
Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF


 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Detection of Amino Acid Substitutions and Calculation of Their Frequencies
2.2. Statistical Analyses
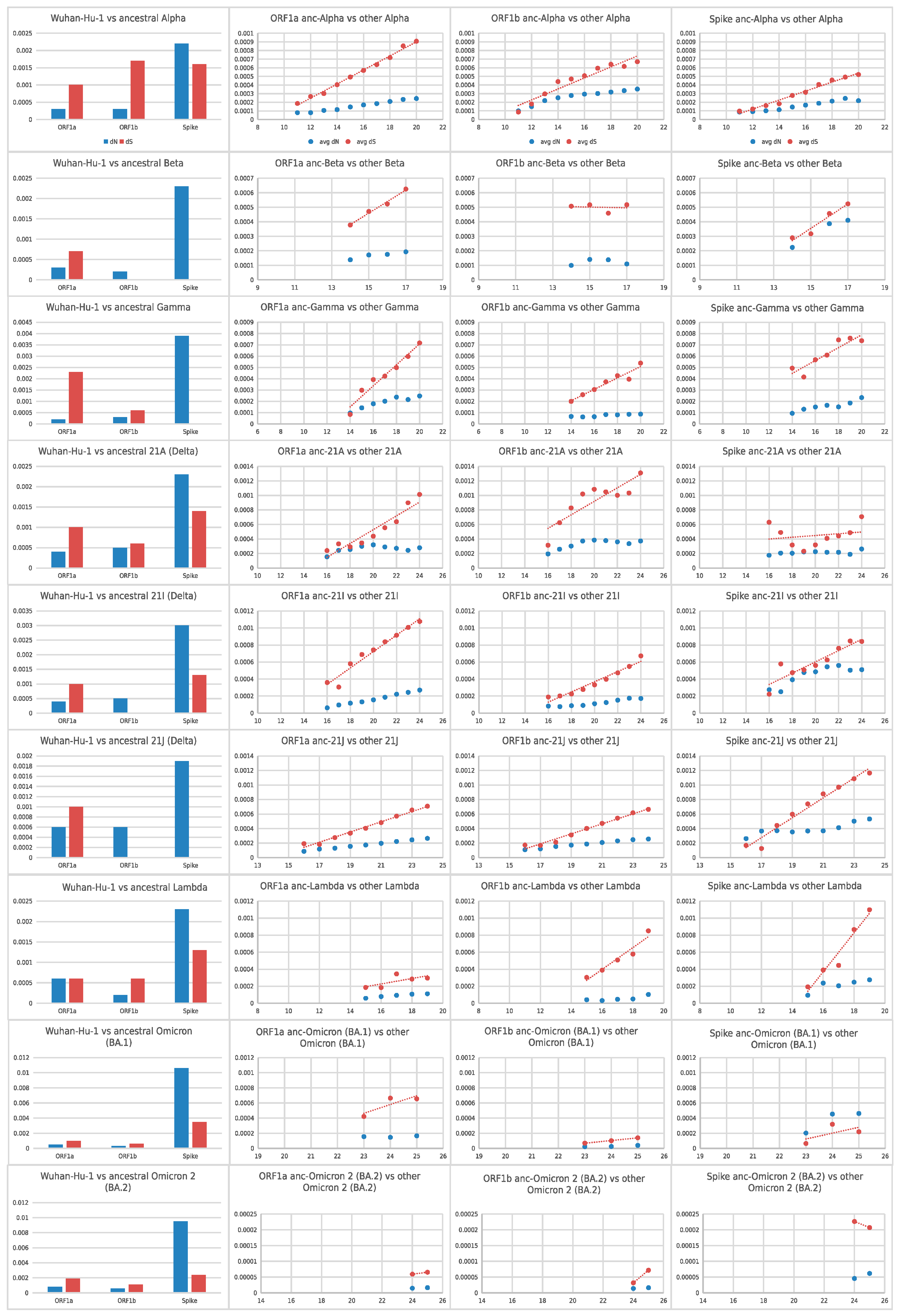
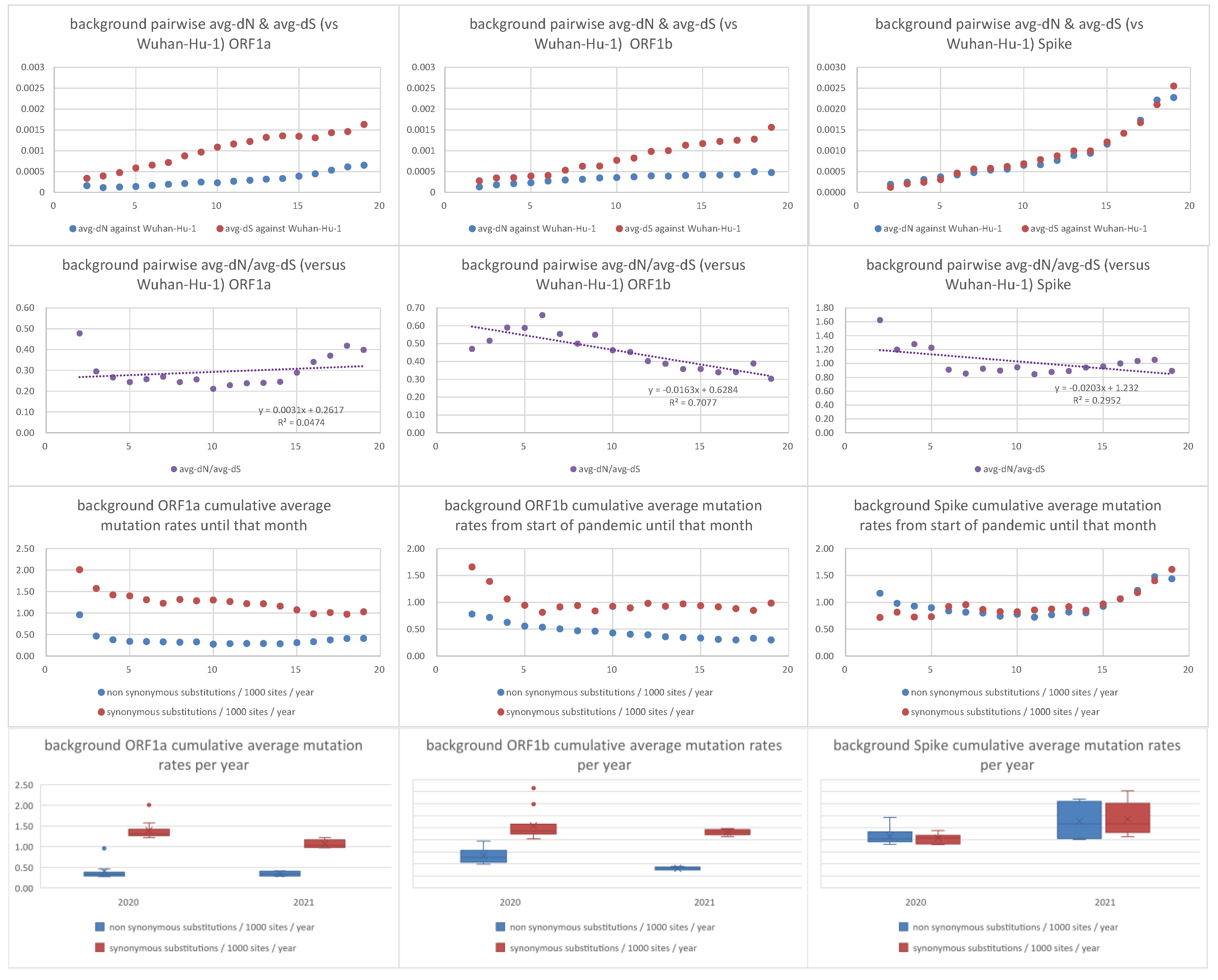
2.3. dN/dS Analyses
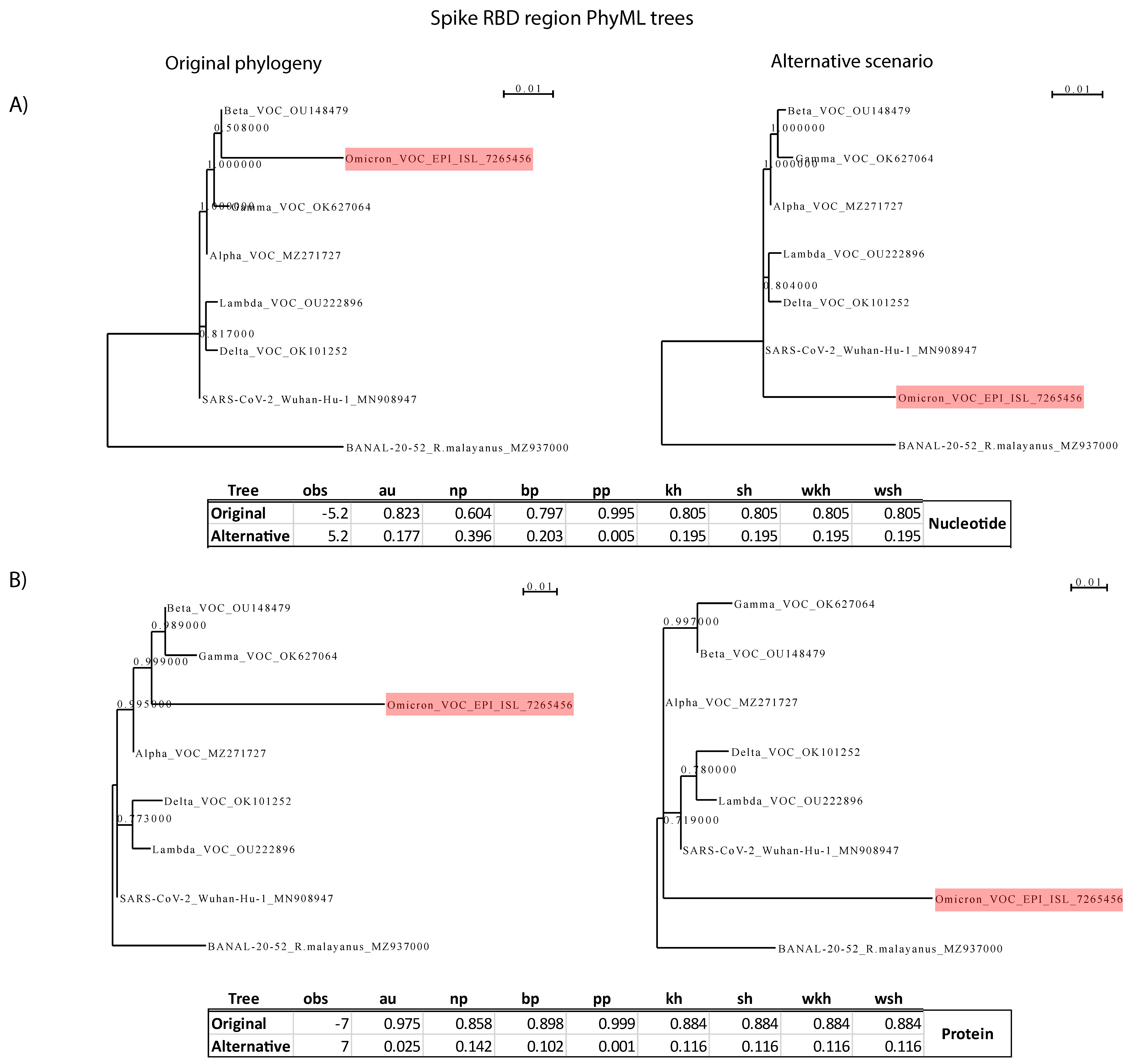
2.4. Test of the Hypothesis That the Omicron’s Highly Mutated Spike Receptor Binding Domain (RBD) Originated from Another Sarbecovirus via Recombination
2.5. Conservation of Substituted Amino Acid Residues in Other Sarbecoviruses
