1. Introduction
Cytomegalovirus (CMV) is a human herpes virus, HHV-5, that is transmitted through saliva or breast milk, trans-placentally, and during organ or hematopoietic cell transplantation. CMV infects more than 50% of the world’s population, and as with other herpes viruses, CMV infects its host for life in a latent form [
1]. While largely asymptomatic in the general population, CMV causes serious disease in neonates and immunocompromised hosts, including recipients of allogeneic hematopoietic cell transplants (HCT) in whom CMV causes pneumonia, gastroenteritis, and retinitis [
1,
2].
Intra-host mathematical modeling of CMV and other viral infections has proven critical to understanding the dynamics of virus-host interactions and allows for the simulation of clinical trials to improve the development of antiviral therapies and vaccines [
3,
4,
5]. However, prior mathematical modeling of CMV has been limited by a lack of availability of untreated viral load data. Ganciclovir was approved for the treatment of AIDS retinitis in 1989 before the widespread adoption of quantitative polymerase chain reaction (PCR) for measurement of CMV viral load [
6,
7,
8]. Thus, serious CMV infections are generally treated with antivirals, precluding studies of untreated natural infection with quantitative PCR. Previous studies analyzing CMV kinetics calculated viral doubling-times and decay half-lives and associated high CMV viral load, viral load slope, and CMV doubling-times with poor outcomes [
9,
10,
11]. Kepler et al. developed a theoretical model from in vitro and literature values for CMV parameters [
12]; Rose and colleagues modeled varying viral load responses to ganciclovir [
13]; Mayer and colleagues developed deterministic and stochastic models of infant infection fit to viral load data from frequent sampling of the oral mucosa [
14,
15].
A mathematical model of CMV viral loads measured in blood from untreated patients is needed to characterize the natural history of CMV accurately. Such a model would quantify the main mechanisms driving the dynamics of CMV in the HCT population after transplant. A data-validated model may allow us to understand how antiviral treatments reduce viral replication in this setting and then further simulate treatment and dosing scenarios to optimize viral suppression and to lower risk of disease after HCT. Here, we present a mathematical model fit to viral load data obtained from frozen serum samples from the placebo group from the only randomized controlled trial of ganciclovir for the early treatment of CMV in bone marrow transplant recipients [
16]. Because these participants were not treated with ganciclovir until they reached the study endpoint of tissue-invasive CMV disease or death, we were able to capture the full dynamic range of CMV viral loads in our model. Following HCT, viral load trajectories and responses to antiviral treatments varied widely among transplant recipients likely due to great variability in immune parameters. Modeling infection in the absence of treatment allowed us to capture the natural variability in these immune parameters and associate their values with clinical outcomes from the trial.
2. Materials and Methods
2.1. Study Approach
We analyzed CMV viral loads from viral episodes that occurred in HCT recipients after transplant to characterize the natural history of CMV in untreated individuals. We developed four mechanistic ordinary differential Equation (ODE) mathematical models of within-host CMV infection each with specific underlying mechanistic assumptions regarding the dynamics of CMV-susceptible cells and CMV-specific immune responses during infection. We used model selection theory to compare multiple instances of these models. Specifically, in the model selection process, within each main model, we chose which parameters could plausibly be zero in a biological setting and set those to zero individually and in combination such that we fit every combination of biologically feasible parameters to determine what version of the model the data supported most strongly. We obtained two parsimonious models with identifiable parameters from the competing list to describe the data. We then used the model with the most biological plausibility to validate the model parameters by assessing their associations with risk factors for CMV infection and disease and the primary endpoint of the clinical trial—the development of CMV disease by 100 days after HCT.
2.2. Clinical Data
Frozen serum samples were saved from participants in the placebo-controlled randomized controlled trial of ganciclovir for early treatment of CMV after HCT [
16,
17]. In this trial, viral cultures were used to screen for CMV in allogeneic HCT recipients who were either CMV seropositive or who had received marrow from CMV seropositive donors. If viral cultures were positive prior to day 80 after transplant and tissue-invasive CMV disease had not already by diagnosed, HCT recipients were randomized to receive ganciclovir or placebo through day 100 post-transplantation. Participants were followed for primary and secondary outcomes of tissue-invasive CMV disease or death, respectively [
16]. In a recent follow-up study to this historic trial, Duke and colleagues obtained these frozen samples from the Fred Hutch Infectious Disease Sciences Biospecimen Repository and selected samples for testing at approximately weekly intervals from day 0 to 100 after transplantation. The University of Washington Molecular Virology Laboratory performed quantitative CMV DNA PCR testing using a laboratory-developed assay with limit of quantification of 71.4 IU/mL and limit of detection of 35.7 IU/mL [
17].
