F.; Damasceno, S.; Fritch, E.J.; Levi, J.A.; Johnson, N.J.; Scholle, F.; Premkumar, L.; Hurst, B.L.; Lee-Montiel, F. Vandetanib Blocks the Cytokine Storm in SARS-CoV-2-Infected Mice. ACS Omega 2022, 7, 31935–31944. [Google Scholar] [CrossRef]
- Puigdomènech, I.; Casartelli, N.; Porrot, F.; Schwartz, O. SAMHD1 restricts HIV-1 cell-to-cell transmission and limits immune detection in monocyte-derived dendritic cells. J. Virol. 2013, 87, 2846–2856. [Google Scholar] [CrossRef] [Green Version]
- Başcı, S.; Ata, N.; Altuntaş, F.; Yiğenoğlu, T.N.; Dal, M.S.; Korkmaz, S.; Namdaroğlu, S.; Baştürk, A.; Hacıbekiroğlu, T.; Doğu, M.H.; et al. Outcome of COVID-19 in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitors. J. Oncol. Pharm. Pract. 2020, 26, 1676–1682. [Google Scholar] [CrossRef]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G., Jr.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.A.; Dyall, J.; Hoenen, T.; Barnes, A.B.; Zhou, H.; Liang, J.Y.; Michelotti, J.; Dewey, W.H.; DeWald, L.E.; Bennett, R.S.; et al. The phosphatidylinositol-3-phosphate 5-kinase inhibitor apilimod blocks filoviral entry and infection. PLoS Negl. Trop. Dis. 2017, 11, e0005540. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.L.; Chou, Y.Y.; Rothlauf, P.W.; Liu, Z.; Soh, T.K.; Cureton, D.; Case, J.B.; Chen, R.E.; Diamond, M.S.; Whelan, S.P.J.; et al. Inhibition of PIKfyve kinase prevents infection by Zaire ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 20803–20813. [Google Scholar] [CrossRef]
- Su, J.; Zheng, J.; Huang, W.; Zhang, Y.; Lv, C.; Zhang, B.; Jiang, L.; Cheng, T.; Yuan, Q.; Xia, N.; et al. PIKfyve inhibitors against SARS-CoV-2 and its variants including Omicron. Signal Transduct. Target. Ther. 2022, 7, 167. [Google Scholar] [CrossRef]
- Assaad, H.S.; Assaad-Khalil, S. Imatinib a Tyrosine Kinase Inhibitor: A potential treatment for SARS-COV-2 induced pneumonia. Alex. J. Med. 2020, 56, 68–72. [Google Scholar] [CrossRef]
- Clark, M.J.; Miduturu, C.; Schmidt, A.G.; Zhu, X.; Pitts, J.D.; Wang, J.; Potisopon, S.; Zhang, J.; Wojciechowski, A.; Hann Chu, J.J.; et al. GNF-2 Inhibits Dengue Virus by Targeting Abl Kinases and the Viral E Protein. Cell Chem. Biol. 2016, 23, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Sisk, J.M.; Frieman, M.B.; Machamer, C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018, 99, 619–630. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Habtemariam, S.; Clementi, E.; Berindan-Neagoe, I.; Cismaru, C.A.; Rasekhian, M.; Banach, M.; Izadi, M.; Bagheri, M.; Bagheri, M.S.; et al. Lessons learned from SARS-CoV and MERS-CoV: FDA-approved Abelson tyrosine-protein kinase 2 inhibitors may help us combat SARS-CoV-2. Arch. Med. Sci. 2020, 16, 519–521. [Google Scholar] [CrossRef]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-C.; Simanjuntak, Y.; Chu, L.-W.; Ping, Y.-H.; Lee, Y.-L.; Lin, Y.-L.; Li, W.-S. Benzenesulfonamide Derivatives as Calcium/Calmodulin-Dependent Protein Kinase Inhibitors and Antiviral Agents against Dengue and Zika Virus Infections. J. Med. Chem. 2020, 63, 1313–1327. [Google Scholar] [CrossRef]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Li, H.; Milton, S.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G., Jr.; Sharma, A.; Ramaiah, A.; Sen, C.; Purkayastha, A.; Kohn, D.B.; Parcells, M.S.; Beck, S.; Kim, H.; Bakowski, M.A.; et al. Antiviral drug screen identifies DNA-damage response inhibitor as potent blocker of SARS-CoV-2 replication. Cell Rep. 2021, 35, 108940. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Mankouri, J.; Harris, M. Viruses and the fuel sensor: The emerging link between AMPK and virus replication. Rev. Med. Virol. 2011, 21, 205–212. [Google Scholar] [CrossRef]