3. Results
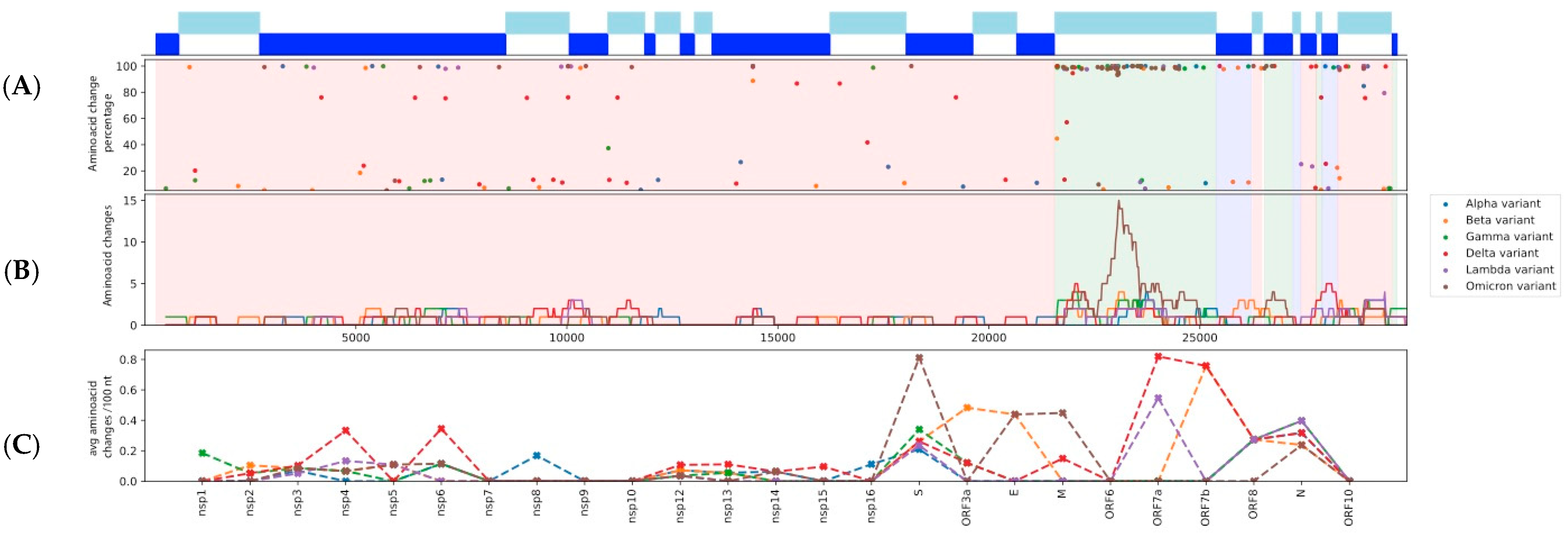
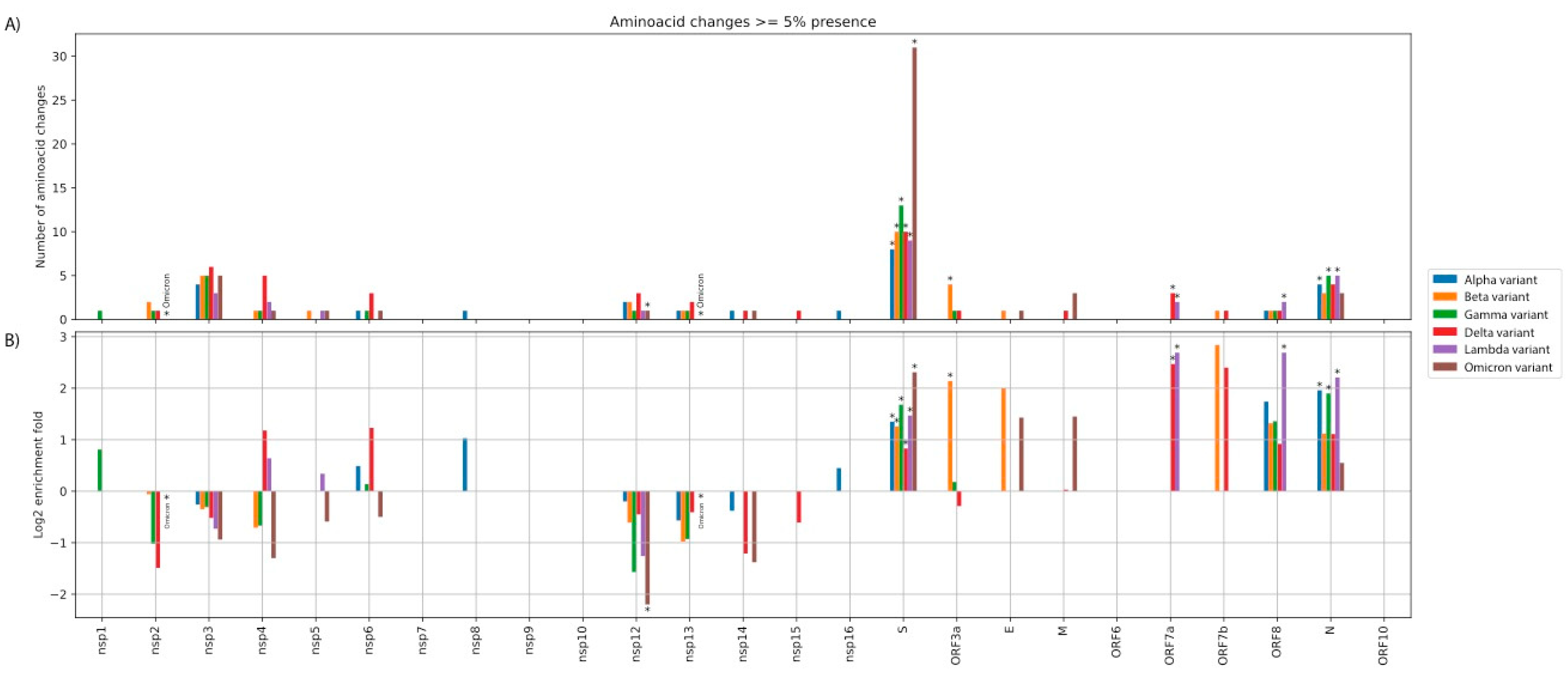
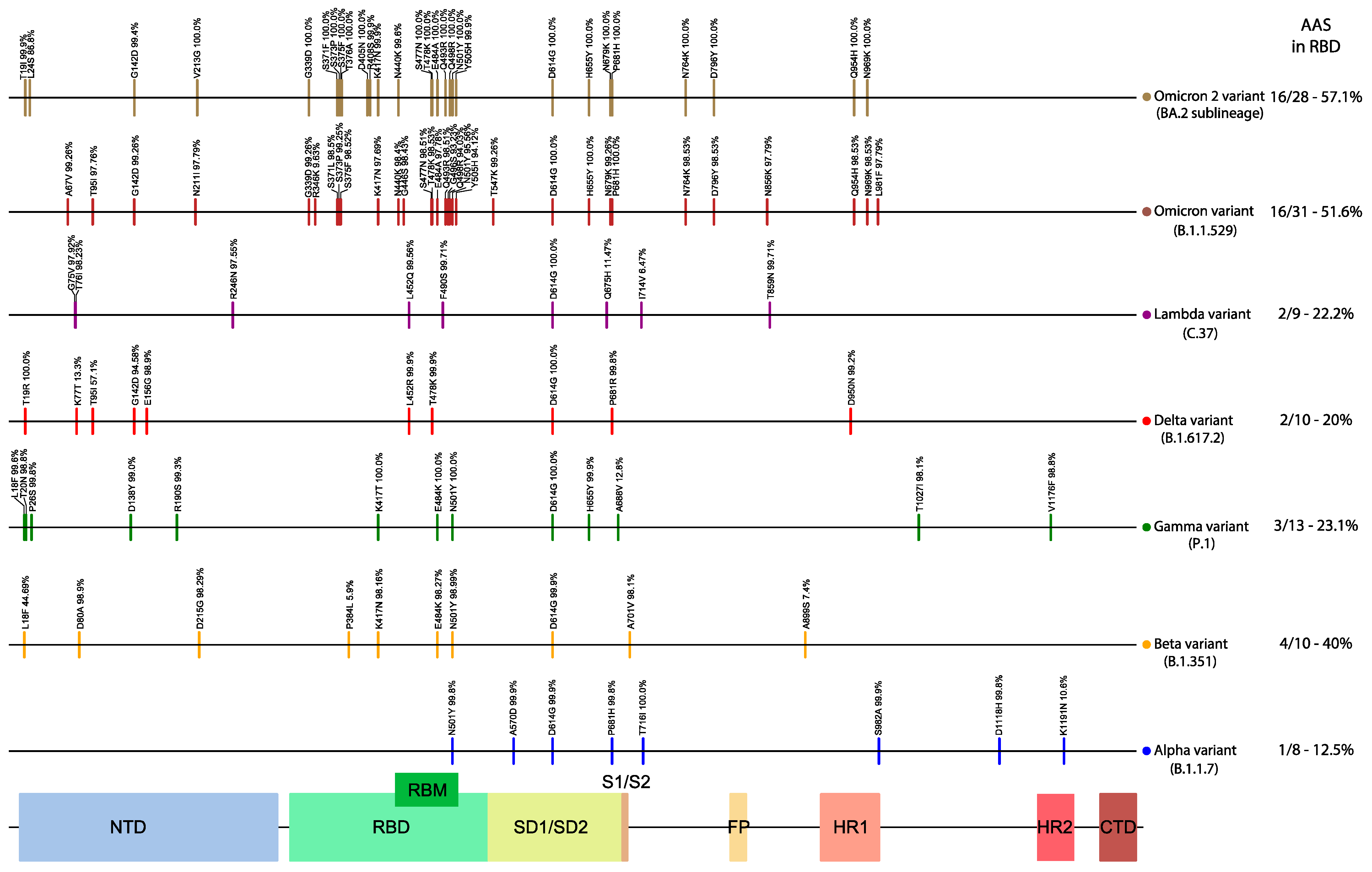
3.1. Distribution and Enrichment of Amino Acid Changes in the nsps/ORFs of Each of the Six Variant Lineages Consistently Highlights the Spike ORF as an Amino Acid Substitution Hotspot
3.2. Positive Selection Affected the Emergence of AAS in the Spike ORF of Each Variant of Concern, Followed by Purifying Selection
3.3. The Omicron Spike-RBD Is Highly Mutated and Probably Diverged by Successive Point Mutations, Rather than by Recombination with Another Sarbecovirus