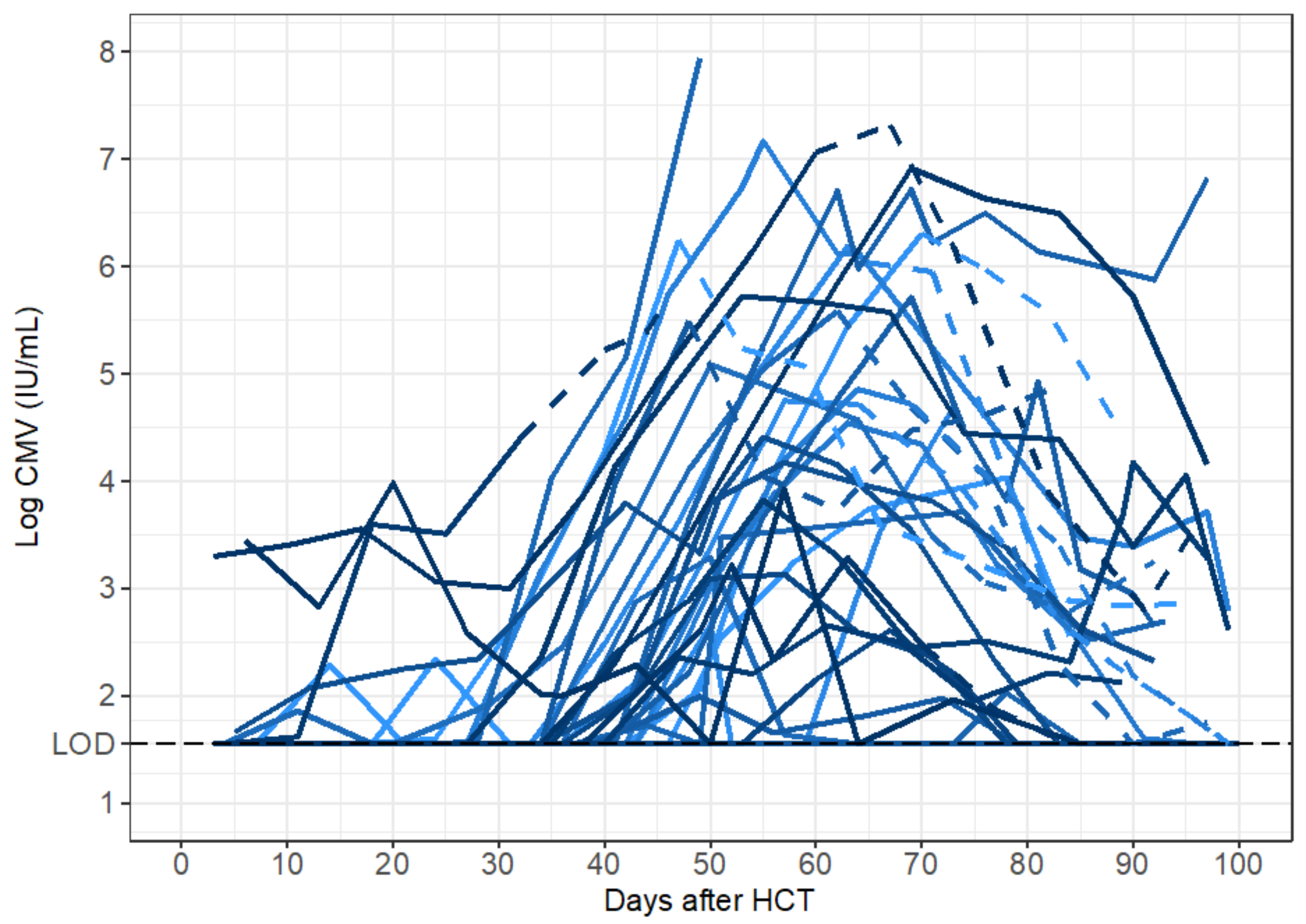
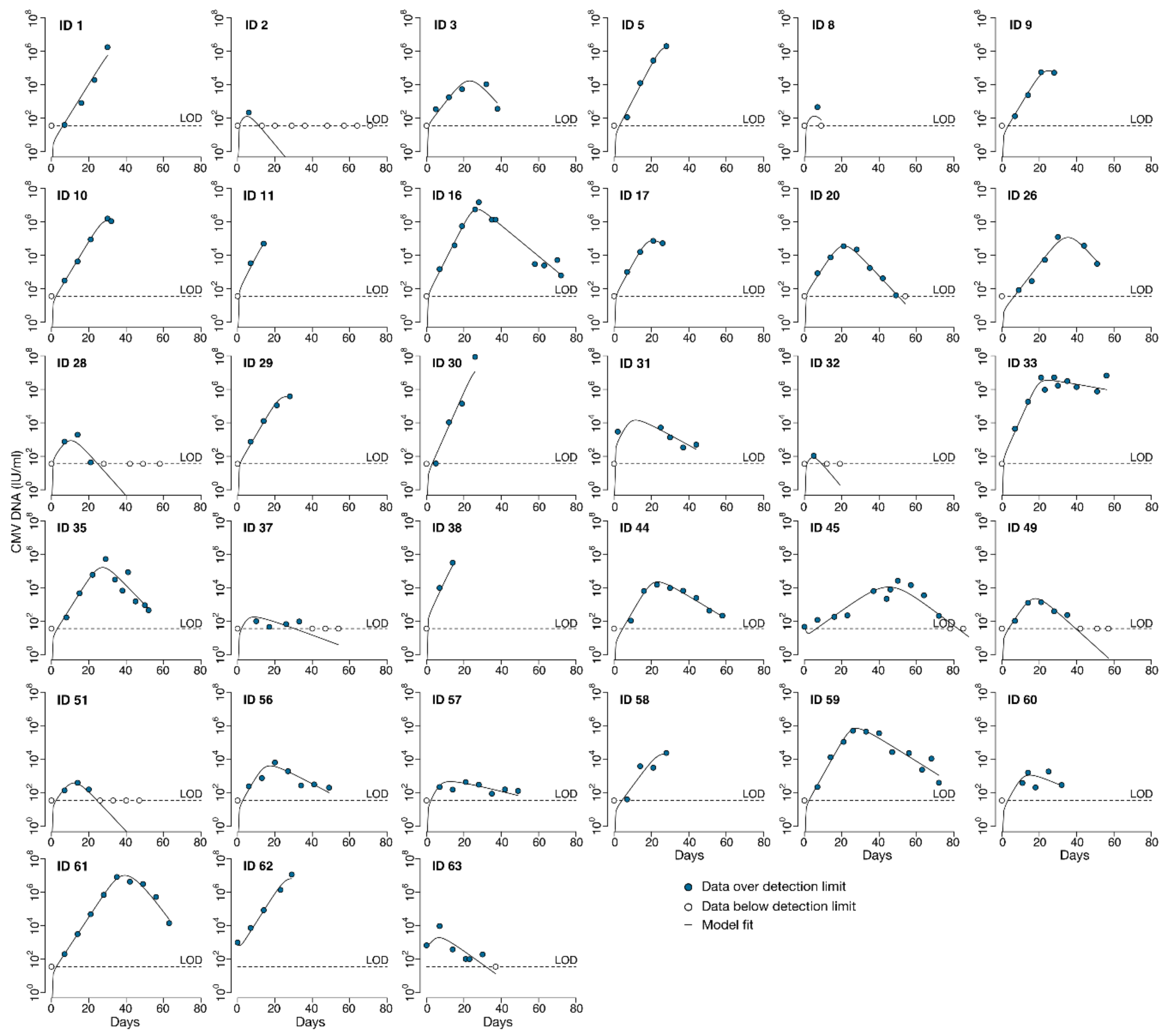
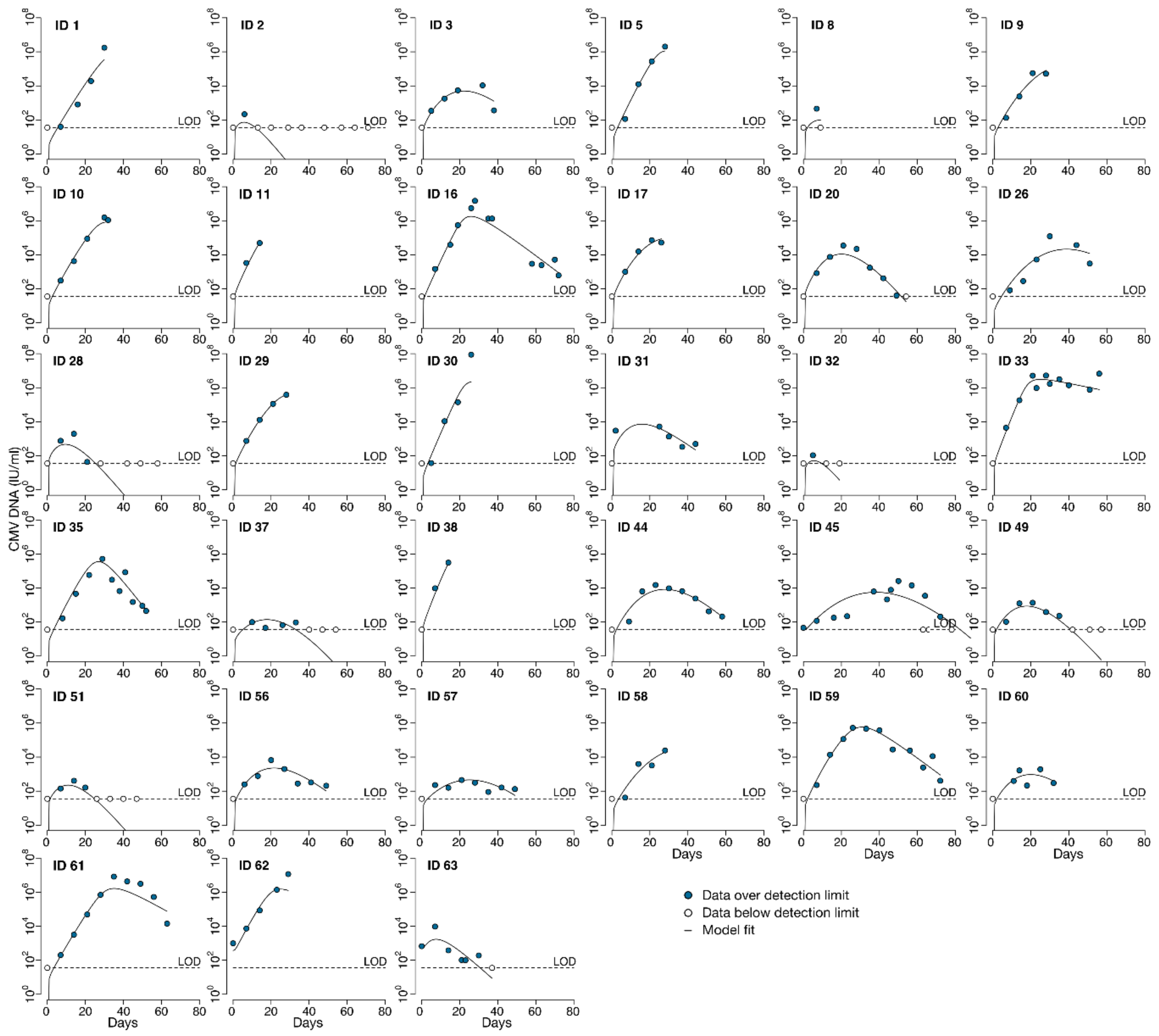
Here, we analyzed viral loads obtained from participants in the placebo group prior to diagnosis of CMV disease (at which point participants received treatment with open-label ganciclovir). For each individual, we did not include undetectable viral loads measured before the first positive aside from the last negative observation. If there was more than one viral episode in an individual with negative samples between episodes, we analyzed