
Targeting Human Osteoarthritic Chondrocytes with Ligand Directed Bacteriophage-Based Particles

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
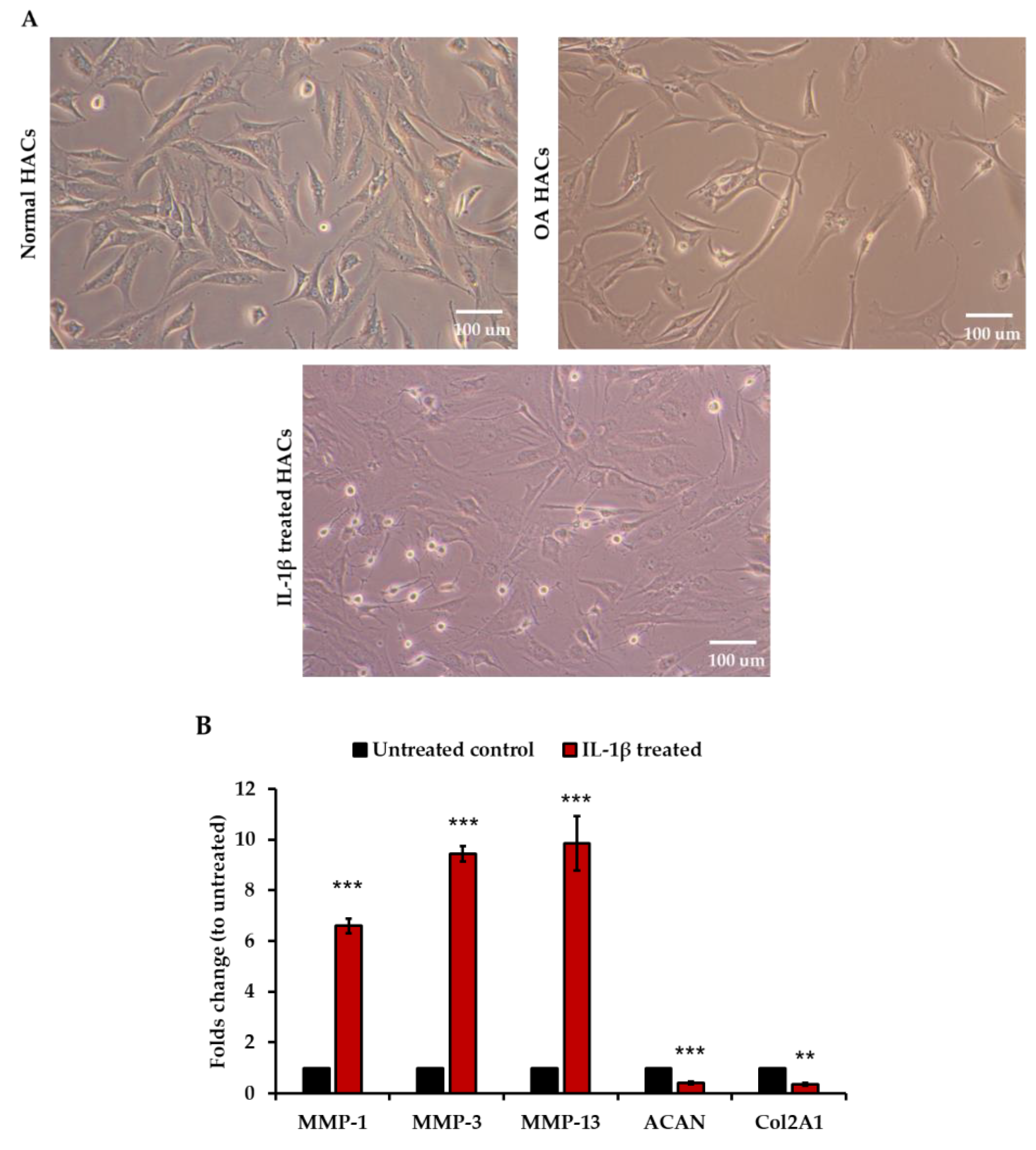
2.1. Cell Culture
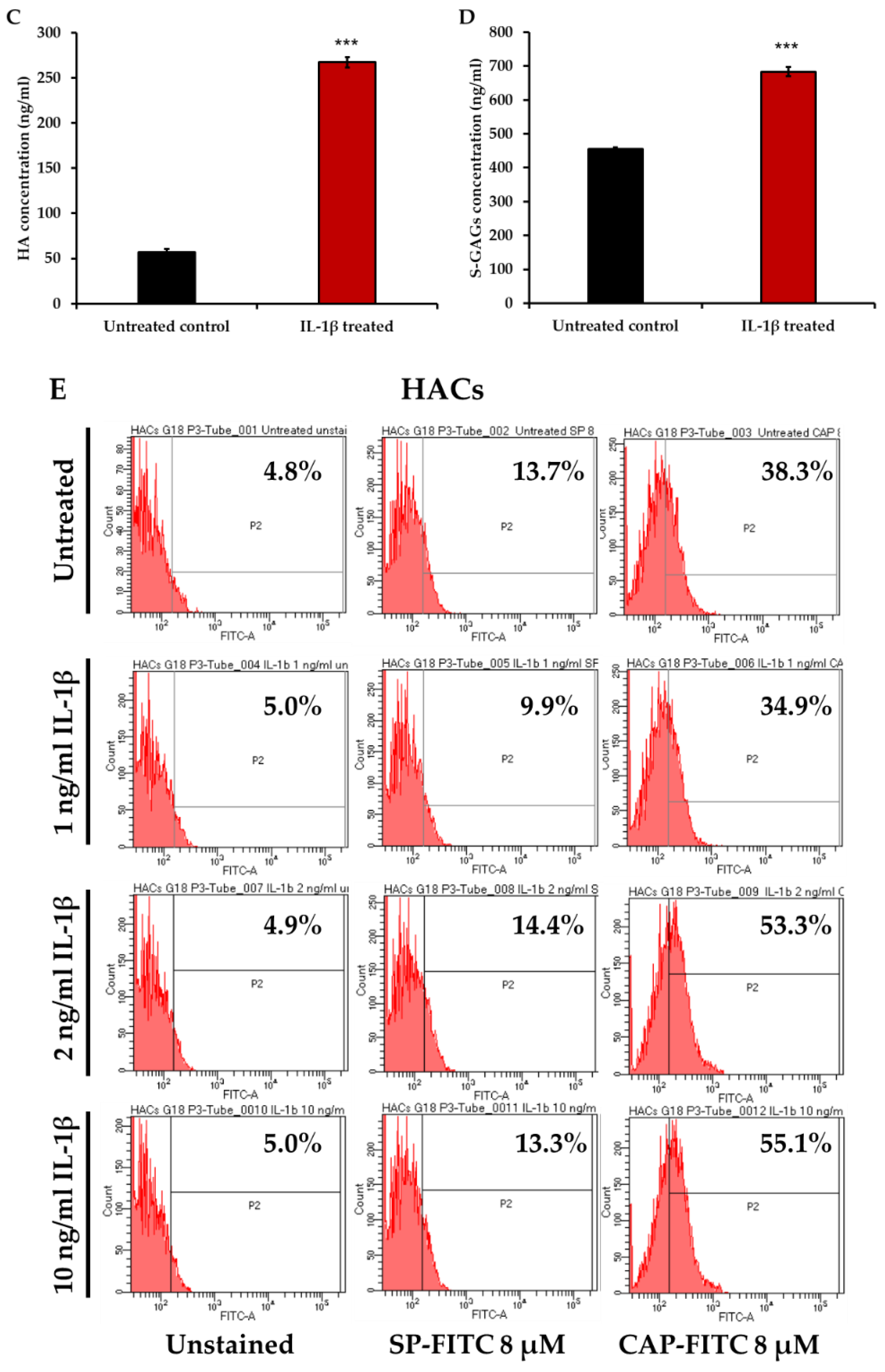
2.2. Determination of CAP Ligand Binding Affinity to HACs versus PHSFs
2.3. IL-1β Treatment
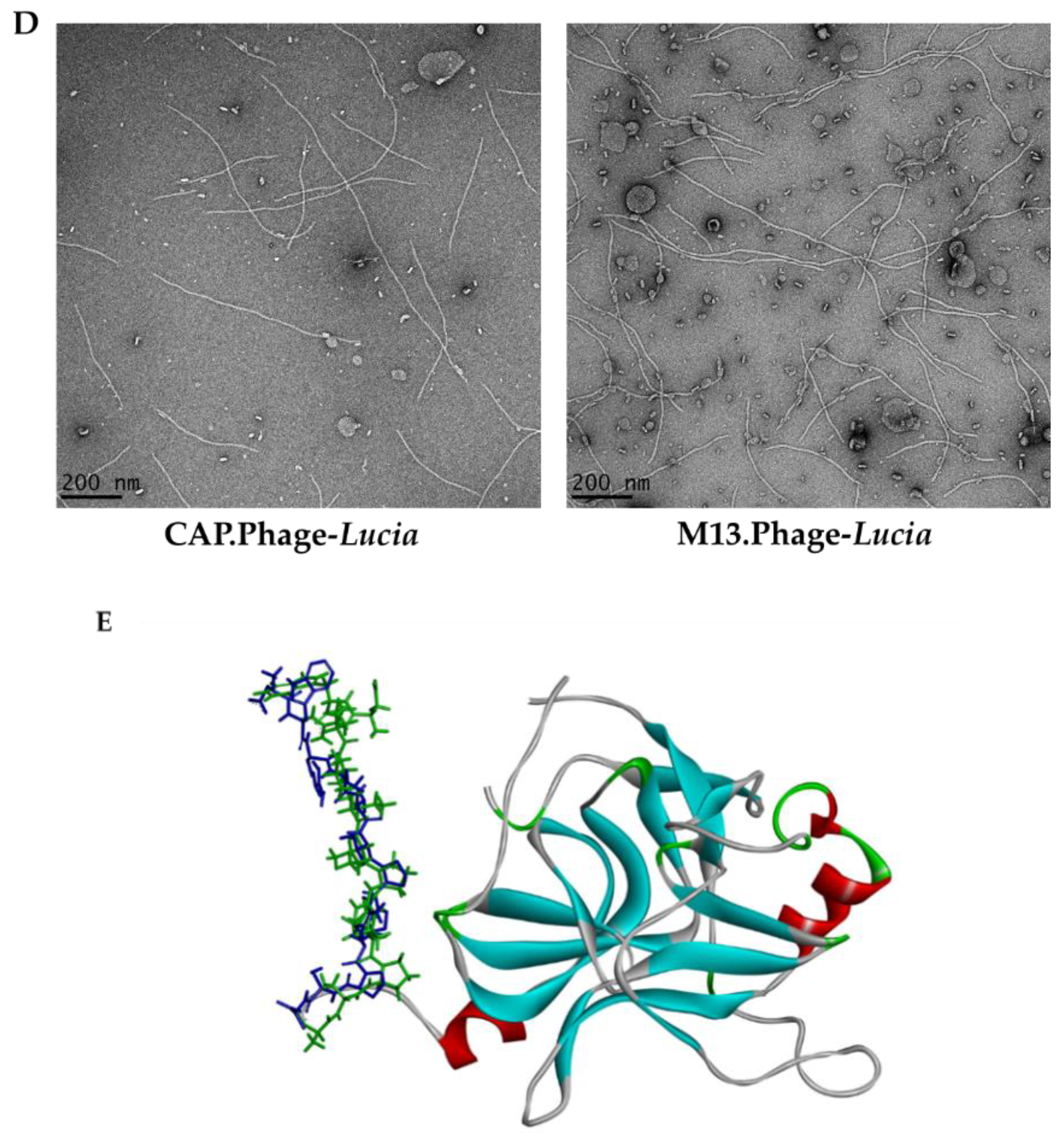
2.4. Molecular Modelling
2.5. RNA Extraction and RT-qPCR
2.6. Hyaluronic Acid (HA) ELISA
2.7. DMMB Assay for Sulfated Glycosaminoglycans (s-GAGs) Levels
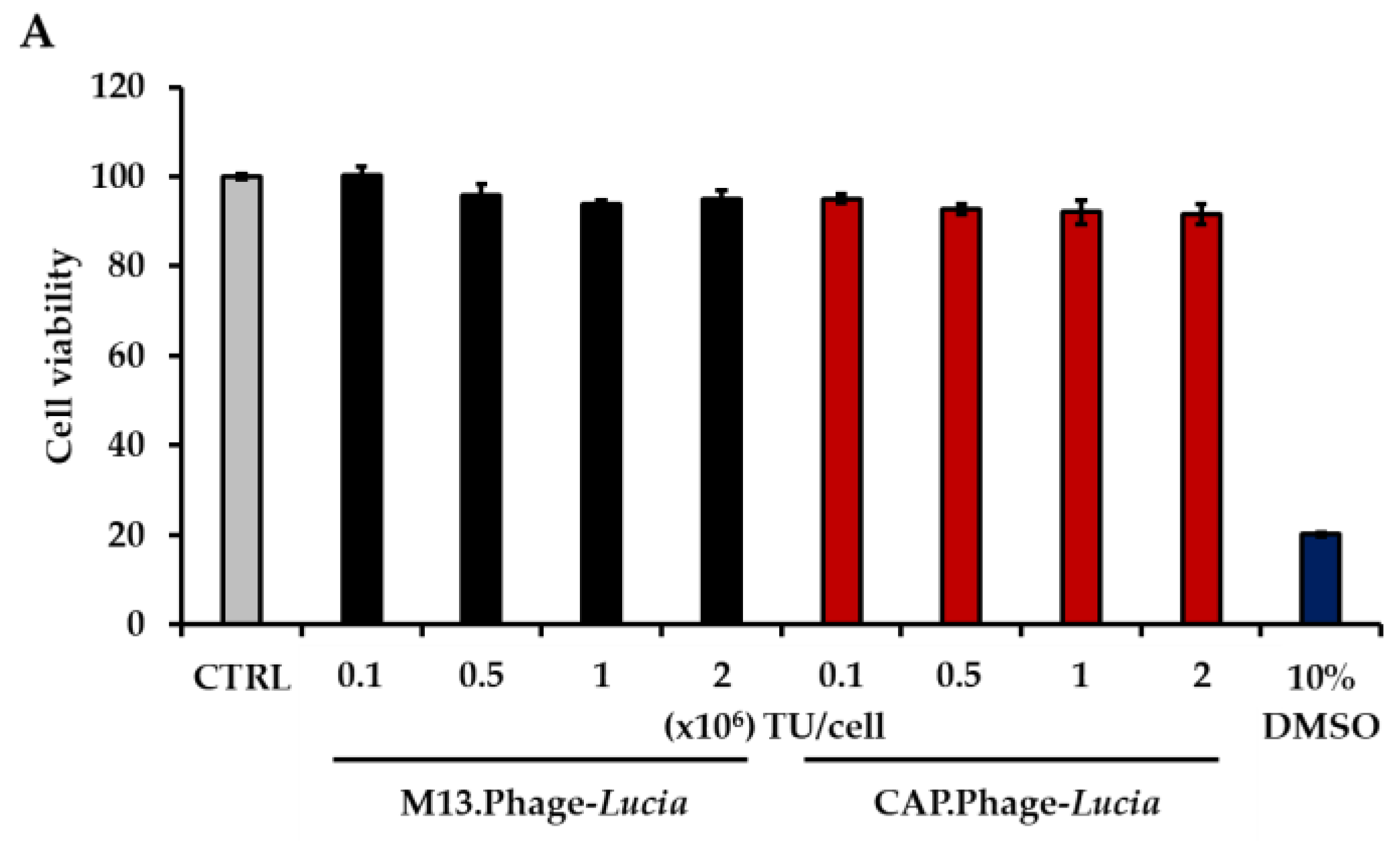
2.8. Cell Viability Assay
2.9. Electron Microscope Imaging of Phage Particles
2.10. Construction and Production of CAP.Phage Particle
2.11. Transduction of Primary HACs with Phage-Lucia
2.12. Statistical Analysis
3. Results
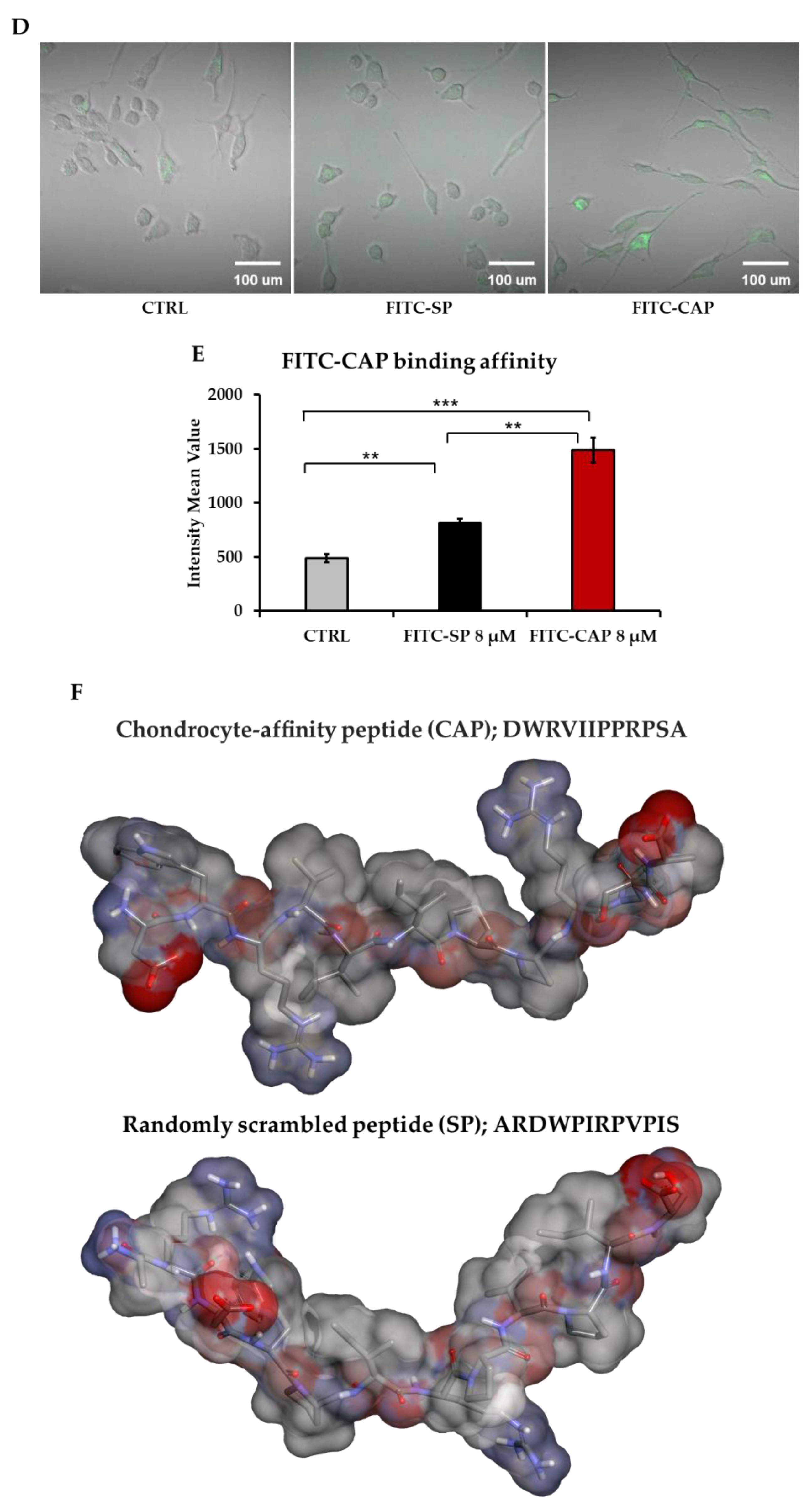
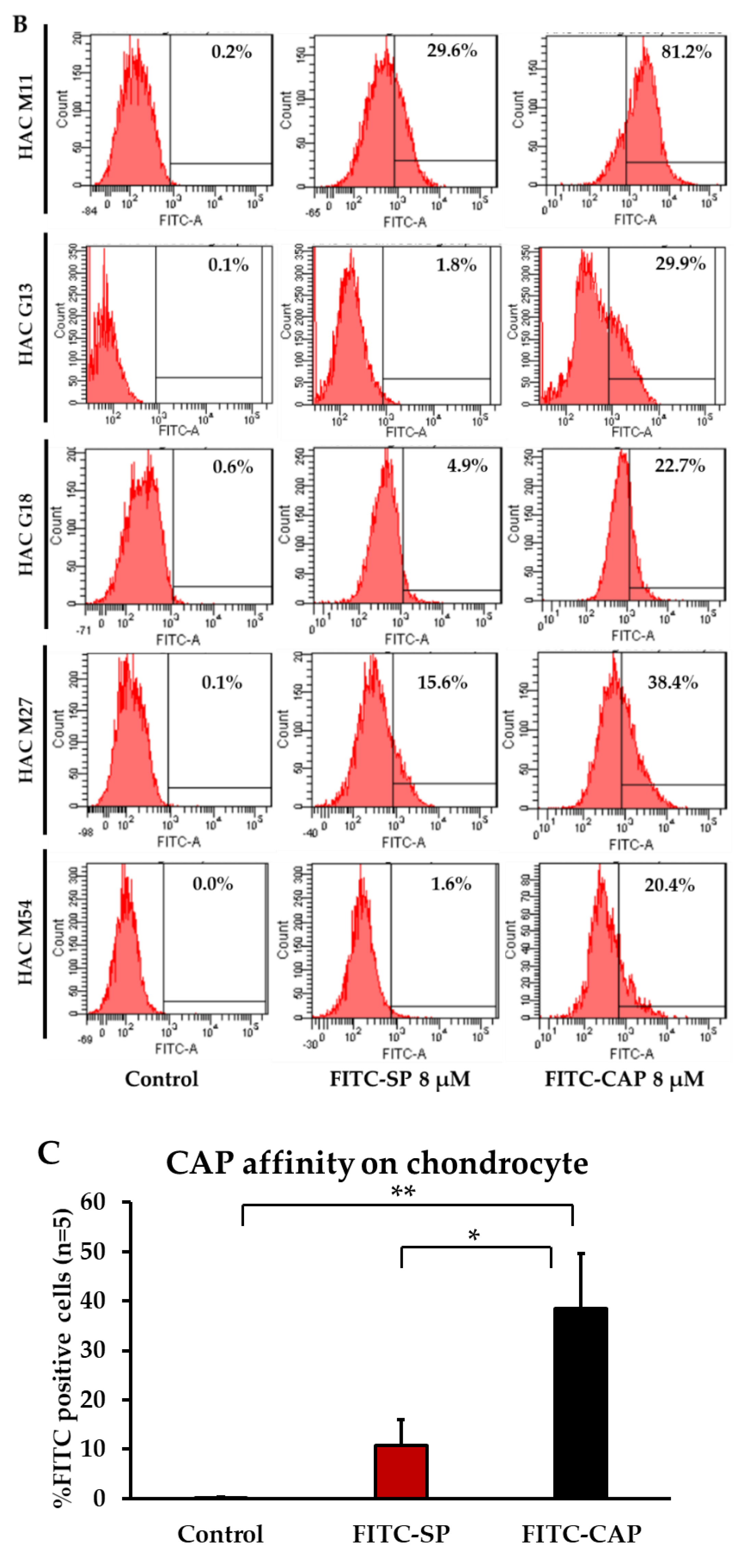
3.1. CAP Peptide Selectively Binds to Primary Human Chondrocytes in a Dose-Dependent Manner
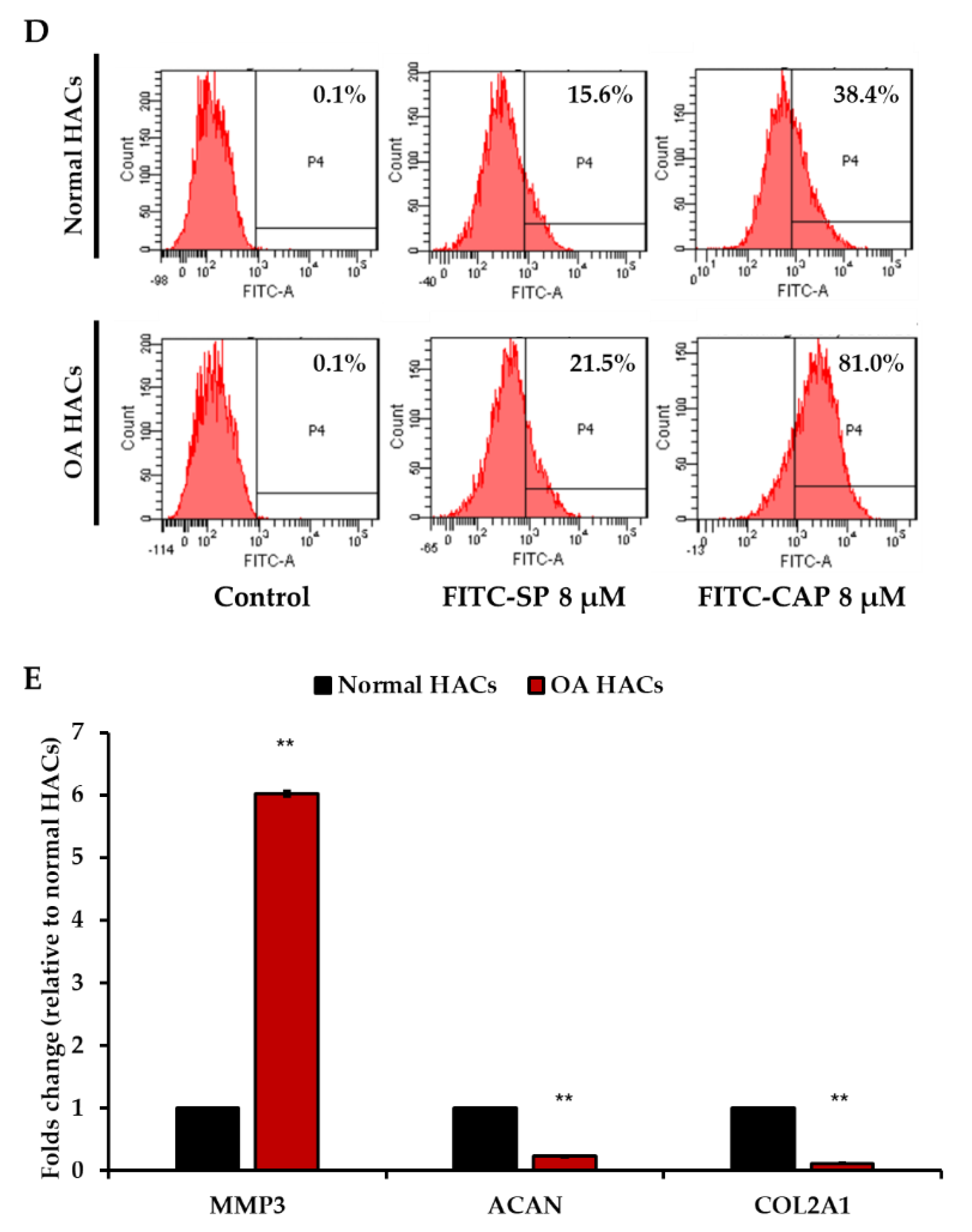
3.2. Pathological Condition of Primary HACs Affects the Affinity of the CAP Ligand
3.3. Increase in CAP Binding in Interleukin-1β (IL-1β)-Induced Inflammatory Primary HACs
3.4. Phage-Based Vector Is a Stable Vehicle for Human Chondrocyte Gene Delivery
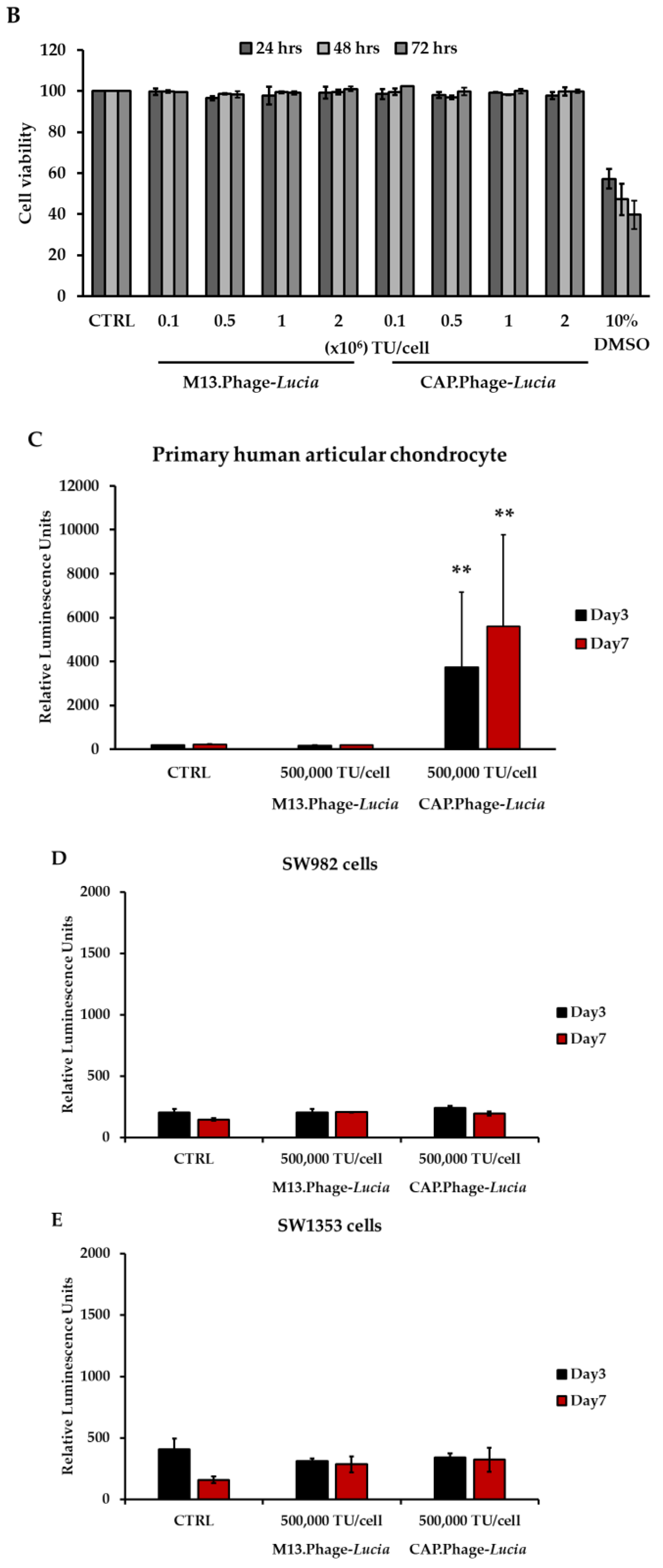
3.5. CAP.Phage Based Vector Is a Non-Toxic and Efficiently Delivered Transgene to Primary HACs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Christensen, K.; Doblhammer, G.; Rau, R.; Vaupel, J. Ageing populations: The challenges ahead. Lancet 2009, 374, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Lodato, E.; Kaplan, W. Antibacterial drug resistance. In Priority Medicines for Europe and the World: 2013 Update Report; World Health Organization: Geneva, Switzerland, 2013; pp. 68–74. [Google Scholar]