3.4. Many Amino Acid Substitutions of SARS-CoV-2 Variants of Concern Are Not Observed in Any Other Sarbecoviruses
3.5. Recurrence of High Frequency AAS in More than One VoC Lineages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Euro. Surveill. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef] [PubMed]
- Ghafari, M.; du Plessis, L.; Raghwani, J.; Bhatt, S.; Xu, B.; Pybus, O.G.; Katzourakis, A. Purifying Selection Determines the Short-Term Time Dependency of Evolutionary Rates in SARS-CoV-2 and PH1N1 Influenza. Mol. Biol. Evol. 2022, 39, msac009. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The Emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef]
- Nussenblatt, V.; Roder, A.E.; Das, S.; de Wit, E.; Youn, J.-H.; Banakis, S.; Mushegian, A.; Mederos, C.; Wang, W.; Chung, M.; et al. Year-Long COVID-19 Infection Reveals within-Host Evolution of SARS-CoV-2 in a Patient with B Cell Depletion. J. Infect. Dis. 2021, jiab622. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern Naming Scheme Conducive for Global Discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Pandit, P.; McArthur, A.G.; Banerjee, A.; Mossman, K. Evolutionary Trajectory of SARS-CoV-2 and Emerging Variants. Virol. J. 2021, 18, 166. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Negi, S.S.; Schein, C.H.; Braun, W. Regional and Temporal Coordinated Mutation Patterns in SARS-CoV-2 Spike Protein Revealed by a Clustering and Network Analysis. Sci. Rep. 2022, 12, 1128. [Google Scholar] [CrossRef] [PubMed]
- WHO Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 2 November 2021).