only the episode with the larger amount of data points. We did not model viral load data from participants who had only undetectable viral loads (n = 2).
2.3. Calculation of Viral Load Kinetics
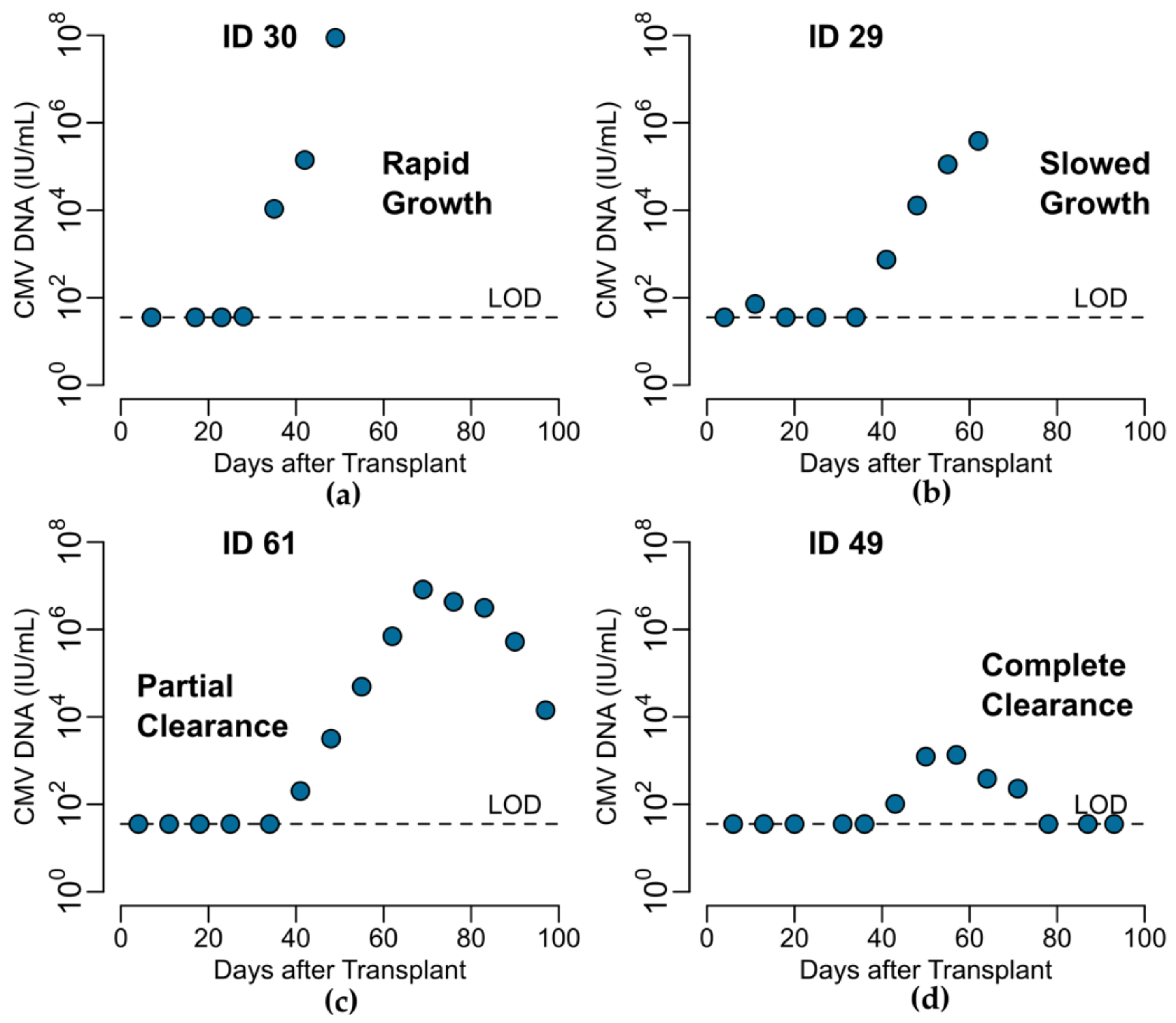
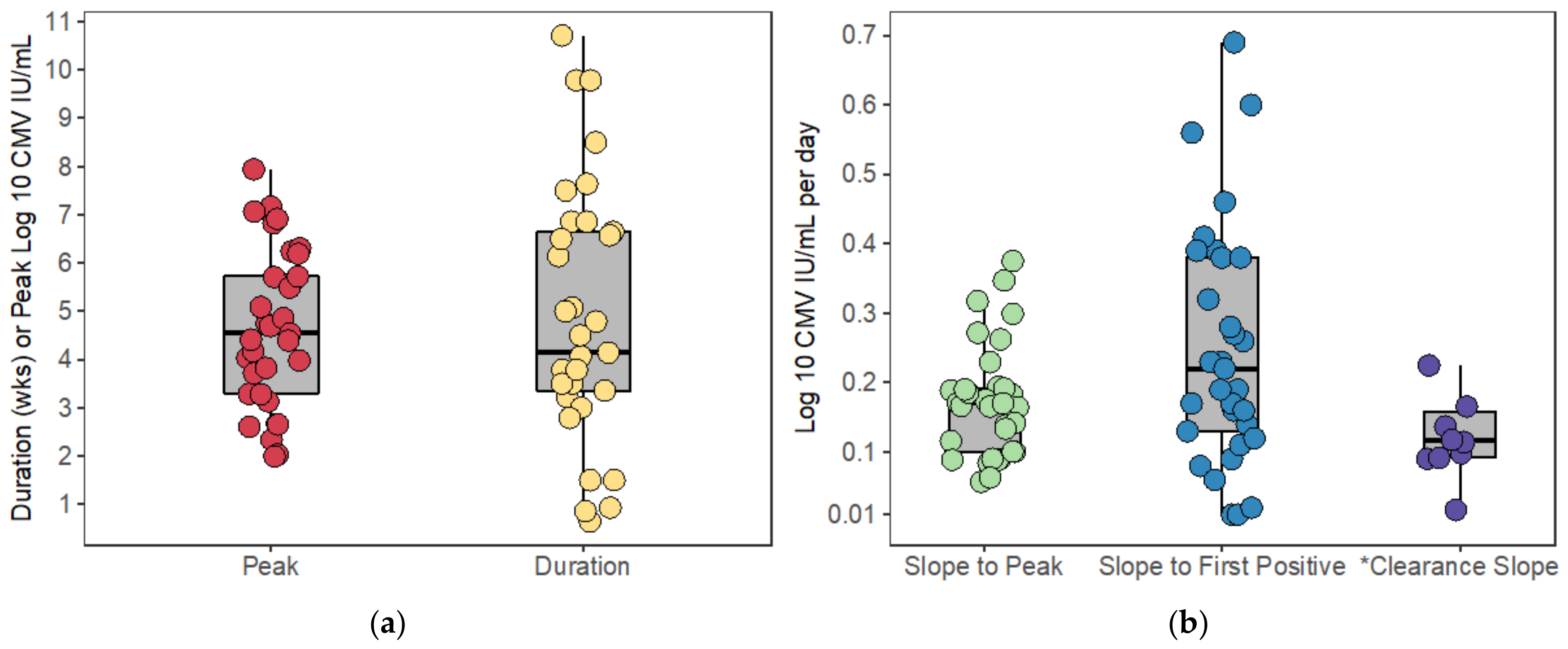
Peak viral load was considered to be the maximum log-10 converted viral load measured during the viral episode. For those participants who had undetectable CMV viral loads after HCT, we included only the last undetectable observation prior to the first positive viral load measured. Because the time when the CMV virus first became detectable between these measurements is unknown, we considered the start of the viral episode to be the midpoint between the last undetectable and first positive viral load. For participants who cleared the virus (i.e., viral load returned to undetectable), we considered the end of the episode to be the midpoint between the last positive and subsequent undetectable viral load. The expansion slope to the first positive was calculated as the difference in log-10 converted viral loads (i.e., viral load value at first positive minus the limit of detection) divided by the difference in times when the first positive viral load was measured and the start of the episode. Likewise, the expansion slope to the peak was calculated as the difference in the peak and the limit of detection divided by the difference in times between when the peak viral load was measured and the start of the episode. The clearance slope was the difference in the limit of detection and the peak viral load divided by the difference in times between when the episode ended and when the peak viral load was measured.