- Kondratowicz, A.S.; Hunt, C.L.; Davey, R.A.; Cherry, S.; Maury, W.J. AMP-Activated Protein Kinase Is Required for the Macropinocytic Internalization of Ebolavirus. Virol. J. 2013, 87, 746–755. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Duan, X.; Yang, L.; Nilsson-Payant, B.E.; Wang, P.; Duan, F.; Tang, X.; Yaron, T.M.; Zhang, T.; Uhl, S.; et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature 2021, 589, 270–275. [Google Scholar] [CrossRef]
- Saeed, M.F.; Kolokoltsov, A.A.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008, 4, e1000141. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Shukla, D. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 2010, 91, 3002–3009. [Google Scholar] [CrossRef]
- Sánchez, E.G.; Quintas, A.; Pérez-Núñez, D.; Nogal, M.; Barroso, S.; Carrascosa, Á.L.; Revilla, Y. African swine fever virus uses macropinocytosis to enter host cells. PLoS Pathog. 2012, 8, e1002754. [Google Scholar] [CrossRef] [Green Version]
- McGowan, E.M.; Haddadi, N.; Nassif, N.T.; Lin, Y. Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19. Int. J. Mol. Sci. 2020, 21, 7189. [Google Scholar] [CrossRef]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Chai, C.; Cox, B.; Yaish, D.; Gross, D.; Rosenberg, N.; Amblard, F.; Shemuelian, Z.; Gefen, M.; Korach, A.; Tirosh, O.; et al. Agonist of RORA Attenuates Nonalcoholic Fatty Liver Progression in Mice via Up-regulation of MicroRNA 122. Gastroenterology 2020, 159, 999–1014.e1019. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Bharate, S.B. Recent Developments in the Use of Kinase Inhibitors for Management of Viral Infections. J. Med. Chem. 2022, 65, 893–921. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Laufer, S. Kinases as Potential Therapeutic Targets for Anti-coronaviral Therapy. J. Med. Chem. 2022, 65, 955–982. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Hu, J.; Zhong, H.A. Advances in the Development of Class I Phosphoinositide 3-Kinase (PI3K) Inhibitors. Curr. Top. Med. Chem. 2016, 16, 1413–1426. [Google Scholar] [CrossRef]
- Sabbah, D.A.; Brattain, M.G.; Zhong, H. Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: Which way shall we go? Curr. Med. Chem. 2011, 18, 5528–5544. [Google Scholar] [CrossRef]
- Hernandez, R.; Brown, D.T. Growth and maintenance of baby hamster kidney (BHK) cells. Curr. Protoc. Microbiol. 2010, 17, A.4H.1–A.4H.7. [Google Scholar] [CrossRef]
- Ammerman, N.C.; Beier-Sexton, M.; Azad, A.F. Growth and maintenance of Vero cell lines. Curr. Protoc. Microbiol. 2008, 11, A.4E.1–A.4E.7. [Google Scholar] [CrossRef] [Green Version]
- Spickler, C.; Lippens, J.; Laberge, M.-K.; Desmeules, S.; Bellavance, É.; Garneau, M.; Guo, T.; Hucke, O.; Leyssen, P.; Neyts, J.; et al. Phosphatidylinositol 4-Kinase III Beta Is Essential for Replication of Human Rhinovirus and Its Inhibition Causes a Lethal Phenotype In Vivo. Antimicrob. Agents Chemother. 2013, 57, 3358–3368. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, S.; Choudhary, S.; Kumar, P.; Tomar, S. Antiviral strategies targeting host factors and mechanisms obliging +ssRNA viral pathogens. Bioorg. Med. Chem. 2021, 46, 116356. [Google Scholar] [CrossRef]
- Lin, K.; Gallay, P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral Res. 2013, 99, 68–77. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Munawar, A.H. A painful lesson from the COVID-19 pandemic: The need for broad-spectrum, host-directed antivirals. J. Transl. Med. 2020, 18, 390. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. What are the pros and cons of the use of host-targeted agents against hepatitis C? Antiviral Res. 2014, 105, 22–25. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Flisiak, R.; Sarin, S.K.; Rasenack, J.; Piratvisuth, T.; Chuang, W.L.; Peng, C.Y.; Foster, G.R.; Shah, S.; Wedemeyer, H.; et al. Alisporivir plus ribavirin, interferon free or in combination with pegylated interferon, for hepatitis C virus genotype 2 or 3 infection. Hepatology 2015, 62, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.P.; Sasse, F.; Brönstrup, M.; Diez, J.; Meyerhans, A. Antiviral drug discovery: Broad-spectrum drugs from nature. Nat. Prod. Rep. 2015, 32, 29–48. [Google Scholar] [CrossRef]