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.S.; Boshier, F.A.T.; et al. Emergence of Genomic Diversity and Recurrent Mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- MacLean, O.A.; Lytras, S.; Weaver, S.; Singer, J.B.; Boni, M.F.; Lemey, P.; Kosakovsky Pond, S.L.; Robertson, D.L. Natural Selection in the Evolution of SARS-CoV-2 in Bats Created a Generalist Virus and Highly Capable Human Pathogen. PLoS Biol. 2021, 19, e3001115. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Time-Dependent Rate Phenomenon in Viruses. J. Virol. 2016, 90, 7184–7195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 Spike D614G Change Enhances Replication and Transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 Spike N439K Variants Maintain Fitness While Evading Antibody-Mediated Immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 Evolution during Treatment of Chronic Infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody Evasion by the P.1 Strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta Spike P681R Mutation Enhances SARS-CoV-2 Fitness over Alpha Variant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; PHE Virology Consortium; de Silva, T.I.; Barclay, W.S. The SARS-CoV-2 Variants Associated with Infections in India, B.1.617, Show Enhanced Spike Cleavage by Furin. bioRxiv 2021. [Google Scholar] [CrossRef]
- Burki, T.K. Omicron Variant and Booster COVID-19 Vaccines. Lancet Respir. Med. 2021, 10, e17. [Google Scholar] [CrossRef]
- Ferguson, N.; Ghani, A.; Cori, A.; Hogan, A.; Hinsley, W.; Volz, E. Report 49: Growth, Population Distribution and Immune Escape of Omicron in England. 2021. Available online: https://www.imperial.ac.uk/mrc-global-infectious-disease-analysis/covid-19/report-49-Omicron/ (accessed on 5 December 2021).
- Martin, D.P.; Lytras, S.; Lucaci, A.G.; Maier, W.; Grüning, B.; Shank, S.D.; Weaver, S.; MacLean, O.A.; Orton, R.J.; Lemey, P.; et al. Selection Analysis Identifies Unusual Clustered Mutational Changes in Omicron Lineage BA.1 That Likely Impact Spike Function. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cameroni, E.; Saliba, C.; Bowen, J.E.; Rosen, L.E.; Culap, K.; Pinto, D.; VanBlargan, L.A.; Marco, A.D.; Zepeda, S.K.; Iulio, J.d.; et al. Broadly Neutralizing Antibodies Overcome SARS-CoV-2 Omicron Antigenic Shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- FDA SARS-CoV-2 Viral Mutations: Impact on COVID-19 Tests. FDA. 2021. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests (accessed on 5 December 2021).
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation Rate of SARS-CoV-2 and Emergence of Mutators during Experimental Evolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jaroszewski, L.; Iyer, M.; Alisoltani, A.; Sedova, M.; Godzik, A. The Interplay of SARS-CoV-2 Evolution and Constraints Imposed by the Structure and Functionality of Its Proteins. PLoS Comput. Biol. 2021, 17, e1009147. [Google Scholar] [CrossRef]
- Garushyants, S.K.; Rogozin, I.B.; Koonin, E.V. Template Switching and Duplications in SARS-CoV-2 Genomes Give Rise to Insertion Variants That Merit Monitoring. Commun. Biol. 2021, 4, 1343. [Google Scholar] [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Shan, K.-J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a Mouse Origin of the SARS-CoV-2 Omicron Variant. J. Genet. Genom. 2021, 48, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Poon, A.F.Y.; Velazquez, R.; Weaver, S.; Hepler, N.L.; Murrell, B.; Shank, S.D.; Magalis, B.R.; Bouvier, D.; Nekrutenko, A.; et al. HyPhy 2.5-A Customizable Platform for Evolutionary Hypothesis Testing Using Phylogenies. Mol. Biol. Evol. 2020, 37, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimodaira, H.; Hasegawa, M. CONSEL: For Assessing the Confidence of Phylogenetic Tree Selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast Selection of Best-Fit Models of Protein Evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytras, S.; Hughes, J.; Martin, D.; de Klerk, A.; Lourens, R.; Kosakovsky Pond, S.L.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. Genome Biol. Evol. 2022, 14, evac018. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat Coronaviruses Related to SARS-CoV-2 and Infectious for Human Cells. Nature 2022. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Richards, F.M. The Interpretation of Protein Structures: Estimation of Static Accessibility. J. Mol. Biol. 1971, 55, 379–400. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Thornton, J.M. PDBsum Extras: SARS-CoV-2 and AlphaFold Models. Protein Sci. 2022, 31, 283–289. [Google Scholar] [CrossRef]