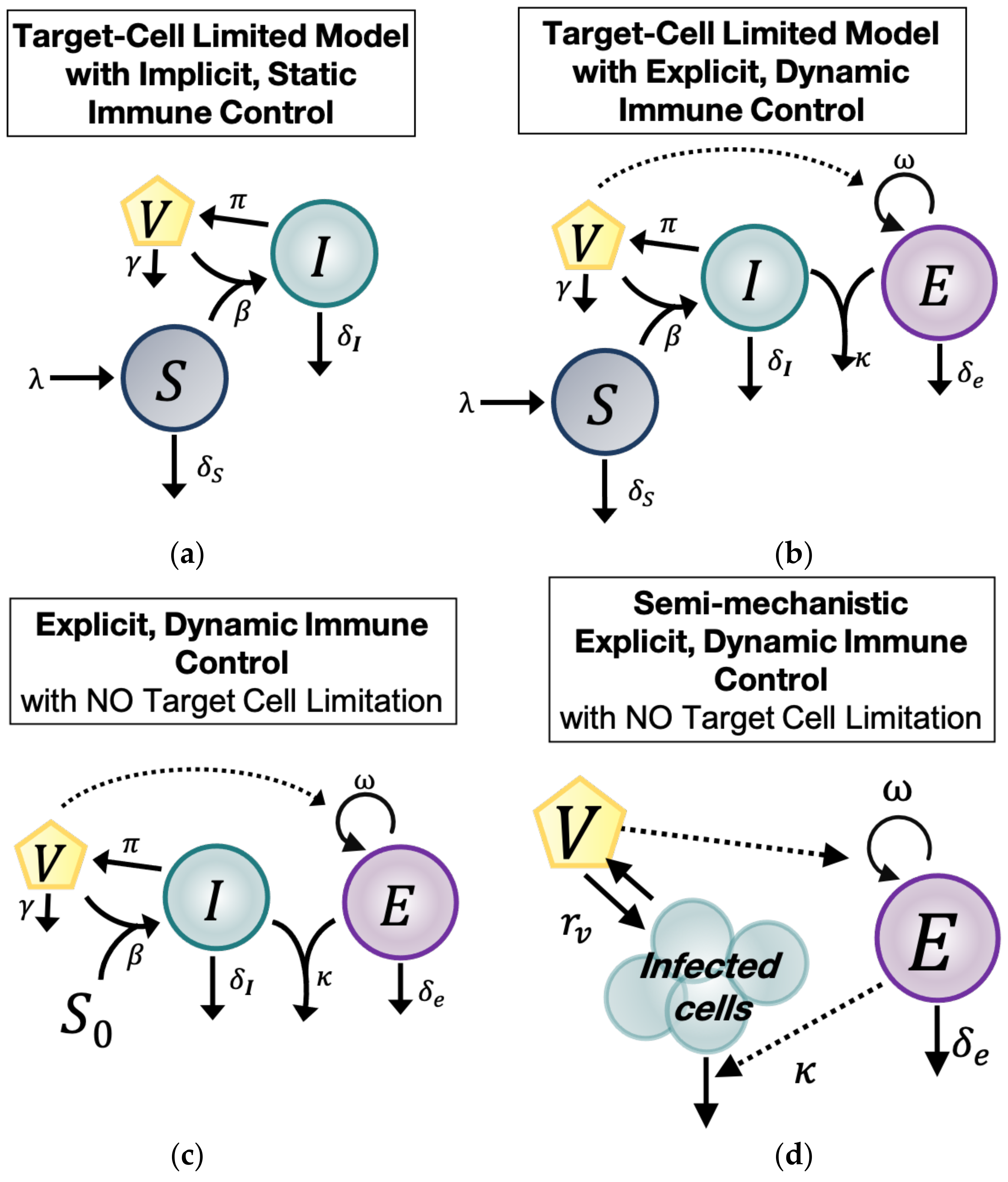
2.4. Model with Target Cell Limitation and Implicit, Static Immune Control (TC, No EIS)
The first of four main ODE models we used to understand the natural history of untreated CMV during HCT was the standard within-host model of virus dynamics (
Figure 1a) [
3]. This model includes three main compartments: Cells susceptible to CMV (
), CMV-infected cells (
, and CMV virions (
). Susceptible cells (
) expand with constant rate
, die at rate
, and are infected by CMV with rate
. CMV-infected cells (
die at rate
, assumed implicitly to include a static immune response against CMV. Finally, CMV-infected cells (
produce at rate
virions (
) that are cleared at rate
. Under these assumptions, the model has the form
2.5. Model with Target Cell Limitation and an Explicit, Dynamic Immune System (TC, EIS)
The second main model (
TC,
EIS) adds an explicit immune response that changes over time (explicit immune system =
EIS) to the target cell-limited model in Equation (1). The model schematic is shown in
Figure 1b. We assumed there is a CMV-specific effector cell compartment (
) that expands in the presence of CMV virions (
) at rate
and decays with rate
[
18,
19,
20,
21,
22]. Effector cells (
) kill CMV-infected cells (
at rate
. Because this model includes an explicit, cytotoxic immune response against CMV, the parameter
in this model may represent an innate response to infection or an intrinsic death rate of the infected cells (
due to viral infection or both. With these assumptions, the
TC,
EIS model is:
2.6. Explicit, Dynamic Immune Control Model without Target Cell Limitation (EIS, No TC)
The third main model (
EIS,
no TC) differs from the second in that we assume the susceptible cell pool (
) is so large that it is not changed significantly during CMV infection. This notion is plausible biologically given the large number of cell types that CMV infects and the high prevalence of those cell types in the human body [
1]. Thus, we assumed in this model that the size of the susceptible cell compartment remains constant with concentration
, allowing us to remove the
S Equation entirely. We simplify further by defining the composite parameter
. Based on this assumption, the
EIS,
no TC model has the form:
The model schematic is shown in
Figure 1c. For parameter identifiability purposes, we rescaled variables and parameters to consider three more related models. First, we rescaled the variable
and introduced the composite parameter
into Equation (3) so that the model takes the form:
Next, for further simplification, we considered the additional scaling
and introduced the composite parameter
. Incorporating these definitions into Equation (4), the model becomes:
Alternatively, we considered the scaling
and
in Equation (4), which results in the following model:
2.7. Semi-Mechanistic, Explicit Immune Control Model (VE)
Finally, because of the possibility of overfitting the above models due to the large number of parameters, we constructed a fourth main model, a semi-mechanistic model for CMV viral and immune dynamics, by assuming that during CMV infection, viral dynamics are in quasi-stationary state with respect to the infected cell compartment (
Figure 1d). Under this assumption,
. We simplified the model in Equation (3) by combining the remaining viral and infected cell terms into one parameter:
. We called
the CMV turnover rate. Under these assumptions, the model is:
To find an identifiable model that fit the data well, we further considered the rescaling
and the composite parameter
, resulting in the model:
Alternatively, with the same goal of identifiability, we introduced the rescaling
and the composite parameter
. Incorporating these assumptions into Equation (7) resulted in the model:
2.8. Population, Nonlinear, Mixed-Effects Approach
To fit the models to the CMV viral load observations, we used a nonlinear, mixed-effects framework. Under this framework, a viral load observation for individual at time is modeled as . Here, represents the solution of the mechanistic model for the variable describing the virus () where is the parameter vector for individual and is the measurement error for the log10-transformed viral load. We assumed that is drawn from a probability distribution with median or fixed effects and random effects . Unless otherwise specified, we modeled parameters as . In other words, the modeled parameters are log-normally distributed among the population with variability denoted by such that .
We modeled the initial value for the variable as for participants who were viremic (i.e., CMV viral loads were detectable) at the start of the modeling interval, which in this data set meant that they were viremic on first measurement after transplant. We estimated as a covariate of , meaning that for the group of participants who were not viremic at the time of transplant (and thus at the time of the start of the modeled viremic episode), but that could be estimated as greater than zero for the group of participants who were viremic at the time of transplant (and at the start of the modeled viremic episode). Including as a covariate of allowed the population distribution for to be bimodal.
For viral load observations below the limit of detection we used the probabilistic model that Monolix software (
www.lixoft.com accessed on 27 October 2021) provides for left-censored data [
23].