- Nair, S.; Austine, J.; Mirza, K. Perspective of orthopedists on pain management in osteoarthritis: A qualitative study. Indian J. Palliat. Care 2016, 22, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Arthritis gene therapy and its tortuous path into the clinic. Transl. Res. 2013, 161, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remant Bahadur, K.C.; Uludağ, H. PEI and its derivatives for gene therapy. In Polymers and Nanomaterials for Gene Therapy; Narain, R., Ed.; Woodhead Publishing: Sawston, UK, 2016; pp. 29–54. [Google Scholar]
- Sharon, D.; Kamen, A. Advancements in the design and scalable production of viral gene transfer vectors. Biotechnol. Bioeng. 2017, 115, 25–40. [Google Scholar] [CrossRef]
- Evans, C.H.; Ghivizzani, S.C.; Robbins, P.D. Arthritis gene therapy is becoming a reality. Nat. Rev. Rheumatol. 2018, 14, 381–382. [Google Scholar] [CrossRef]
- Pi, Y.; Zhang, X.; Shi, J.; Zhu, J.; Chen, W.; Zhang, C.; Gao, W.; Zhou, C.; Ao, Y. Targeted delivery of non-viral vectors to cartilage in vivo using a chondrocyte-homing peptide identified by phage display. Biomaterials 2011, 32, 6324–6332. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune responses to viral gene therapy vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Raper, S.E.; Chirmule, N.; Lee, F.; Wivel, N.A.; Bagg, A.; Gao, G.-P.; Wilson, J.; Batshaw, M.L. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 2003, 80, 148–158. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C., III; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef]
