
Pseudorabies Virus Inhibits Expression of Liver X Receptors to Assist Viral Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cells, Viruses, and Plasmids
2.3. Chemicals and Antibodies
2.4. Cell Viability Assays
2.5. Flow Cytometry Assay
2.6. Immunoblotting Analysis
2.7. Cell Surface Biotinylation Assay
2.8. Quantitative Real-Time PCR (qRT-PCR)
2.9. RNA Interference (RNAi)
2.10. Fluorescence Recovery after Photobleaching (FRAP)
2.11. Histological Analysis
2.12. Statistical Analysis
3. Results
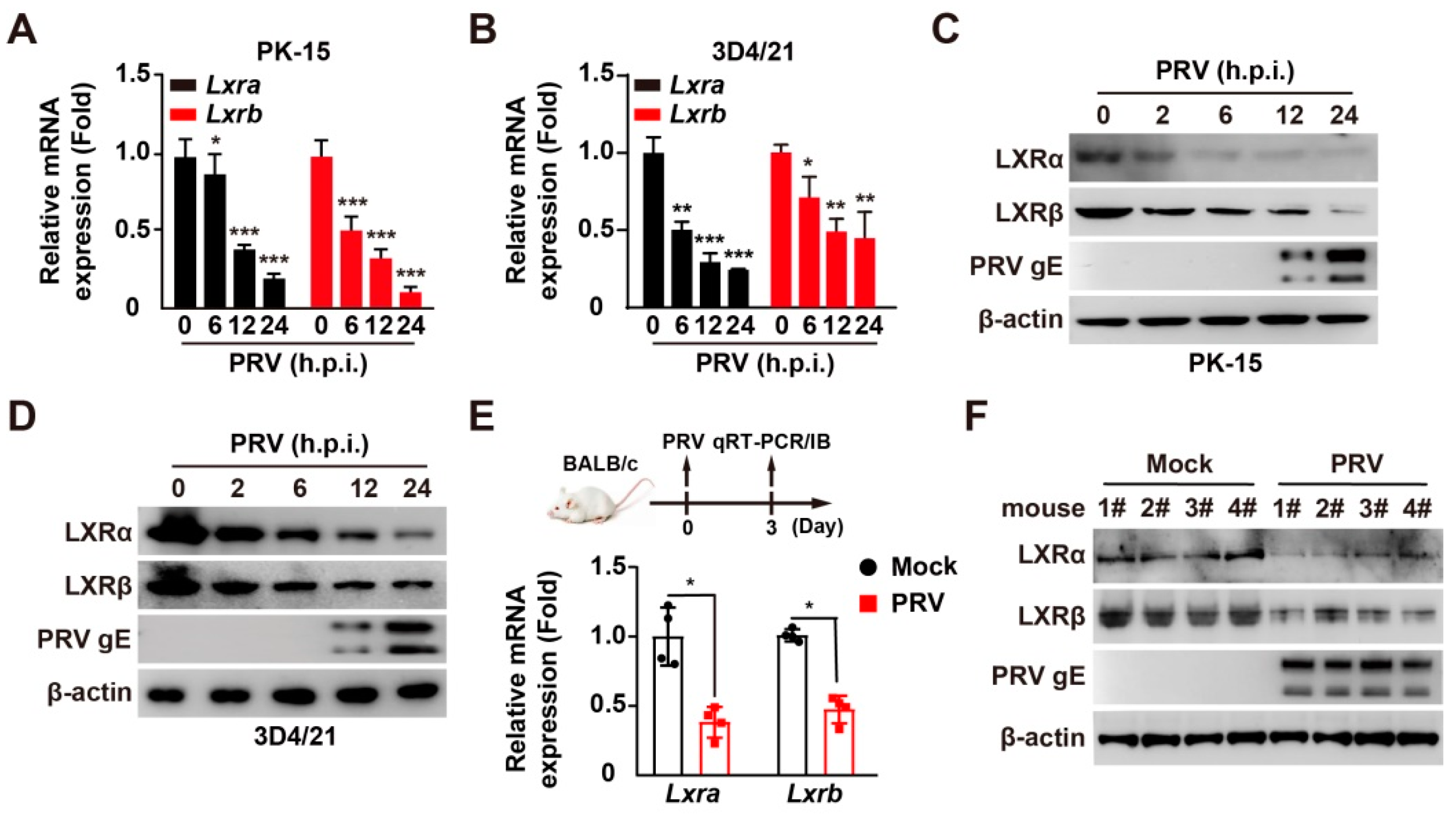
3.1. PRV Infection Inhibits LXRα and LXRβ Expression In Vitro and In Vivo
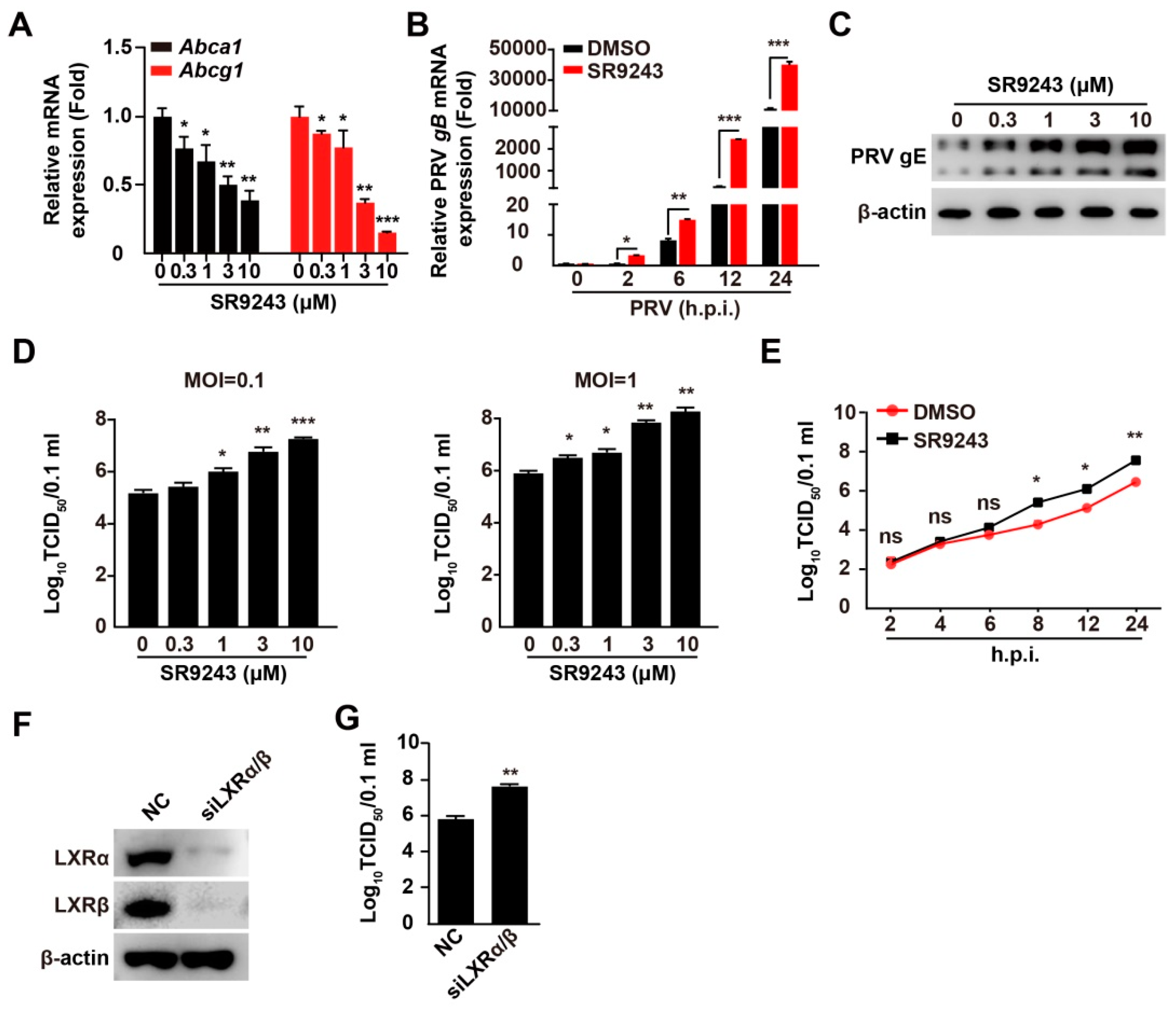
3.2. Inhibition of LXR Increases PRV Infection
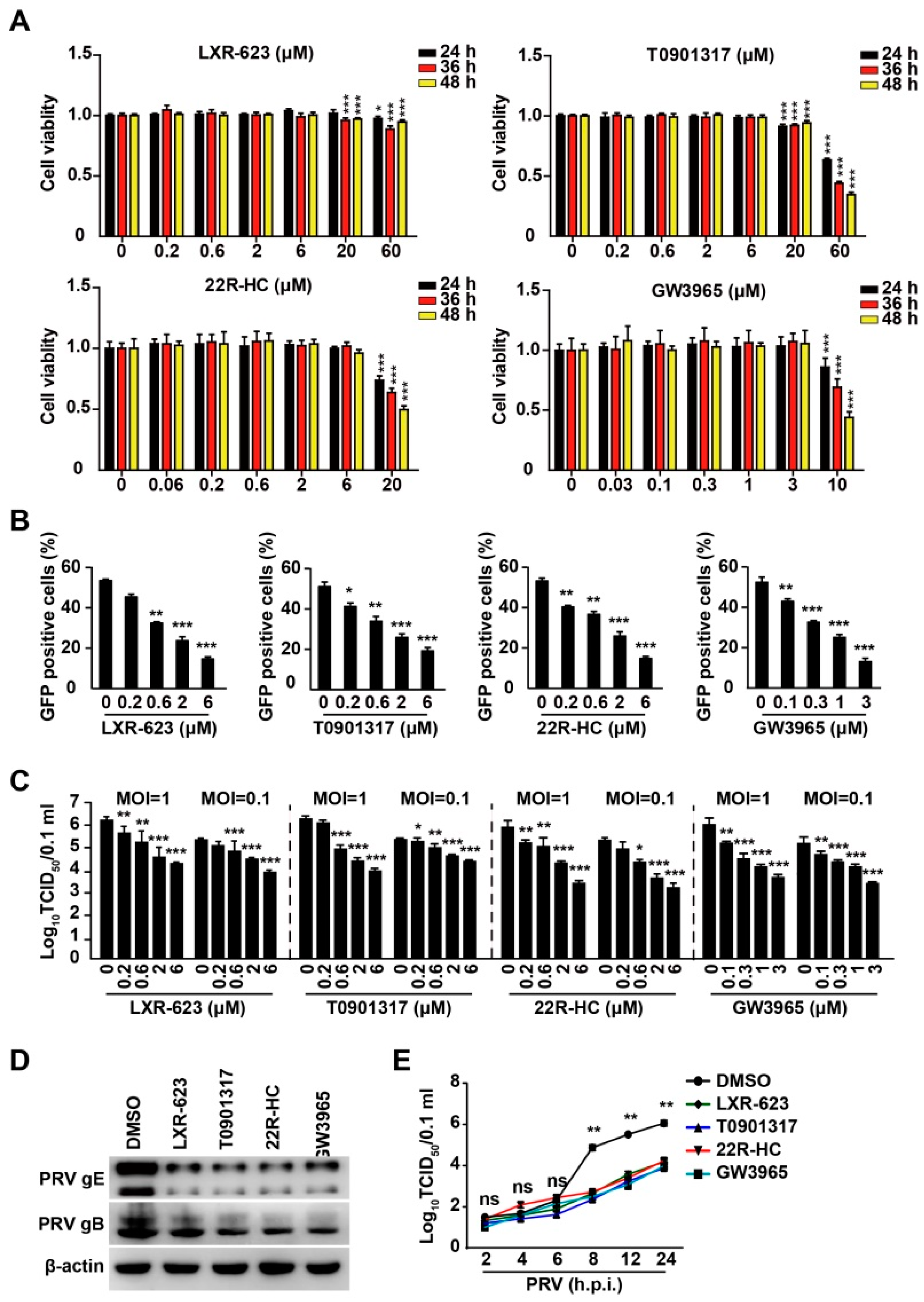
3.3. Activation of LXR by Their Agonists Inhibits PRV Infection
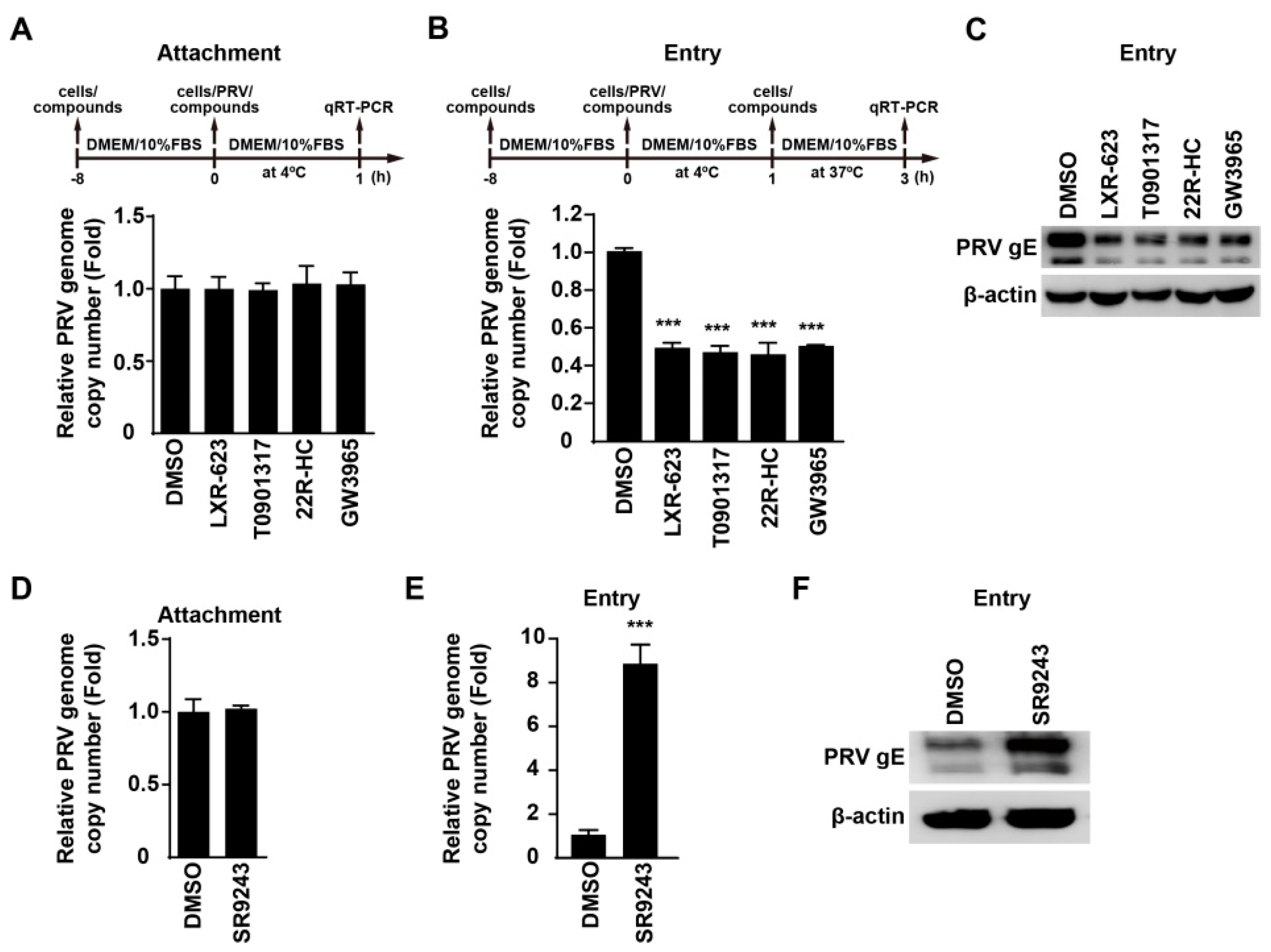
3.4. Activation of LXR Inhibits PRV Entry
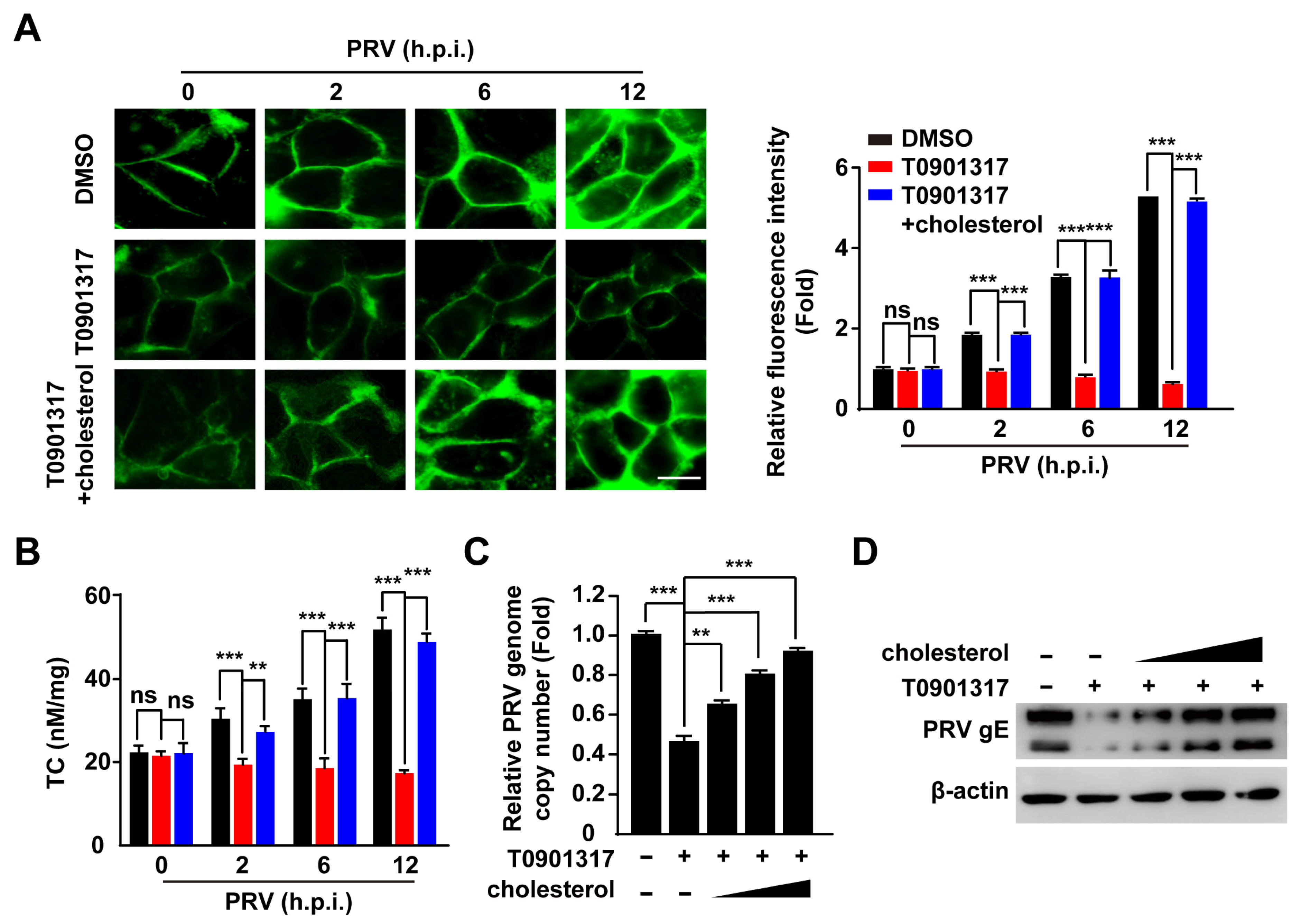
3.5. PRV Infection Increases Cellular Cholesterol Content That Is Inhibited by LXR Activation
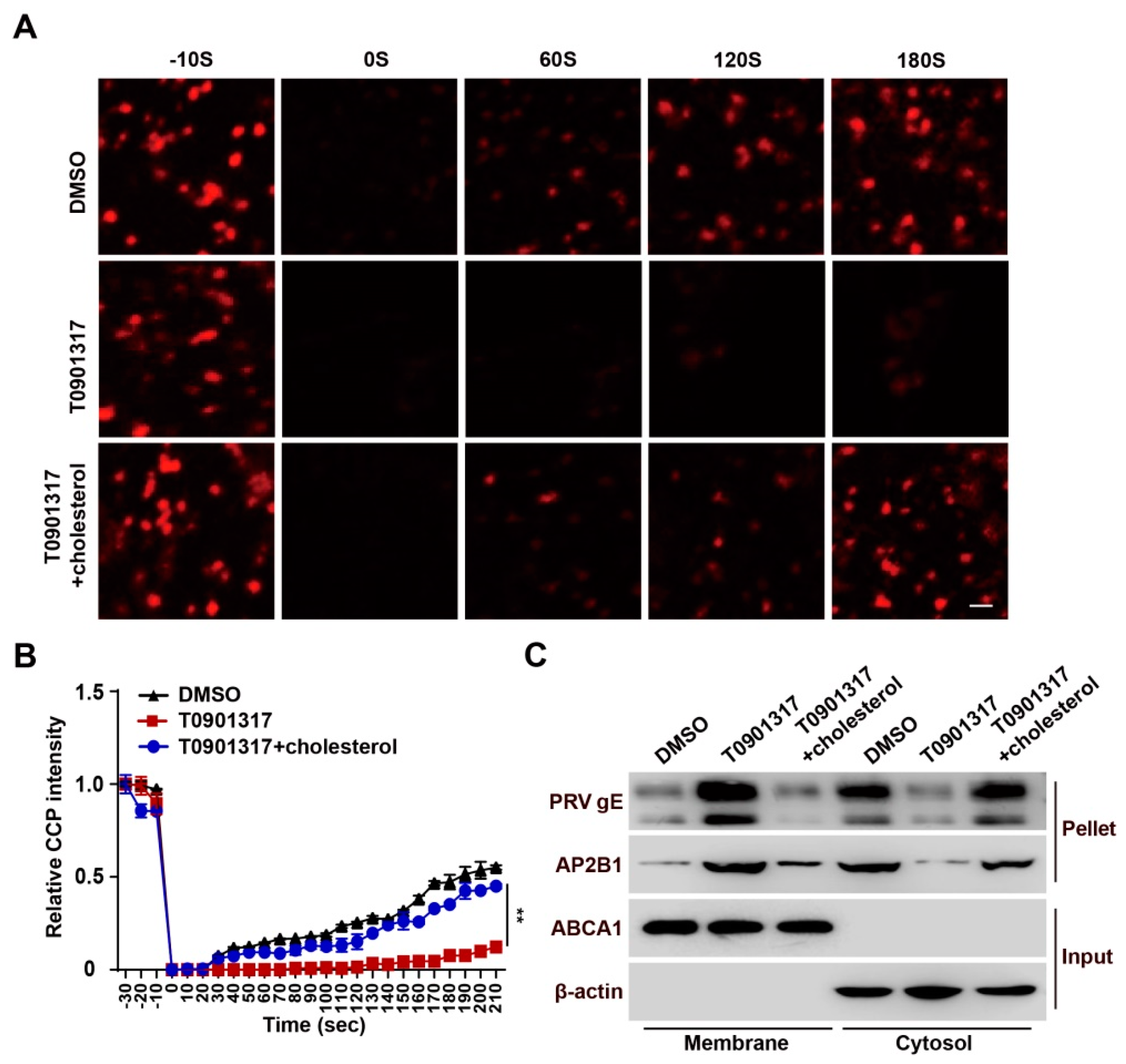
3.6. T0901317 Inhibit CCP Dynamics through Reducing Cellular Cholesterol