- Bader, T.; Fazili, J.; Madhoun, M.; Aston, C.; Hughes, D.; Rizvi, S.; Seres, K.; Hasan, M. Fluvastatin inhibits hepatitis C replication in humans. Am. J. Gastroenterol. 2008, 103, 1383–1389. [Google Scholar] [CrossRef]
- Sezaki, H.; Suzuki, F.; Akuta, N.; Yatsuji, H.; Hosaka, T.; Kobayashi, M.; Suzuki, Y.; Arase, Y.; Ikeda, K.; Miyakawa, Y.; et al. An open pilot study exploring the efficacy of fluvastatin, pegylated interferon and ribavirin in patients with hepatitis C virus genotype 1b in high viral loads. Intervirology 2009, 52, 43–48. [Google Scholar] [CrossRef]
- Malekinejad, Z.; Baghbanzadeh, A.; Nakhlband, A.; Baradaran, B.; Jafari, S.; Bagheri, Y.; Raei, F.; Montazersaheb, S.; Farahzadi, R. Recent clinical findings on the role of kinase inhibitors in COVID-19 management. Life Sci. 2022, 306, 120809. [Google Scholar] [CrossRef]
- Kung, C.; Shokat, K.M. Small-molecule kinase-inhibitor target assessment. Chembiochem 2005, 6, 523–526. [Google Scholar] [CrossRef]
- Sevecka, M.; MacBeath, G. State-based discovery: A multidimensional screen for small-molecule modulators of EGF signaling. Nat. Methods 2006, 3, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Lazo, J.S.; Brady, L.S.; Dingledine, R. Building a pharmacological lexicon: Small molecule discovery in academia. Mol. Pharmacol. 2007, 72, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H., 3rd; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Natarajan, M.; Lin, K.M.; Hsueh, R.C.; Sternweis, P.C.; Ranganathan, R. A global analysis of cross-talk in a mammalian cellular signalling network. Nat. Cell Biol. 2006, 8, 571–580. [Google Scholar] [CrossRef]
- Ventura, A.C.; Sepulchre, J.A.; Merajver, S.D. A hidden feedback in signaling cascades is revealed. PLoS Comput. Biol. 2008, 4, e1000041. [Google Scholar] [CrossRef] [Green Version]
- Wynn, M.L.; Ventura, A.C.; Sepulchre, J.A.; García, H.J.; Merajver, S.D. Kinase inhibitors can produce off-target effects and activate linked pathways by retroactivity. BMC Syst. Biol. 2011, 5, 156. [Google Scholar] [CrossRef] [Green Version]
- Janes, K.A.; Albeck, J.G.; Gaudet, S.; Sorger, P.K.; Lauffenburger, D.A.; Yaffe, M.B. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science 2005, 310, 1646–1653. [Google Scholar] [CrossRef] [Green Version]
- Miller-Jensen, K.; Janes, K.A.; Brugge, J.S.; Lauffenburger, D.A. Common effector processing mediates cell-specific responses to stimuli. Nature 2007, 448, 604–608. [Google Scholar] [CrossRef]
- Kemp, M.L.; Wille, L.; Lewis, C.L.; Nicholson, L.B.; Lauffenburger, D.A. Quantitative network signal combinations downstream of TCR activation can predict IL-2 production response. J. Immunol. 2007, 178, 4984–4992. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, B.; Smith, A.M.; Fernandes, J.D.; Frankel, A.D. Oligomeric viral proteins: Small in size, large in presence. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 379–394. [Google Scholar] [CrossRef] [Green Version]
- Schlicksup, C.J.; Zlotnick, A. Viral structural proteins as targets for antivirals. Curr. Opin. Virol. 2020, 45, 43–50. [Google Scholar] [CrossRef]
- Legiewicz, M.; Badorrek, C.S.; Turner, K.B.; Fabris, D.; Hamm, T.E.; Rekosh, D.; Hammarskjöld, M.L.; Le Grice, S.F. Resistance to RevM10 inhibition reflects a conformational switch in the HIV-1 Rev response element. Proc. Natl. Acad. Sci. USA 2008, 105, 14365–14370. [Google Scholar] [CrossRef] [Green Version]
