
Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Genetic Analysis
2.2. Clinical Evaluation and Diagnostic Tests
2.3. Neuropathological Evaluation
2.4. Biochemical Analysis
2.4.1. Brain Homogenate Processing
2.4.2. PrP Deglycosylation
2.4.3. PK Titration Curves
2.4.4. Western Blot
3. Results
3.1. Molecular Genetic Analysis
3.2. Clinical Features
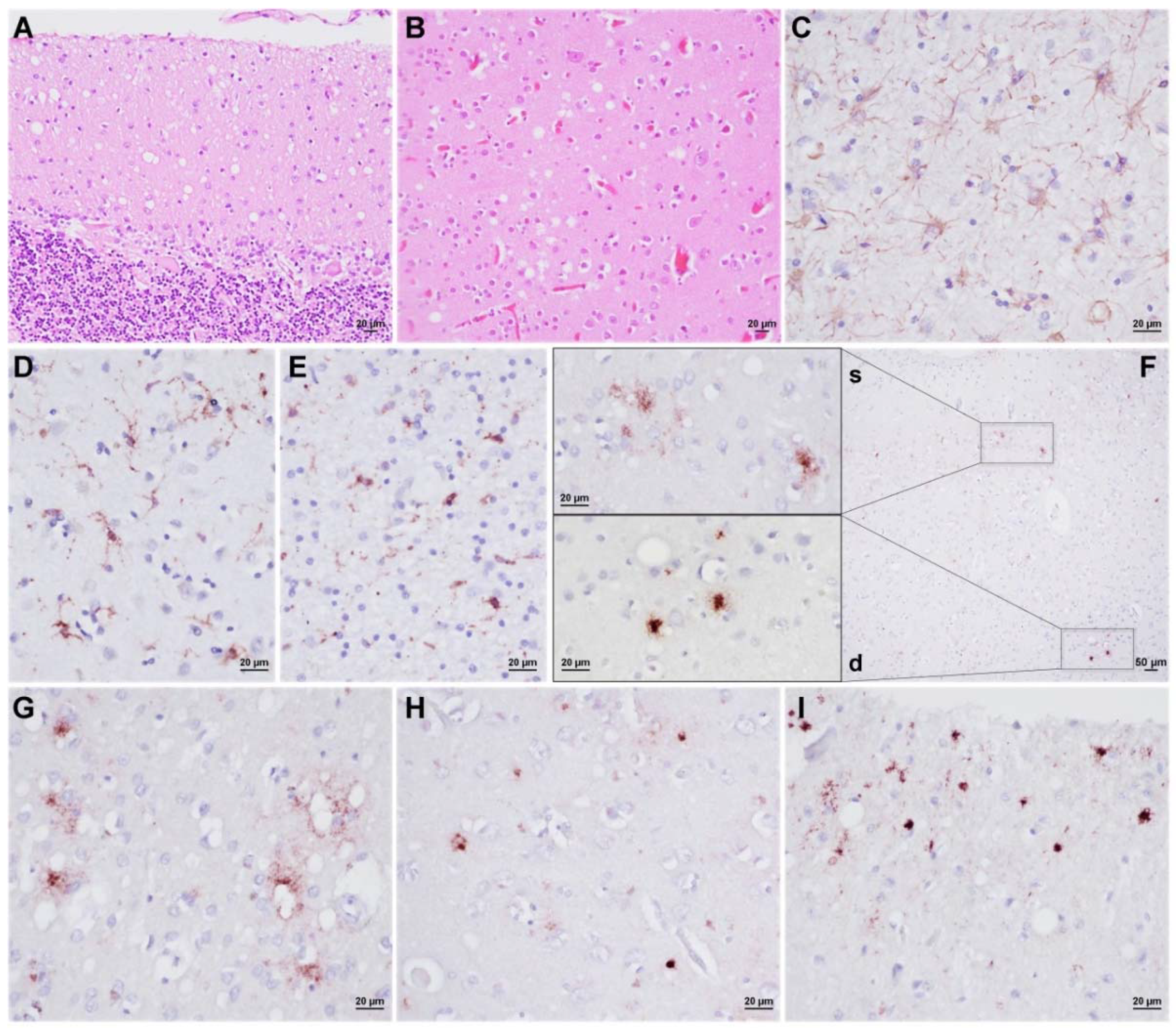
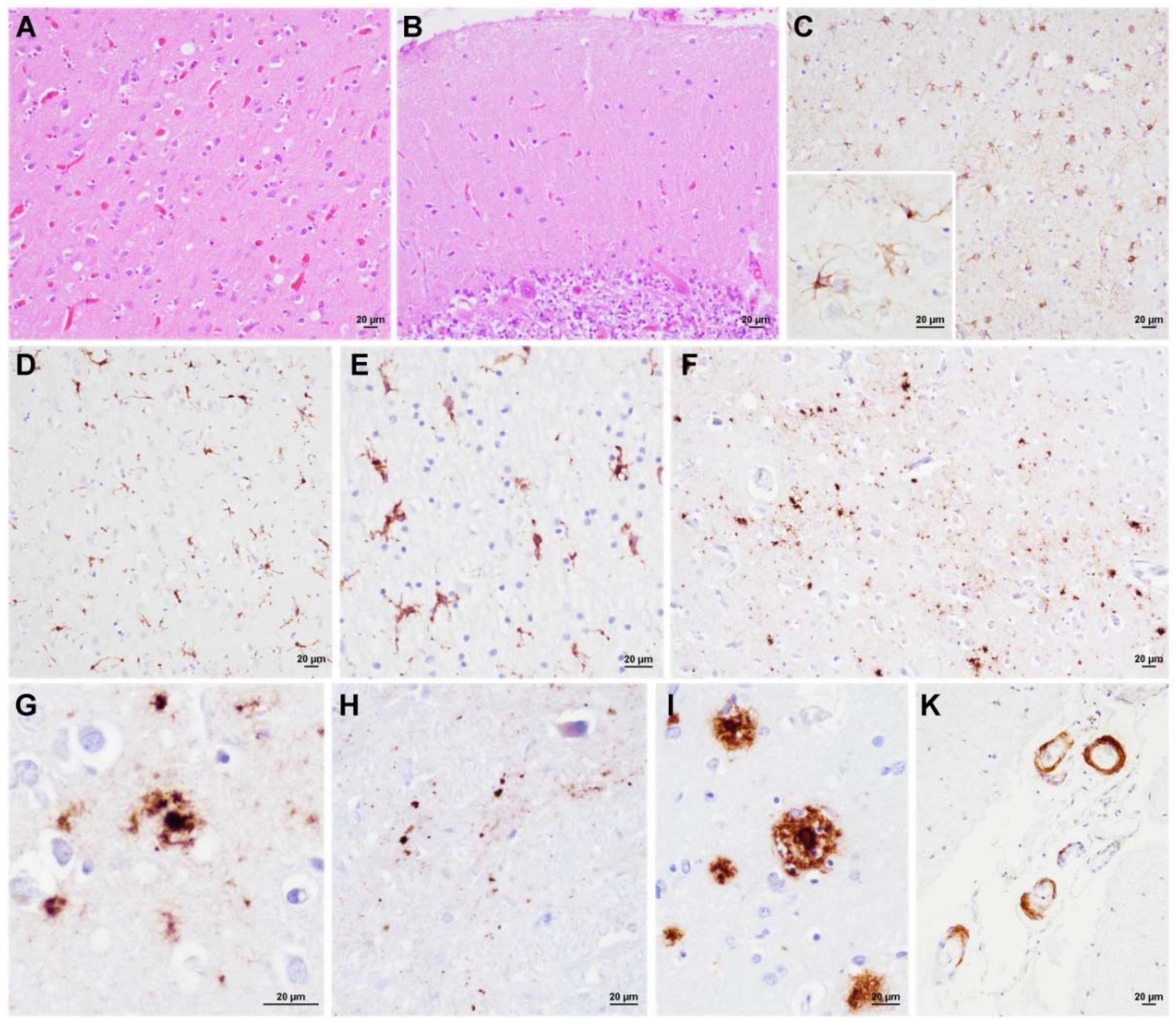
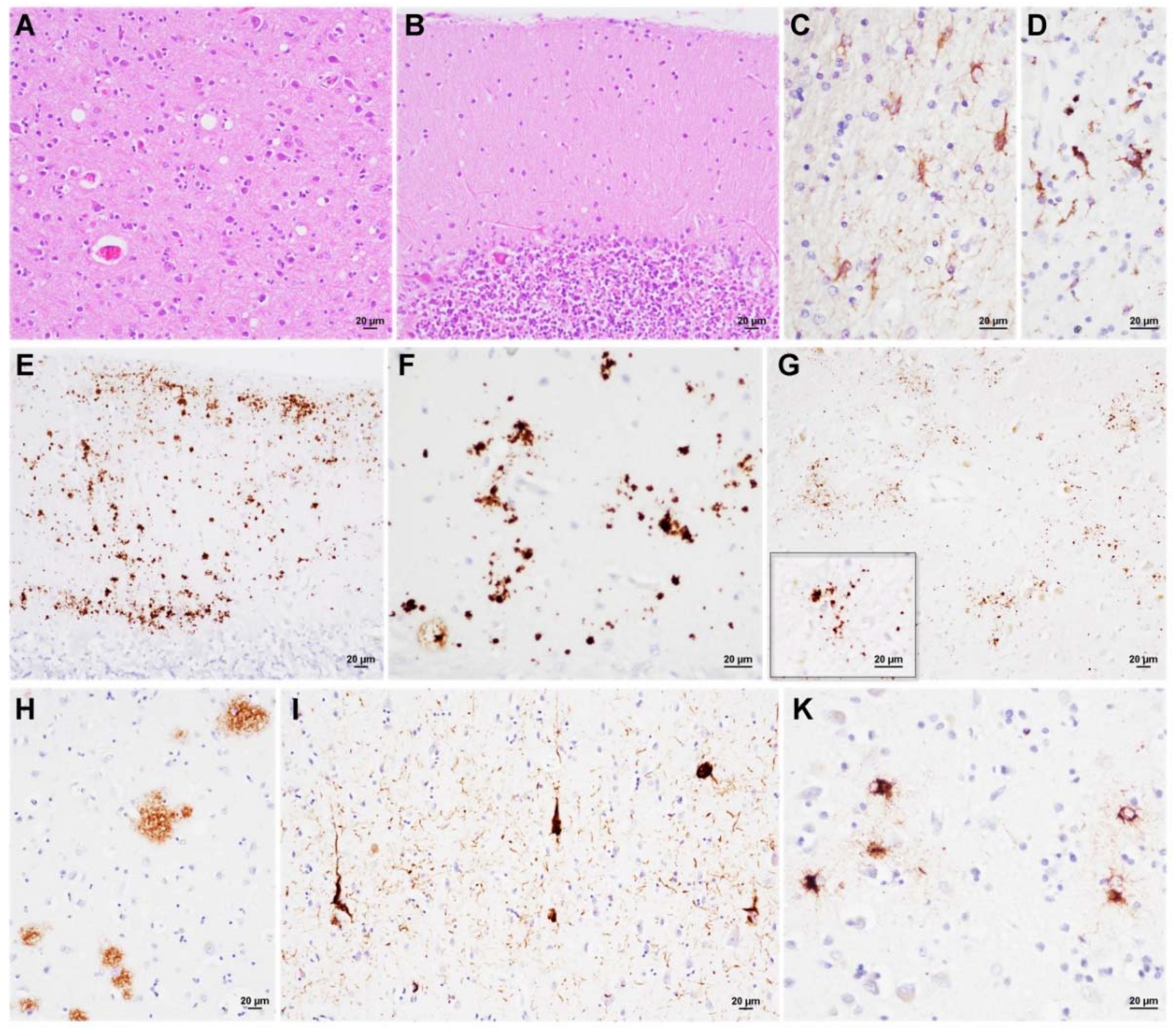
3.3. Neuropathology
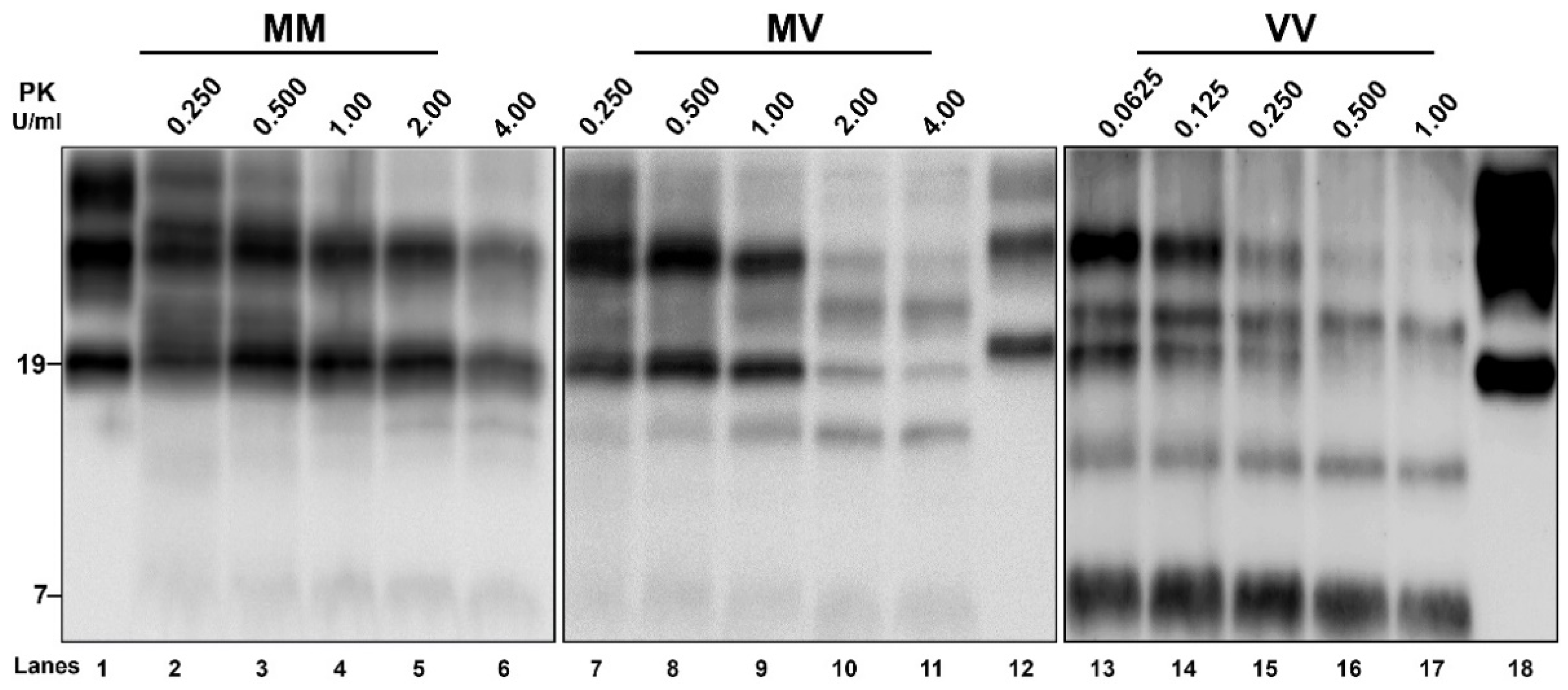
3.4. Biochemical PrPSc Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobb, N.J.; Surewicz, W.K. Prion Diseases and Their Biochemical Mechanisms. Biochemistry 2009, 48, 2574–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baiardi, S.; Rossi, M.; Capellari, S.; Parchi, P. Recent Advances in the Histo-Molecular Pathology of Human Prion Disease. Brain Pathol. 2019, 29, 278–300. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W. Sporadic and Infectious Human Prion Diseases. Cold Spring Harb. Perspect. Med. 2017, 7, a024364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capellari, S.; Strammiello, R.; Saverioni, D.; Kretzschmar, H.; Parchi, P. Genetic Creutzfeldt-Jakob Disease and Fatal Familial Insomnia: Insights into Phenotypic Variability and Disease Pathogenesis. Acta Neuropathol. 2011, 121, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of Sporadic Creutzfeldt-Jakob Disease Based on Molecular and Phenotypic Analysis of 300 Subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Parchi, P.; Strammiello, R.; Notari, S.; Giese, A.; Langeveld, J.P.M.; Ladogana, A.; Zerr, I.; Roncaroli, F.; Cras, P.; Ghetti, B.; et al. Incidence and Spectrum of Sporadic Creutzfeldt-Jakob Disease Variants with Mixed Phenotype and Co-Occurrence of PrPSc Types: An Updated Classification. Acta Neuropathol. 