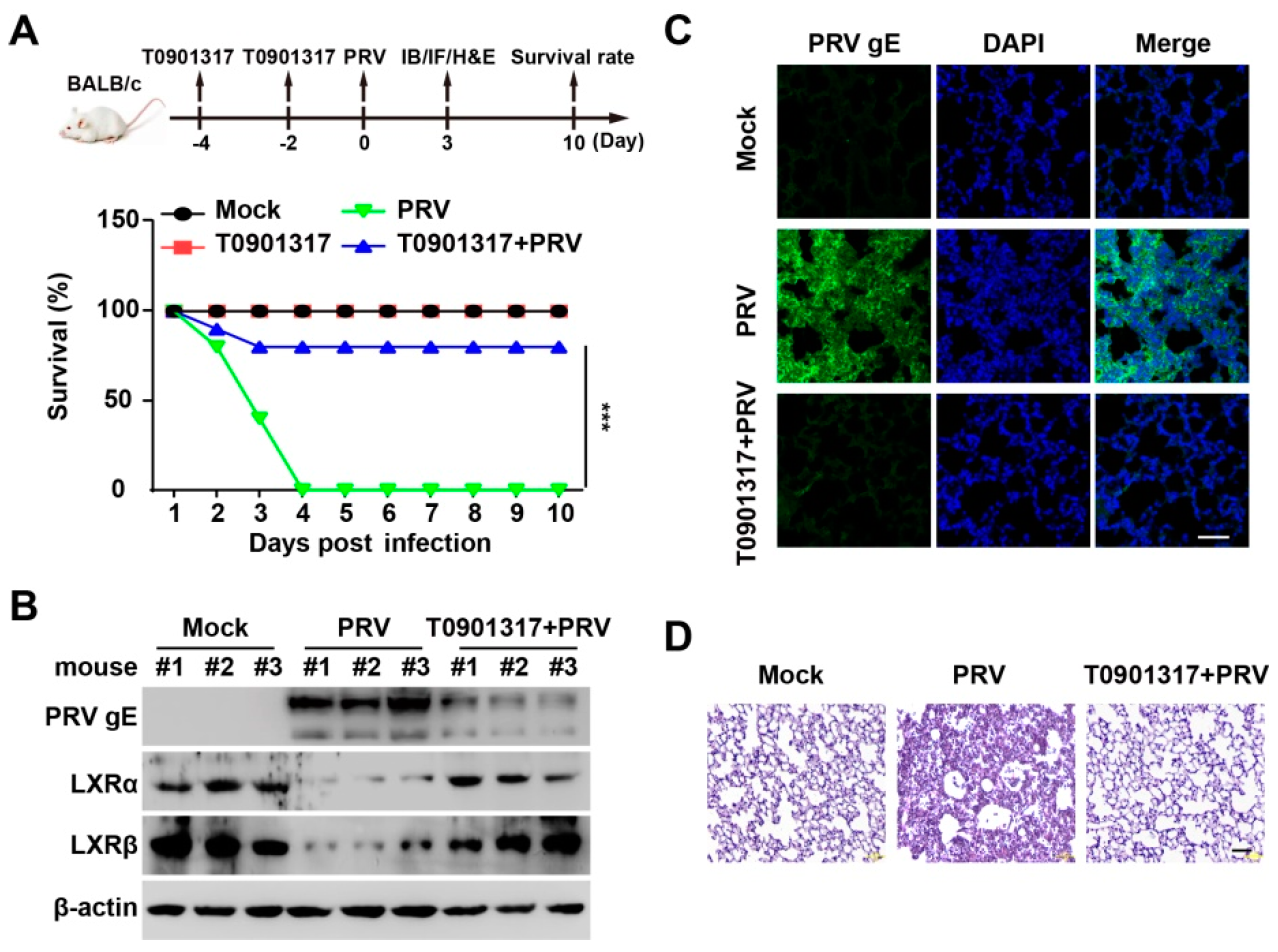
3.7. T0901317 Prevents PRV Infection In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniakowski, G.; Samorek-Salamonowicz, E. Animal herpesviruses and their zoonotic potential for cross-species infection. Ann. Agric. Environ. Med. 2015, 22, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Ai, J.W.; Weng, S.S.; Cheng, Q.; Cui, P.; Li, Y.J.; Wu, H.L.; Zhu, Y.M.; Xu, B.; Zhang, W.H. Human Endophthalmitis Caused By Pseudorabies Virus Infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Guan, H.; Li, C.; Li, Y.; Wang, S.; Zhao, X.; Zhao, Y.; Liu, Y. Characteristics of human encephalitis caused by pseudorabies virus: A case series study. Int. J. Infect. Dis. 2019, 87, 92–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, G.; Lu, J.; Zhang, W.; Gao, G.F. Pseudorabies virus: A neglected zoonotic pathogen in humans? Emerg. Microbes Infect. 2019, 8, 150–154. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.F.; Rader, D.J. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ. Res. 2005, 96, 1221–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 2018, 14, 452–463. [Google Scholar] [CrossRef]