- Locarnini, S.; Bowden, S. Drug resistance in antiviral therapy. Clin. Liver Dis. 2010, 14, 439–459. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, C.; Liu, Z.; Zou, G.; Li, J.; Lu, M. Host Genetic Determinants of Hepatitis B Virus Infection. Front. Genet. 2019, 10, 696. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
An overview of antiviral drugs according to (a) drug type, (b) top mechanisms of action, and (c) top targets. * Others in b and c indicate that there are other mechanisms and targets that are not shown in the figure because they represent a large number of categories with very small percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 23 December 2022, ©2022 Clarivate. All rights reserved.
Figure 1.
An overview of antiviral drugs according to (a) drug type, (b) top mechanisms of action, and (c) top targets. * Others in b and c indicate that there are other mechanisms and targets that are not shown in the figure because they represent a large number of categories with very small percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 23 December 2022, ©2022 Clarivate. All rights reserved.

Figure 2.
An overview of antiviral drugs under active development for viral infections according to (a) drug type, (b) top mechanisms of action, (c) top targets, and (d) developmental status. * Others in (b,c) indicate that there are other mechanisms and targets not shown in the figure because they represent a large number of categories with tiny percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 24 December 2022, ©2022 Clarivate. All rights reserved.
Figure 2.
An overview of antiviral drugs under active development for viral infections according to (a) drug type, (b) top mechanisms of action, (c) top targets, and (d) developmental status. * Others in (b,c) indicate that there are other mechanisms and targets not shown in the figure because they represent a large number of categories with tiny percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 24 December 2022, ©2022 Clarivate. All rights reserved.

Figure 3.
An overview of antiviral drugs modulating signal transduction according to (a) top targets, (b) top therapeutic groups, (c) top organizations, and (d) drug types. * Others in (a–c) indicate that there are other targets, therapeutic groups and mechanisms that are not shown in the figure because they represent a large number of categories with very small percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 24 December 2022, ©2022 Clarivate. All rights reserved.
Figure 3.
An overview of antiviral drugs modulating signal transduction according to (a) top targets, (b) top therapeutic groups, (c) top organizations, and (d) drug types. * Others in (a–c) indicate that there are other targets, therapeutic groups and mechanisms that are not shown in the figure because they represent a large number of categories with very small percentages. Data source: Cortellis Drug Discovery Intelligence [37], https://www.cortellis.com/drugdiscovery/, accessed on 24 December 2022, ©2022 Clarivate. All rights reserved.

Figure 4.
Protein-protein interactions network of kinases explored as diagnostic biomarkers for viral infections. Network nodes are human kinases that are viral disease biomarkers, according to the Cortellis Drug Discovery Intelligence databases [37]. The network was generated using Cytoscape version 3.9.1. Network nodes were colored based on the top five enriched KEGG pathways [73] shown in the color key beneath the network. Blue nodes indicate that the gene/gene product is not part of the top five enriched KEGG pathways shown underneath the network. The pathway prediction false discovery rate (FDR) is reported for each pathway. CDDI [37] was on https://www.cortellis.com/drugdiscovery/, accessed on 26 December 2022, ©2022 Clarivate. All rights reserved.
Figure 4.