2009, 118, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Notari, S.; Appleby, B.S.; Gambetti, P. Variably Protease-Sensitive Prionopathy. Handb. Clin. Neurol. 2018, 153, 175–190. [Google Scholar] [CrossRef]
- Gambetti, P.; Dong, Z.; Yuan, J.; Xiao, X.; Zheng, M.; Alshekhlee, A.; Castellani, R.; Cohen, M.; Barria, M.A.; Gonzalez-Romero, D.; et al. A Novel Human Disease with Abnormal Prion Protein Sensitive to Protease. Ann. Neurol. 2008, 63, 697–708. [Google Scholar] [CrossRef]
- Saverioni, D.; Notari, S.; Capellari, S.; Poggiolini, I.; Giese, A.; Kretzschmar, H.A.; Parchi, P. Analyses of Protease Resistance and Aggregation State of Abnormal Prion Protein across the Spectrum of Human Prions. J. Biol. Chem. 2013, 288, 27972–27985. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.-Q.; Puoti, G.; Xiao, X.; Yuan, J.; Qing, L.; Cali, I.; Shimoji, M.; Langeveld, J.P.M.; Castellani, R.; Notari, S.; et al. Variably Protease-Sensitive Prionopathy: A New Sporadic Disease of the Prion Protein. Ann. Neurol. 2010, 68, 162–172. [Google Scholar] [CrossRef]
- Aizpurua, M.; Selvackadunco, S.; Yull, H.; Kipps, C.M.; Ironside, J.W.; Bodi, I. Variably Protease-Sensitive Prionopathy Mimicking Frontotemporal Dementia. Neuropathology 2019, 39, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Head, M.W.; van Gool, W.A.; Baas, F.; Yull, H.; Ironside, J.W.; Rozemuller, A.J.M. The First Case of Protease-Sensitive Prionopathy (PSPr) in The Netherlands: A Patient with an Unusual