- Chen, M.; Beaven, S.; Tontonoz, P. Identification and characterization of two alternatively spliced transcript variants of human liver X receptor alpha. J. Lipid Res. 2005, 46, 2570–2579. [Google Scholar] [CrossRef] [Green Version]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid X receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Lorizate, M.; Krausslich, H.G. Role of lipids in virus replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.X.; Sun, Y.J.; Zhan, Y.; Qu, Y.R.; Wang, H.X.; Luo, M.; Liao, Y.; Qiu, X.S.; Ding, C.; Fan, H.J.; et al. The LXR ligand GW3965 inhibits Newcastle disease virus infection by affecting cholesterol homeostasis. Arch. Virol. 2016, 161, 2491–2501. [Google Scholar] [CrossRef]
- Jiang, H.; Badralmaa, Y.; Yang, J.; Lempicki, R.; Hazen, A.; Natarajan, V. Retinoic acid and liver X receptor agonist synergistically inhibit HIV infection in CD4+ T cells by up-regulating ABCA1-mediated cholesterol efflux. Lipids Health Dis. 2012, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Bocchetta, S.; Maillard, P.; Yamamoto, M.; Gondeau, C.; Douam, F.; Lebreton, S.; Lagaye, S.; Pol, S.; Helle, F.; Plengpanich, W.; et al. Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection. PLoS ONE 2014, 9, e92140. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Wu, D.; Hu, H.; Young, J.A.T.; Yan, Z.; Gao, L. Activation of the Liver X Receptor Pathway Inhibits HBV Replication in Primary Human Hepatocytes. Hepatology 2020, 72, 1935–1948. [Google Scholar] [CrossRef]
- Lange, P.T.; Schorl, C.; Sahoo, D.; Tarakanova, V.L. Liver X Receptors Suppress Activity of Cholesterol and Fatty Acid Synthesis Pathways To Oppose Gammaherpesvirus Replication. mBio 2018, 9, e01115-18. [Google Scholar] [CrossRef] [Green Version]
- Papageorgiou, A.P.; Heggermont, W.; Rienks, M.; Carai, P.; Langouche, L.; Verhesen, W.; De Boer, R.A.; Heymans, S. Liver X receptor activation enhances CVB3 viral replication during myocarditis by stimulating lipogenesis. Cardiovasc. Res. 2015, 107, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Wang, Y.; Fikrig, E. Inhibition of Chikungunya Virus Replication in Primary Human Fibroblasts by Liver X Receptor Agonist. Antimicrob. Agents Chemother. 2019, 63, e01220-19. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chang, H.; Yang, X.; Zhao, Y.; Chen, L.; Wang, X.; Liu, H.; Wang, C.; Zhao, J. Antiviral Activity of Porcine Interferon Regulatory Factor 1 against Swine Viruses in Cell Culture. Viruses 2015, 7, 5908–5918. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, Z.F.; Wang, L.; Gao, B.; Pang, D.W.; Wang, H.Z.; Zhang, Z.L. A microfluidic platform for real-time and in situ monitoring of virus infection process. Biomicrofluidics 2012, 6, 34122. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chu, B.; Du, L.; Han, Y.; Zhang, X.; Fan, S.; Wang, Y.; Yang, G. Molecular cloning and functional characterization of porcine cyclic GMP-AMP synthase. Mol. Immunol. 2015, 65, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.L.; Ming, S.L.; Wang, C.F.; Shi, L.J.; Su, B.Q.; Wu, H.T.; Zeng, L.; Han, Y.Q.; Liu, Z.H.; et al. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 2020, 16, e1008429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffett, K.; Solt, L.A.; El-Gendy Bel, D.; Kamenecka, T.M.; Burris, T.P. A liver-selective LXR inverse agonist that suppresses hepatic steatosis. ACS Chem. Biol. 2013, 8, 559–567. [Google Scholar] [CrossRef]