- Przystal, J.M.; Waramit, S.; Pranjol, Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M.; et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492. [Google Scholar] [CrossRef] [PubMed]
- Suwan, K.; Yata, T.; Waramit, S.; Przystal, J.M.; Stoneham, C.A.; Bentayebi, K.; Asavarut, P.; Chongchai, A.; Pothachareon, P.; Lee, K.-Y.; et al. Next-generation of targeted AAVP vectors for systemic transgene delivery against cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 18571–18577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moye, Z.D.; Woolston, J.; Sulakvelidze, A. Bacteriophage applications for food production and processing. Viruses 2018, 10, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Accelrys Software Inc. Material Studio; Accelrys Software Inc.: San Diego, CA, USA, 2011. [Google Scholar]
- Pothacharoen, P.; Najarus, S.; Settakorn, J.; Mizumoto, S.; Sugahara, K.; Kongtawelert, P. Effects of sesamin on the biosynthesis of chondroitin sulfate proteoglycans in human articular chondrocytes in primary culture. Glycoconj. J. 2013, 31, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Hajitou, A.; Rangel, R.; Trepel, M.; Soghomonyan, S.; Gelovani, J.G.; Alauddin, M.M.; Pasqualini, R.; Arap, W. Design and construction of targeted AAVP vectors for mammalian cell transduction. Nat. Protoc. 2007, 2, 523–531. [Google Scholar] [CrossRef]