- Milewska, A.; Kindler, E.; Vkovski, P.; Zeglen, S.; Ochman, M.; Thiel, V.; Rajfur, Z.; Pyrc, K. APOBEC3-Mediated Restriction of RNA Virus Replication. Sci. Rep. 2018, 8, 5960. [Google Scholar] [CrossRef]
- Wang, Y.; Mao, J.-M.; Wang, G.-D.; Luo, Z.-P.; Yang, L.; Yao, Q.; Chen, K.-P. Human SARS-CoV-2 Has Evolved to Reduce CG Dinucleotide in Its Open Reading Frames. Sci. Rep. 2020, 10, 12331. [Google Scholar] [CrossRef]
- Ou, Z.; Ouzounis, C.; Wang, D.; Sun, W.; Li, J.; Chen, W.; Marlière, P.; Danchin, A. A Path toward SARS-CoV-2 Attenuation: Metabolic Pressure on CTP Synthesis Rules the Virus Evolution. Genome Biol. Evol. 2020, 12, 2467–2485. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Non-Homologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2022, 39, msab292. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.-M.; O’Donnell, A.C.; Ochman, H. Recombination Events Are Concentrated in the Spike Protein Region of Betacoronaviruses. PLoS Genet. 2020, 16, e1009272. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2021, 38, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.A.; Brown, J.; Pedersen, B.S.; Quinlan, A.R.; Elde, N.C. Extensive Recombination-Driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens. bioRxiv 2021. [Google Scholar] [CrossRef]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; De Maio, N.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-Scale Phylogenomics Reveals Elevated Recombination Rates in the SARS-CoV-2 Spike Region. bioRxiv 2021. [Google Scholar] [CrossRef]
- VanInsberghe, D.; Neish, A.S.; Lowen, A.C.; Koelle, K. Recombinant SARS-CoV-2 Genomes Circulated at Low Levels over the First Year of the Pandemic. Virus Evol. 2021, 7, veab059. [Google Scholar] [CrossRef]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and Transmission of Interlineage Recombinants in the SARS-CoV-2 Pandemic. Cell 2021, 184, 5179–5188.e8. [Google Scholar] [CrossRef]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid Detection of Inter-Clade Recombination in SARS-CoV-2 with Bolotie. Genetics 2021, 218, iyab074. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An Oral SARS-CoV-2 Mpro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Molla, A.; Korneyeva, M.; Gao, Q.; Vasavanonda, S.; Schipper, P.J.; Mo, H.M.; Markowitz, M.; Chernyavskiy, T.; Niu, P.; Lyons, N.; et al. Ordered Accumulation of Mutations in HIV Protease Confers Resistance to Ritonavir. Nat. Med. 1996, 2, 760–766. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The Emergence and Ongoing Convergent Evolution of the SARS-CoV-2 N501Y Lineages. Cell 2021, 184, 5189–5200.e7. [Google Scholar] [CrossRef] [PubMed]
- Chaintoutis, S.C.; Thomou, Z.; Mouchtaropoulou, E.; Tsiolas, G.; Chassalevris, T.; Stylianaki, I.; Lagou, M.; Michailidou, S.; Moutou, E.; Koenen, J.J.H.; et al. Outbreaks of SARS-CoV-2 in Naturally Infected Mink Farms: Impact, Transmission Dynamics, Genetic Patterns, and Environmental Contamination. PLoS Pathog. 2021, 17, e1009883. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, M.I.; Wolf, Y.I.; Koonin, E.V. On the Feasibility of Saltational Evolution. Proc. Natl. Acad. Sci. USA 2019, 116, 21068–21075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, E. Beyond Omicron: What’s next for COVID’s Viral Evolution. Nature 2021, 600, 204–207. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where Did “weird” Omicron Come From? Science 2021, 374, 1179. [Google Scholar] [CrossRef] [PubMed]
- Karim, F.; Moosa, M.; Gosnell, B.; Cele, S.; Giandhari, J.; Pillay, S.; Tegally, H.; Wilkinson, E.; San, J.; Msomi, N.; et al. Persistent SARS-CoV-2 Infection and Intra-Host Evolution in Association with Advanced HIV Infection. MedRxiv 2021. [Google Scholar] [CrossRef]
- Mallapaty, S. Where Did Omicron Come from? Three Key Theories. Nature 2022, 602, 26–28. [Google Scholar] [CrossRef]
- Rottier, P.J.M.; Nakamura, K.; Schellen, P.; Volders, H.; Haijema, B.J. Acquisition of Macrophage Tropism during the Pathogenesis of Feline Infectious Peritonitis Is Determined by Mutations in the Feline Coronavirus Spike Protein. J. Virol. 2005, 79, 14122–14130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, P.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Winkler, M.S.; Lier, M.; Schulz, S.; Jäck, H.-M.; Cossmann, A.; et al. The Spike Protein of SARS-CoV-2 Variant A. 30 Is Heavily Mutated and Evades Vaccine-Induced Antibodies with High Efficiency. Cell Mol. Immunol. 2021, 18, 2673–2675. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef]