- Xu, P.; Li, D.; Tang, X.; Bao, X.; Huang, J.; Tang, Y.; Yang, Y.; Xu, H.; Fan, X. LXR agonists: New potential therapeutic drug for neurodegenerative diseases. Mol. Neurobiol. 2013, 48, 715–728. [Google Scholar] [CrossRef]
- Li, G.; Su, B.; Fu, P.; Bai, Y.; Ding, G.; Li, D.; Wang, J.; Yang, G.; Chu, B. NPC1-regulated dynamic of clathrin-coated pits is essential for viral entry. Sci. China Life Sci. 2022, 65, 341–361. [Google Scholar] [CrossRef]
- Ketter, E.; Randall, G. Virus Impact on Lipids and Membranes. Annu. Rev. Virol. 2019, 6, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Oosterveer, M.H.; Grefhorst, A.; Groen, A.K.; Kuipers, F. The liver X receptor: Control of cellular lipid homeostasis and beyond Implications for drug design. Prog. Lipid Res. 2010, 49, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A.; Kennedy, M.A.; Mak, P.A. LXRs: Oxysterol-activated nuclear receptors that regulate genes controlling lipid homeostasis. Vascul. Pharmacol. 2002, 38, 249–256. [Google Scholar] [CrossRef]
- Kim, H.Y.; Cho, H.K.; Kim, H.H.; Cheong, J. Oxygenated derivatives of cholesterol promote hepatitis B virus gene expression through nuclear receptor LXRalpha activation. Virus Res. 2011, 158, 55–61. [Google Scholar] [CrossRef]
- Mlera, L.; Offerdahl, D.K.; Dorward, D.W.; Carmody, A.; Chiramel, A.I.; Best, S.M.; Bloom, M.E. The liver X receptor agonist LXR 623 restricts flavivirus replication. Emerg. Microbes Infect. 2021, 10, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, Z.; Ma, X.; Yang, X.; Chen, Y.; Sun, L.; Ma, C.; Miao, Q.R.; Hajjar, D.P.; Han, J.; et al. 25-Hydroxycholesterol activates the expression of cholesterol 25-hydroxylase in an LXR-dependent mechanism. J. Lipid Res. 2018, 59, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zeng, L.; Zhang, L.; Guo, Z.Z.; Lu, S.F.; Ming, S.L.; Li, G.L.; Wan, B.; Tian, K.G.; Yang, G.Y.; et al. Cholesterol 25-hydroxylase acts as a host restriction factor on pseudorabies virus replication. J. Gen. Virol. 2017, 98, 1467–1476. [Google Scholar] [CrossRef]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Acebes, M.A.; Gonzalez-Magaldi, M.; Sandvig, K.; Sobrino, F.; Armas-Portela, R. Productive entry of type C foot-and-mouth disease virus into susceptible cultured cells