- Aurnhammer, C.; Haase, M.; Muether, N.; Hausl, M.; Rauschhuber, C.; Huber, I.; Nitschko, H.; Busch, U.; Sing, A.; Ehrhardt, A.; et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods 2012, 23, 18–28. [Google Scholar] [CrossRef]
- Daheshia, M.; Yao, J.Q. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J. Rheumatol. 2008, 35, 2306–2312. [Google Scholar] [CrossRef]
- Inoue, H.; Takamori, M.; Nagata, N.; Nishikawa, T.; Oda, H.; Yamamoto, S.; Koshihara, Y. An investigation of cell proliferation and soluble mediators induced by interleukin 1beta in human synovial fibroblasts: Comparative response in osteoarthritis and rheumatoid arthritis. Inflamm. Res. 2001, 50, 65–72. [Google Scholar]
- Smith, M.D.; Triantafillou, S.; Parker, A.; Youssef, P.P.; Coleman, M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol. 1997, 24, 365–371. [Google Scholar]
- Zheng, C.; Goldsmith, C.M.; O’Connell, B.; Baum, B.J. Adenoviral vector cytotoxicity depends in part on the transgene encoded. Biochem. Biophys. Res. Commun. 2000, 274, 767–771. [Google Scholar] [CrossRef]
- Jin, L.; Zeng, X.; Liu, M.; Deng, Y.; He, N. Current progress in gene delivery technology based on chemical methods and nano-carriers. Theranostics 2014, 4, 240–255. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Kafil, V.; Omidi, Y. Cytotoxic impacts of linear and branched polyethylenimine nanostructures in A431 cells. BioImpacts 2011, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage therapy: A renewed approach to combat antibiotic-resistant bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.I. Phage therapy of human bacterial