- Pan, R.; Kindler, E.; Cao, L.; Zhou, Y.; Zhang, Z.; Liu, Q.; Ebert, N.; Züst, R.; Sun, Y.; Gorbalenya, A.E.; et al. N7-Methylation of the Coronavirus RNA Cap Is Required for Maximal Virulence by Preventing Innate Immune Recognition. mBio 2022, 13, e0366221. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Excluding Indels | Alpha | Beta | Gamma | Delta | Lambda | Omicron (BA.1) | All VoC/VoI | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | ≥50% | Total | ≥50% | Total | ≥50% | Total | ≥50% | Total | ≥50% | Total | ≥50% | Total | ≥50% | |
| Nucleotide substitutions | 42 | 26 | 43 | 17 | 49 | 31 | 58 | 32 | 40 | 27 | 66 | 59 | 253 | 150 |
| Synonymous nucleotide substitutions | 14 | 7 | 10 | 2 | 17 | 9 | 14 | 3 | 13 | 6 | 14 | 10 | 74 | 31 |
| Non synonymous nucleotide substitutions | 28 | 19 | 32 | 15 | 32 | 22 | 44 | 29 | 27 | 21 | 52 | 49 | 178 | 119 |
| Aminoacid changes | 24 | 16 | 32 | 15 | 31 | 21 | 43 | 28 | 25 | 19 | 48 | 45 | 164 | 109 |
| Recurrent amino acid changes in our analysis | 2 | 3 | 2 | 5 | 3 | 8 | 10 | |||||||
| Amino acid changes absent in other Sarbecoviruses | 18 | 12 | 22 | 10 | 19 | 14 | 30 | 19 | 18 | 15 | 30 | 29 | 105 | 69 |
| Spike nucleotide substitutions | 9 | 8 | 12 | 7 | 14 | 12 | 11 | 10 | 13 | 9 | 36 | 35 | 76 | 63 |
| Spike non-synonymous nucleotide substitutions | 8 | 7 | 10 | 7 | 13 | 12 | 11 | 10 | 11 | 8 | 34 | 33 | 69 | 60 |
| Spike synonymous nucleotide substitutions | 1 | 1 | 2 | 0 | 1 | 0 | 0 | 0 | 2 | 1 | 2 | 2 | 7 | 3 |
| Spike average pairwise dN/dS vs Wuhan-Hu-1 | 1.2 | 6.53 | 5.93 | 1.19–1.63 | 1.29 | 2.79 | N/A | |||||||
| Spike aminoacid changes | 8 | 7 | 10 | 7 | 13 | 12 | 10 | 9 | 9 | 7 | 31 | 30 | 61 | 53 |
| All accessory ORFs nucleotide substitutions | 5 | 4 | 6 | 2 | 3 | 2 | 7 | 4 | 4 | 0 | 3 | 3 | 28 | 15 |
| All accessory ORFs aminoacid changes | 1 | 1 | 6 | 2 | 2 | 2 | 6 | 4 | 3 | 0 | 0 | 0 | 18 | 9 |
| Our Analysis | Recurrent van Dorp 2020 | Reccurent Rochman 2021 | ORF_aa_Position | Ref. aa | Alpha | Beta | Gamma | Delta | Lambda | Omicron | Sarbeco. Cons. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Homoplasy | No | No | ORF1ab_2287; nsp3_1469 | P | X | X | X | S:75.43 | S:97.94 | X | A |
| Homoplasy | No | No | ORF1ab_3255; nsp4_492 | T | X | X | X | I:76.13 | I:99.71 | I:100.0 | A |
| Inherited | YES | YES | ORF1ab_4715; nsp12_323 | P | L:99.5 | L:88.69 | L:99.9 | L:99.7 | L:99.71 | L:100.0 | A |
| Homoplasy | No | No | Spike_95 | T | X | X | X | I:57.1 | X | I:97.76 | |
| Homoplasy | No | No | Spike_142 | G | X | X | X | D:94.58 | X | D:99.26 | A |
| Homoplasy | No | No | Spike_417 | K | X | N:98.16 | T:100.0 | X | X | N:97.69 | A |
| Homoplasy | No | No | Spike_478 | T | X | X | X | K:99.9 | X | K:98.53 | |
| Homoplasy | No | No | Spike_484 | E | X | K:98.27 | K:100.0 | X | X | A:97.78 | A |
| Homoplasy | No | YES | Spike_501 | N | Y:99.8 | Y:98.99 | Y:100.0 | X | X | Y:95.56 | A |
| Inherited | YES | YES | Spike_614 | D | G:99.9 | G:99.9 | G:100.0 | G:100.0 | G:100.0 | G:100.0 | A |
| Inherited | No | No | Spike_655 | H | X | X | Y:99.9 | X | X | Y:100.0 | |
| Homoplasy | No | No | Spike_681 | P | H:99.8 | X | X | R:99.8 | X | H:100.0 | |
| Homoplasy | YES | No | Nucleocapsid 13 | P | X | S:14.4 | X | X | L:98.53 | L:97.04 | A |
| Inherited | YES | YES | Nucleocapsid 203 | R | K:98.0 | X | K:99.29 | M:99.8 | K:100.0 | K:98.53 | A |
| Inherited | YES | YES | Nucleocapsid 204 | G | R:84.78 | X | R:99.49 | X | R:100.0 | R:98.53 | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolaidis, M.; Papakyriakou, A.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G.; Amoutzias, G.D. Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF. Viruses 2022, 14, 707. https://doi.org/10.3390/v14040707
Nikolaidis M, Papakyriakou A, Chlichlia K, Markoulatos P, Oliver SG, Amoutzias GD. Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF. Viruses. 2022; 14(4):707. https://doi.org/10.3390/v14040707
Chicago/Turabian StyleNikolaidis, Marios, Athanasios Papakyriakou, Katerina Chlichlia, Panayotis Markoulatos, Stephen G. Oliver, and Grigorios D. Amoutzias. 2022. "Comparative Analysis of SARS-CoV-2 Variants of Concern, Including Omicron, Highlights Their Common and Distinctive Amino Acid Substitution Patterns, Especially at the Spike ORF" Viruses 14, no. 4: 707. https://doi.org/10.3390/v14040707