requires clathrin and is dependent on the presence of plasma membrane cholesterol. Virology 2007, 369, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyers, L.; Zwickl, H.; Blaas, D. Human rhinovirus type 2 is internalized by clathrin-mediated endocytosis. J. Virol. 2003, 77, 5360–5369. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, G.-L.; Qi, Y.-L.; Li, L.-Y.; Wang, L.-F.; Wang, C.-R.; Niu, X.-R.; Liu, T.-X.; Wang, J.; Yang, G.-Y.; et al. Pseudorabies Virus Inhibits Expression of Liver X Receptors to Assist Viral Infection. Viruses 2022, 14, 514. https://doi.org/10.3390/v14030514
Wang Y, Li G-L, Qi Y-L, Li L-Y, Wang L-F, Wang C-R, Niu X-R, Liu T-X, Wang J, Yang G-Y, et al. Pseudorabies Virus Inhibits Expression of Liver X Receptors to Assist Viral Infection. Viruses. 2022; 14(3):514. https://doi.org/10.3390/v14030514
Chicago/Turabian StyleWang, Yi, Guo-Li Li, Yan-Li Qi, Li-Yun Li, Lu-Fang Wang, Cong-Rong Wang, Xin-Rui Niu, Tao-Xue Liu, Jiang Wang, Guo-Yu Yang, and et al. 2022. "Pseudorabies Virus Inhibits Expression of Liver X Receptors to Assist Viral Infection" Viruses 14, no. 3: 514. https://doi.org/10.3390/v14030514