infections: A systematic review. In Human Viruses: Diseases, Treatments and Vaccines; Springer: Cham, Switzerland, 2021; pp. 663–692. [Google Scholar]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Rose, J.; Söder, S.; Skhirtladze, C.; Schmitz, N.; Gebhard, P.; Sesselmann, S.; Aigner, T. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthr. Cartil. 2012, 20, 1020–1028. [Google Scholar] [CrossRef] [Green Version]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, M.; Zhou, Y.; Polson, S.W.; Wan, L.Q.; Wang, M.; Han, L.; Wang, L.; Lu, X.L. Identification of chondrocyte genes and signaling pathways in response to acute joint inflammation. Sci. Rep. 2019, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Attur, M.G.; Dave, M.N.; Clancy, R.M.; Patel, I.R.; Abramson, S.B.; Amin, A.R. Functional genomic analysis in arthritis-affected cartilage: Yin-yang regulation of inflammatory mediators by α5β1 and αVβ3 integrins. J. Immunol. 2000, 164, 2684–2691. [Google Scholar] [CrossRef] [Green Version]
- Jeremiasse, B.; Matta, C.; Fellows, C.R.; Boocock, D.J.; Smith, J.R.; Liddell, S.; Lafeber, F.; Van Spil, W.E.; Mobasheri, A. Alterations in the chondrocyte surfaceome in response to pro-inflammatory cytokines. BMC Mol. Cell Biol. 2020, 21, 1–18. [Google Scholar] [CrossRef]
- Yata, T.; Lee, E.L.Q.; Suwan, K.; Syed, N.; Asavarut, P.; Hajitou, A. Modulation of extracellular matrix in cancer is associated with enhanced tumor cell targeting by bacteriophage vectors. Mol. Cancer 2015, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Aghebati-Maleki, L.; Bakhshinejad, B.; Baradaran, B.; Motallebnezhad, M.; Aghebati-Maleki, A.; Nickho, H.; Yousefi, M.; Majidi, J. Phage