GSS-like Clinical Phenotype. J. Neurol. Neurosurg. Psychiatry 2010, 81, 1052–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, M.W.; Yull, H.M.; Ritchie, D.L.; Langeveld, J.P.; Fletcher, N.A.; Knight, R.S.; Ironside, J.W. Variably Protease-Sensitive Prionopathy in the UK: A Retrospective Review 1991–2008. Brain 2013, 136, 1102–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Pascual, M.; Rossi, M.; Gámez, J.; Lladó, A.; Valls, J.; Grau-Rivera, O.; Ávila Polo, R.; Llorens, F.; Zerr, I.; Ferrer, I.; et al. Variably Protease-Sensitive Prionopathy Presenting within ALS/FTD Spectrum. Ann. Clin. Transl. Neurol. 2018, 5, 1297–1302. [Google Scholar] [CrossRef] [Green Version]
- Baiardi, S.; Rossi, M.; Mammana, A.; Appleby, B.S.; Barria, M.A.; Calì, I.; Gambetti, P.; Gelpi, E.; Giese, A.; Ghetti, B.; et al. Phenotypic Diversity of Genetic Creutzfeldt-Jakob Disease: A Histo-Molecular-Based Classification. Acta Neuropathol. 2021, 142, 707–728. [Google Scholar] [CrossRef]
- Baiardi, S.; Abu-Rumeileh, S.; Rossi, M.; Zenesini, C.; Bartoletti-Stella, A.; Polischi, B.; Capellari, S.; Parchi, P. Antemortem CSF Aβ42/Aβ40 Ratio Predicts Alzheimer’s Disease Pathology Better than Aβ42 in Rapidly Progressive Dementias. Ann. Clin. Transl. Neurol. 2019, 6, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-Specific and Surrogate CSF Biomarkers in Creutzfeldt-Jakob Disease: Diagnostic Accuracy in Relation to Molecular Subtypes and Analysis of Neuropathological Correlates of p-Tau and Aβ42 Levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, A.; Baiardi, S.; Hughson, A.G.; McKenzie, N.; Moda, F.; Rossi, M.; Capellari, S.; Green, A.; Giaccone, G.; Caughey, B.; et