Protein-protein interactions network of kinases explored as diagnostic biomarkers for viral infections. Network nodes are human kinases that are viral disease biomarkers, according to the Cortellis Drug Discovery Intelligence databases [37]. The network was generated using Cytoscape version 3.9.1. Network nodes were colored based on the top five enriched KEGG pathways [73] shown in the color key beneath the network. Blue nodes indicate that the gene/gene product is not part of the top five enriched KEGG pathways shown underneath the network. The pathway prediction false discovery rate (FDR) is reported for each pathway. CDDI [37] was on https://www.cortellis.com/drugdiscovery/, accessed on 26 December 2022, ©2022 Clarivate. All rights reserved.

Table 1.
Two classification systems for viruses based on the virus genome and Baltimore classes.
| Viruses Classification Systems | |
|---|---|
| Virus Genomes | Baltimore Classes |
|
|
Table 2.
A stepwise summary of gene expression and gene replication mechanisms among different classes of viruses [13,22,33,34,35,36].
| Virus Class | mRNA and Protein Synthesis | Gene Replication Location and Enzymes |
|---|---|---|
| BCI—dsDNA |
|
|
| BCII—ssDNA |
| ssDNA viruses use host enzymes to synthesize dsDNA replication intermediate; followed by ssDNA progeny formation |
| BCIII—dsRNA |
|
|
| BCIV—(+) ssRNA |
|
|
| BCV—(−) ssRNA |
|
|
| BCVI—RT (+) ssRNA |
|
|
| BCVII—RT dsDNA |
|
|
BC: Baltimore Class; ds: double-stranded; ss: single-stranded; RT: reverse transcribing; (+): positive-sense; (−): negative-sense; hRNApol-II: host RNA polymerase II; RdRp: viral RNA-dependent RNA polymerase; RTz: viral reverse transcriptase enzyme.
Table 4.
Important antiviral kinase inhibitors reported in the biomedical literature.
| Compound | Target | Virus | Mechanism of Antivirus |
|---|---|---|---|
 U0126 (1) MEK1 IC50 = 72 nM MEK2 IC50 = 58 nM | MEK 1/2- MAPK | IBV IAV | Inhibits viral RNP release |
 SP600125 (2) JNK1/2 IC50 = 40 nM JNK3 IC50 = 90 nM | JNK1- MAPK | Astrovirus JEV MERS-CoV | Inhibits viral entry Decreases inflammatory cytokines |
 SB203580 (3) SAPK2a/p38 IC50 = 50 nM | p38-MAPK | MERS-CoV | Inhibits viral entry |
 Selumetinib (4) MEK1/2 IC50 = 14 nM | MEK 1/2- MAPK | MERS-CoV | Antiproliferative activity |
 Trametinib (5) MEK1/2 IC50 = 2 nM | MEK 1/2- MAPK | MERS-CoV | Antiproliferative activity |
 Papaverine (6) IC50 = 2.0-36.4 μM | MEK & ERK-MAPK | IAV Paramyxovirus | Inhibits viral RNP release |
 ATR-002 (7) MEK1 IC50 = 5.73 nM | MEK1-MAPK | IAV IBV | Antiproliferative activity |
 Palbociclib (8, PD-0332991) CDK 4 IC50 = 11 nM CDK 6 IC50 = 16 nM | CDK4/CDK6 | HIV-1 HSV-1 | CDK4: Antiproliferative activity CDK6: Inhibits reverse transcription |
 Ribociclib (9, LEE011) Breast cancer Novartis, 2017 | CDK4/CDK6 | SARS-CoV-2 | Antiproliferative activity |
 Abemaciclib (10, LY2835219) Breast cancer Lilly, 2017 SARS-CoV-2 (IC50 = 6.6 mM) | CDK4/CDK6 | SARS-CoV-2 | Antiproliferative activity |