display as a promising approach for vaccine development. J. Biomed. Sci. 2016, 23, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalewska-Piątek, B.; Piątek, R. Bacteriophages as potential tools for use in antimicrobial therapy and vaccine development. Pharmaceuticals 2021, 14, 331. [Google Scholar] [CrossRef] [PubMed]
- Krysiak-Baltyn, K.; Martin, G.J.O.; Gras, S.L. Computational modelling of large scale phage production using a two-stage batch process. Pharmaceuticals 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameau, E.; Pedregal, A.; Glover, C. Cost modelling comparison of adherent multi-trays with suspension and fixed-bed bioreactors for the manufacturing of gene therapy products. Cell Gene Ther. Insights 2019, 5, 1663–1674. [Google Scholar] [CrossRef]
- Kimura, T.; Ferran, B.; Tsukahara, Y.; Shang, Q.; Desai, S.; Fedoce, A.; Pimentel, D.R.; Luptak, I.; Adachi, T.; Ido, Y.; et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Negrini, M.; Wang, G.; Heuer, A.; Björklund, T.; Davidsson, M. AAV production everywhere: A simple, fast, and reliable protocol for in-house AAV vector production based on chloroform extraction. Curr. Protoc. Neurosci. 2020, 93. [Google Scholar] [CrossRef] [PubMed]
- Midoux, P.; Kichler, A.; Boutin, V.; Maurizot, A.J.-C.; Monsigny, M. Membrane permeabilization and efficient gene transfer by a peptide containing several histidines. Bioconjugate Chem. 1998, 9, 260–267. [Google Scholar] [CrossRef]