al. High Diagnostic Value of Second Generation CSF RT-QuIC across the Wide Spectrum of CJD Prions. Sci. Rep. 2017, 7, 10655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Armstrong, J.; Capellari, S.; Parchi, P.; Arzberger, T.; Bell, J.; Budka, H.; Ströbel, T.; Giaccone, G.; Rossi, G.; et al. Effects of Formalin Fixation, Paraffin Embedding, and Time of Storage on DNA Preservation in Brain Tissue: A BrainNet Europe Study. Brain Pathol. 2007, 17, 297–303. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A Beta-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Arzberger, T.; Al-Sarraj, S.; Bodi, I.; Bogdanovic, N.; Braak, H.; Bugiani, O.; Del-Tredici, K.; Ferrer, I.; Gelpi, E.; et al. Staging of Neurofibrillary Pathology in Alzheimer’s Disease: A Study of the BrainNet Europe Consortium. Brain Pathol. 2008, 18, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Alafuzoff, I.; Ince, P.G.; Arzberger, T.; Al-Sarraj, S.; Bell, J.; Bodi, I.; Bogdanovic, N.; Bugiani, O.; Ferrer, I.; Gelpi, E.; et al. Staging/Typing of Lewy Body Related Alpha-Synuclein Pathology: A Study of the BrainNet Europe Consortium. Acta Neuropathol. 2009, 117, 635–652. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE): Consensus Working Group Report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Saverioni, D.; Di Bari, M.; Baiardi, S.; Lemstra, A.W.; Pirisinu, L.; Capellari, S.; Rozemuller, A.; Nonno, R.; Parchi, P. Atypical Creutzfeldt-Jakob Disease with PrP-Amyloid Plaques in White Matter: Molecular Characterization and Transmission to Bank Voles Show the M1 Strain Signature. Acta Neuropathol. Commun. 