 (R)-Roscovitine (11) CDK2 IC50 = 0.7 μM CDK5 IC50 = 0.2 μM | CDK2/CDK5 | HCMV | Inhibits DNA synthesis |
 Flavopiridol (12) CDK1 IC50 = 30 nM CDK2 IC50 = 170 nM CDK4 IC50 = 100 nM CDK6 IC50 = 60 nM | CDK1/CDK2/CDK4 | HIV-1 IAV | Antiproliferative activity Synergistic activity with Dinaciclib (15) |
 Alsterpaullone (13) CDK1 IC50 = 35 nM CDK2 IC50 = 15 nM | CDK1/CDK2 | HIV-1 | Antiproliferative activity |
 FIT-039 (14) CDK9 IC50 = 5.8 μM | CDK9 | HSV-1 HSV-2 HCMV HAdV-5 | Antiproliferative activity Anti-transcription Antiproliferative activity |
 Dinaciclib (15) CDK1 IC50 = 3 nM | CDK1 | SARS-CoV-2 | Antiproliferative activity |
 PHA-690509 (16) CDK2 IC50 = 31 nM | CDK2 | ZIKV | Antiproliferative activity |
 Genistein (17) EGFR IC50 = 0.6 μM | RTKs | IAV HIV-1 Arenavirus HSV-1 | Inhibits viral entry Antiproliferative activity |
 Gefitinib (18, Iressa®, ZD1839) EGFR IC50 = 33 nM GAK Kd = 13 nM AAK1 kd > 3000 nM | EGFR | TGEV IAV Rhinovirus | Inhibits viral entry Inhibits infection |
| FDA- Approved Anticancer Drug, AstraZeneca, 2015 | |||
 AG1478 (19) EGFR IC50 = 3 nM | EGFR | TGEV | Inhibits viral intake by IPEC |
 Erlotinib (20, Tarceva®, CP-358774, R 1415, NSC 718781, OSI 774) EGFR IC50 = 2 nM GAK Kd = 3.1 nM AAK1 kd = 1.2 μM | EGFR GAK/AAK1 | HCV HCV | Inhibits viral entry Inhibits viral entry Inhibits viral assembly |
| FDA- Approved Anticancer Drug, Genentech, 2004 | |||
 Sorafenib (21, Nexavar®, BAY43-9006) PDGFR IC50 = 20 nM | PDGFR | SARS-CoV-2 DNA virus HBV | Antiproliferative activity |
| FDA- Approved Anticancer Drug, Onyx, 2005 | |||
 R428 (23) AxI IC50 = 14 nM | AxI | ZIKV | Inhibits viral entry Activates IFN-1 |
 AG879 (24) TRK-A IC50 = 10 μM | TRK-A | IAV | Antiproliferative activity |
 Tyrphostin A9 (25) PDGFR IC50 = 0.5 μM | PDGFR | IAV | Antiproliferative activity |
 Sunitinib (26, SU-11248, Sutent®) AAK1 Kd = 11 nM GAK Kd = 20 nM | AAK1 GAK | HCV | Inhibits viral entry Inhibits viral assembly |
| FDA- Approved Anticancer Drug, Pfizer, 2006 | |||
 Baricitinib (27, LY3009104, Olumiant®) AAK1 Kd = 8.2 nM GAK Kd = 120 nM | AAK1 GAK | SARS-CoV-2 | Inhibits viral entry Inhibits viral assembly |
| FDA- Approved Antirheumatic Agent, Lilly, 2018 | |||
 Dasatinib (28, BMS-354825, Sprycel®) AAK1 Kd > 3000 nM GAK Kd = 2.6 nM ABL IC50 = 2.8 nM | Src Src Fyn Lck ABL | DENV HCV DENV HIV-1 MERS-CoV SARS-CoVMERS-CoV SARS-CoV | Inhibits viral assembly Inhibits viral secretion Potentiates IC50 (210 fold) in Huh7.5.1 in combination with Sofosbuvir Antiproliferative Inhibits viral entry Inhibits reverse transcription |
| FDA- Approved Anticancer Drug, Bristol- Myers Squibb, 2006 | |||
 Saracatinib (29) Src IC50 = 2.7 nM | Src Fyn | DENV DENV MERS-CoV HCoV-229E HCoV-OC43 | Inhibits viral assembly Antiproliferative |
 Bosutinib (30, SKI-606, Bosulif®) Src IC50 = 1.2 nM | Src | SARS-CoV-2 | Inhibits viral entry |
| FDA- Approved Anticancer Drug, Pfizer, 2012 | |||