- Castan, L.; Da Silva, C.; Dos Santos, R.; Molina, E. Comparative study of cytotoxicity and genotoxicity of commercial Jeffamines® and polyethylenimine in CHO-K1 cells. Toxicol. Lett. 2016, 259, S182. [Google Scholar] [CrossRef]
- Kargaard, A.; Sluijter, J.P.; Klumperman, B. Polymeric siRNA gene delivery–transfection efficiency versus cytotoxicity. J. Control. Release 2019, 316, 263–291. [Google Scholar] [CrossRef]
- Patil, S.; Gao, Y.-G.; Lin, X.; Li, Y.; Dang, K.; Tian, Y.; Zhang, W.-J.; Jiang, S.-F.; Qadir, A.; Qian, A.-R. The development of functional non-viral vectors for gene delivery. Int. J. Mol. Sci. 2019, 20, 5491. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.W.; Noh, M.J.; Choi, K.B.; Lee, K.H. Initial phase I safety of retrovirally transduced human chondrocytes expressing transforming growth factor-beta-1 in degenerative arthritis patients. Cytotherapy 2012, 14, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, C.W.; Park, S.H.; Cho, J.J.; Kim, T.W.; Noh, M.J.; Lee, M.C. A phase IIA clinical study of tissuegene-C (TG-C) in patients with osteoarthritis. Osteoarthr. Cartil. 2012, 20, S27–S28. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Cho, J.; Kim, T.; Park, Y.; Jeong, E.; Lee, K.-H. Tissuegene-C (TG-C) improved clinical scores in patients with osteoarthritis: A phase 2B study. Osteoarthr. Cartil. 2014, 22, S194. [Google Scholar] [CrossRef] [Green Version]