2017, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, Y.; Kaku-Ushiki, Y.; Iwamaru, Y.; Muramoto, T.; Kitamoto, T.; Yokoyama, T.; Mohri, S.; Tagawa, Y. A Novel Anti-Prion Protein Monoclonal Antibody and Its Single-Chain Fragment Variable Derivative with Ability to Inhibit Abnormal Prion Protein Accumulation in Cultured Cells. Microbiol. Immunol. 2010, 54, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Piccardo, P.; Seiler, C.; Dlouhy, S.R.; Young, K.; Farlow, M.R.; Prelli, F.; Frangione, B.; Bugiani, O.; Tagliavini, F.; Ghetti, B. Proteinase-K-Resistant Prion Protein Isoforms in Gerstmann-Sträussler-Scheinker Disease (Indiana Kindred). J. Neuropathol. Exp. Neurol. 1996, 55, 1157–1163. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.G.; Ferrer, I.; Grinberg, L.T.; Alafuzoff, I.; Attems, J.; Budka, H.; Cairns, N.J.; Crary, J.F.; Duyckaerts, C.; Ghetti, B.; et al. Aging-Related Tau Astrogliopathy (ARTAG): Harmonized Evaluation Strategy. Acta Neuropathol. 2016, 131, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.-Q.; Gambetti, P.; Xiao, X.; Yuan, J.; Langeveld, J.; Pirisinu, L. Prions in Variably Protease-Sensitive Prionopathy: An Update. Pathogens 2013, 2, 457–471. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xiao, X.; Ding, M.; Yuan, J.; Foutz, A.; Moudjou, M.; Kitamoto, T.; Langeveld, J.P.M.; Cui, L.; Zou, W.-Q. Further Characterization of Glycoform-Selective Prions of Variably Protease-Sensitive Prionopathy. Pathogens 2021, 10, 513. [Google Scholar] [CrossRef]