 Ponatinib (31, AP24534, Iclusig®) Src IC50 = 5.4 nM | Src | SARS-CoV-2 | Inhibits cytokines release |
| FDA- Approved Anticancer Drug, Ariad, 2012 | |||
 Vandetanib (32, ZD6474, Caprelsa®) Src IC50 = 0.79 nM | Src | SARS-CoV-2 HCoV-229E | Antiproliferative Antiproliferative |
| FDA- Approved Anticancer Drug, AstraZeneca, 2011 | |||
 Apilimod (33, LAM-002A, STA-5326) Pikfyve IC50 = 14 nM | Pikfyve | EBOV SARS-CoV-2 | Inhibits viral entry |
| FDA- Approved Anticancer Drug, AI Therapeutics, Inc. | |||
 YM-201636 (34) Pikfyve IC50 = 33 nM | Pikfyve | EBOV | Inhibits viral entry |
 Vacuolin-1 (35) Pikfyve Kd = 9 nM | Pikfyve | EBOV | Inhibits viral entry |
 Methyl 5-[2-(5-nitro-2-furyl) vinyl]-2-furoate (36) GRK2 IC50 = 126 μM | GRK2 | IAV | Inhibits viral entry Antiproliferative |
 Imatinib (37, Gleevec®, Glivec®) ABL IC50 = 220 nM | ABL | MERS-CoV SARS-CoV | Inhibits viral entry |
| FDA- Approved Anticancer Drug, Novartis, 2001 | |||
 Nilotinib (38, AMN107, Tasigna®) ABL IC50 = 30 nM | ABL | SARS-CoV | Antiproliferative |
| FDA- Approved Anticancer Drug, Novartis, 2007 | |||
 GNF-2 (39) ABL IC50 = 138 nM | ABL | DENV MERS-CoV SARS-CoV | Inhibits viral entry Antiproliferative Inhibits viral entry |
 GNF-2 (40) ABL IC50 = 220 nM | ABL | MERS-CoV SARS-CoV | Inhibits viral entry |
 BSA9 (41) CaMKII IC50 = 0.79 μM | CaMKII | DENV ZIKV | Inhibits viral entry |
 Berzosertib (42) ATR Ki = 0.2 nM | ATR inhibitor | MERS-CoV SARS-CoV | Antiproliferative |
 Dorsomorphin (43) AMPK Ki = 109 nM | AMPK | EBOV SARS-CoV-2 | Antiproliferative Antiviral activity |
 LY294002 (44) PI3Kα (IC50 = 0.50 μM) PI3Kβ (IC50 = 0.97 μM) PI3Kδ (IC50 = 0.57 μM) | PI3K | ASFV EBOV HSV-1 | Inhibits viral entry |
 Opaganib (45) SphK2 Ki = 9.8 μM | SphK2 | SARS-CoV-2 | Antiproliferative |
Table 5.
The pros and cons of targeting viral proteins or host proteins.
| Drug Target | Pros | Cons |
|---|---|---|
| Viral Proteins |
|
|
| Host Proteins |
|
|
Table 6.
Types of kinase inhibitors.
| Kinase Inhibitor Type | Characteristics |
|---|---|
| Type I |
|
| Type II |
|
| Type III-VI |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hajjo, R.; Sabbah, D.A.; Abusara, O.H.; Kharmah, R.; Bardaweel, S. Targeting Human Proteins for Antiviral Drug Discovery and Repurposing Efforts: A Focus on Protein Kinases. Viruses 2023, 15, 568. https://doi.org/10.3390/v15020568
AMA Style
Hajjo R, Sabbah DA, Abusara OH, Kharmah R, Bardaweel S. Targeting Human Proteins for Antiviral Drug Discovery and Repurposing Efforts: A Focus on Protein Kinases. Viruses. 2023; 15(2):568. https://doi.org/10.3390/v15020568
Chicago/Turabian StyleHajjo, Rima, Dima A. Sabbah, Osama H. Abusara, Reham Kharmah, and Sanaa Bardaweel. 2023. "Targeting Human Proteins for Antiviral Drug Discovery and Repurposing Efforts: A Focus on Protein Kinases" Viruses 15, no. 2: 568. https://doi.org/10.3390/v15020568
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.