- Silacci, M.; Baenziger-Tobler, N.; Lembke, W.; Zha, W.; Batey, S.; Bertschinger, J.; Grabulovski, D. Linker length matters, fynomer-Fc fusion with an optimized linker displaying picomolar IL-17A inhibition potency. J. Biol. Chem. 2014, 289, 14392–14398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, J.S.; Jiang, S.; Galimidi, R.P.; Keeffe, J.R.; Bjorkman, P.J. Design and characterization of structured protein linkers with differing flexibilities. Protein Eng. Des. Sel. 2014, 27, 325–330. [Google Scholar] [CrossRef]
- Peng, Y.; Zeng, W.; Ye, H.; Han, K.H.; Dharmarajan, V.; Novick, S.; Wilson, I.A.; Griffin, P.R.; Friedman, J.M.; Lerner, R.A. A general method for insertion of functional proteins within proteins via combinatorial selection of permissive junctions. Chem. Biol. 2015, 22, 1134–1143. [Google Scholar] [CrossRef] [Green Version]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chongchai, A.; Waramit, S.; Wongwichai, T.; Kampangtip, J.; Phitak, T.; Kongtawelert, P.; Hajitou, A.; Suwan, K.; Pothacharoen, P. Targeting Human Osteoarthritic Chondrocytes with Ligand Directed Bacteriophage-Based Particles. Viruses 2021, 13, 2343. https://doi.org/10.3390/v13122343
Chongchai A, Waramit S, Wongwichai T, Kampangtip J, Phitak T, Kongtawelert P, Hajitou A, Suwan K, Pothacharoen P. Targeting Human Osteoarthritic Chondrocytes with Ligand Directed Bacteriophage-Based Particles. Viruses. 2021; 13(12):2343. https://doi.org/10.3390/v13122343
Chicago/Turabian StyleChongchai, Aitthiphon, Sajee Waramit, Tunchanok Wongwichai, Jirawan Kampangtip, Thanyaluck Phitak, Prachya Kongtawelert, Amin Hajitou, Keittisak Suwan, and Peraphan Pothacharoen. 2021. "Targeting Human Osteoarthritic Chondrocytes with Ligand Directed Bacteriophage-Based Particles" Viruses 13, no. 12: 2343. https://doi.org/10.3390/v13122343