- Makarava, N.; Chang, J.C.-Y.; Baskakov, I.V. Region-Specific Sialylation Pattern of Prion Strains Provides Novel Insight into Prion Neurotropism. Int. J. Mol. Sci. 2020, 21, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghetti, B.; Piccardo, P.; Zanusso, G. Dominantly Inherited Prion Protein Cerebral Amyloidoses—A Modern View of Gerstmann-Sträussler-Scheinker. Handb. Clin. Neurol. 2018, 153, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Notari, S.; Xiao, X.; Espinosa, J.C.; Cohen, Y.; Qing, L.; Aguilar-Calvo, P.; Kofskey, D.; Cali, I.; Cracco, L.; Kong, Q.; et al. Transmission Characteristics of Variably Protease-Sensitive Prionopathy. Emerg. Infect. Dis. 2014, 20, 2006–2014. [Google Scholar] [CrossRef] [Green Version]
- Diack, A.B.; Ritchie, D.L.; Peden, A.H.; Brown, D.; Boyle, A.; Morabito, L.; Maclennan, D.; Burgoyne, P.; Jansen, C.; Knight, R.S.; et al. Variably Protease-Sensitive Prionopathy, a Unique Prion Variant with Inefficient Transmission Properties. Emerg. Infect. Dis. 2014, 20, 1969–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, D.D.; Wrona, A.; Foutz, A.; Blevins, J.; Glisic, K.; Person, M.; Maddox, R.A.; Belay, E.D.; Schonberger, L.B.; Tatsuoka, C.; et al. Diagnosis of Prion Diseases by RT-QuIC Results in Improved Surveillance. Neurology 2020, 95, e1017–e1026. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Head, M.W.; Lowrie, S.; Chohan, G.; Knight, R.; Scoones, D.J.; Ironside, J.W. Variably Protease-Sensitive Prionopathy in a PRNP Codon 129 Heterozygous UK Patient with Co-Existing Tau, α Synuclein and Aβ Pathology. Acta Neuropathol. 2010, 120, 821–823. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Rahimi, J.; Ströbel, T.; Lutz, M.I.; Regelsberger, G.; Streichenberger, N.; Perret-Liaudet, A.; Höftberger, R.; Liberski, P.P.; Budka, H.; et al. Tau Pathology in Creutzfeldt-Jakob Disease Revisited. Brain Pathol. 2017, 27, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Capellari, S.; Powers, J.; Petersen, R.; Gambetti, P.; Parchi, P. Novel Molecular and Clinico-Pathologic Phenotype of Prion Disease in an American. Neurology 1998, 50, A235. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Codon 129 Genotype | Age at Onset | Disease Duration | Presenting Features | EEG | MRI | CSF 14-3-3 | CSF RT-QuIC | Alternative Diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| #1 | Met/Met | 67 | 2.5 | Gait unsteadiness | Non-specific | Na | Na | Na | MSA |
| #2 | Met/Val | 79 | 0.5 | Memory loss, behavioral changes | Non-specific | Non-specific T2 white matter hyperintensities | Na | Na | AD |
| #3 | Val/Val | 72 | 3.0 | Memory loss, behavioral changes | Non-specific | Non-specific T2- white matter hyperintensities | + | + | CJD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baiardi, S.; Mammana, A.; Rossi, M.; Ladogana, A.; Carlà, B.; Gambetti, P.; Capellari, S.; Parchi, P. Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129. Viruses 2022, 14, 367. https://doi.org/10.3390/v14020367
Baiardi S, Mammana A, Rossi M, Ladogana A, Carlà B, Gambetti P, Capellari S, Parchi P. Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129. Viruses. 2022; 14(2):367. https://doi.org/10.3390/v14020367
Chicago/Turabian StyleBaiardi, Simone, Angela Mammana, Marcello Rossi, Anna Ladogana, Benedetta Carlà, Pierluigi Gambetti, Sabina Capellari, and Piero Parchi. 2022. "Phenotypic Heterogeneity of Variably Protease-Sensitive Prionopathy: A Report of Three Cases Carrying Different Genotypes at PRNP Codon 129" Viruses 14, no. 2: 367. https://doi.org/10.3390/v14020367