2.9. Model Fitting
We explored the ODE models as described above by fitting versions of each model assuming some parameters were equal to zero and estimating the remaining ones, including initial conditions of state variables, as shown in
Table A1,
Table A2,
Table A5 and
Table A7. Certain parameters were estimated for each model and were never fixed at a value of zero because their values must be non-zero in order to sustain infection (
) or an immune response to infection (
). However, in the time frame modeled, susceptible cells may or may not proliferate (
) or die (
), and infected cells (
) or effector immune cells (
) may or may not die at significant rates. Thus, we assign these parameters values of zero individually and in combination such that we fit all combinations of these parameters for each of the four main models that include these parameters. Thus, we explored a total of 38 individual models. For each model, we obtained the Maximum Likelihood Estimation (MLE) of the measurement error standard deviation
, the MLE of the vector of fixed effects
, and the MLE of the vector of standard deviations of the random effects
for each parameter using the Stochastic Approximation of the Expectation Maximization (SAEM) algorithm embedded in the Monolix software. We ran the SAEM algorithm five times (i.e., assessments) for each model using randomly selected initial values for the estimated parameters. For all model fits we assumed
as the time of last negative viral load after HCT or for those whose first viral load after transplant was positive,
coincided with the first viral load measured.
2.10. Model Selection
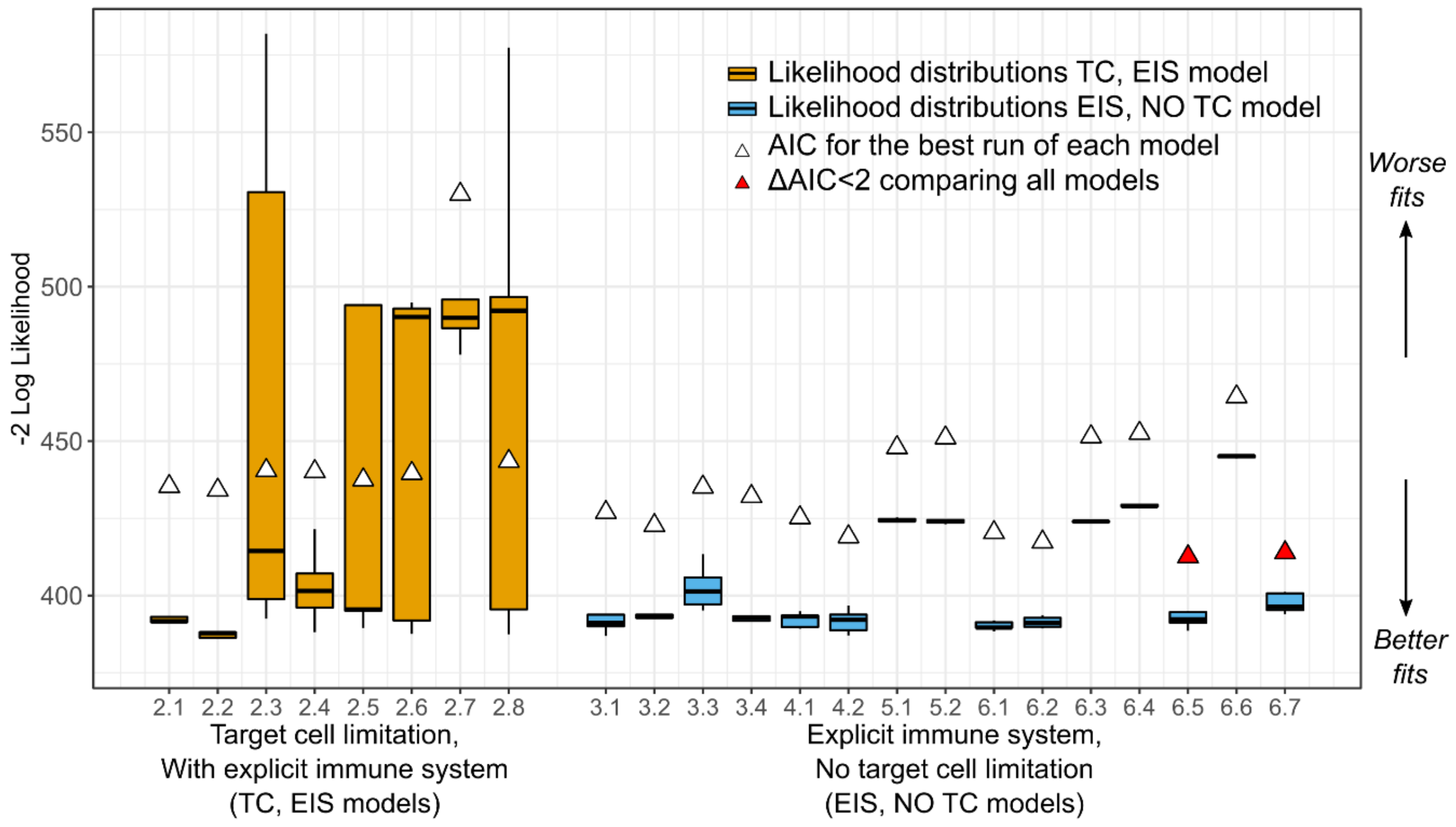
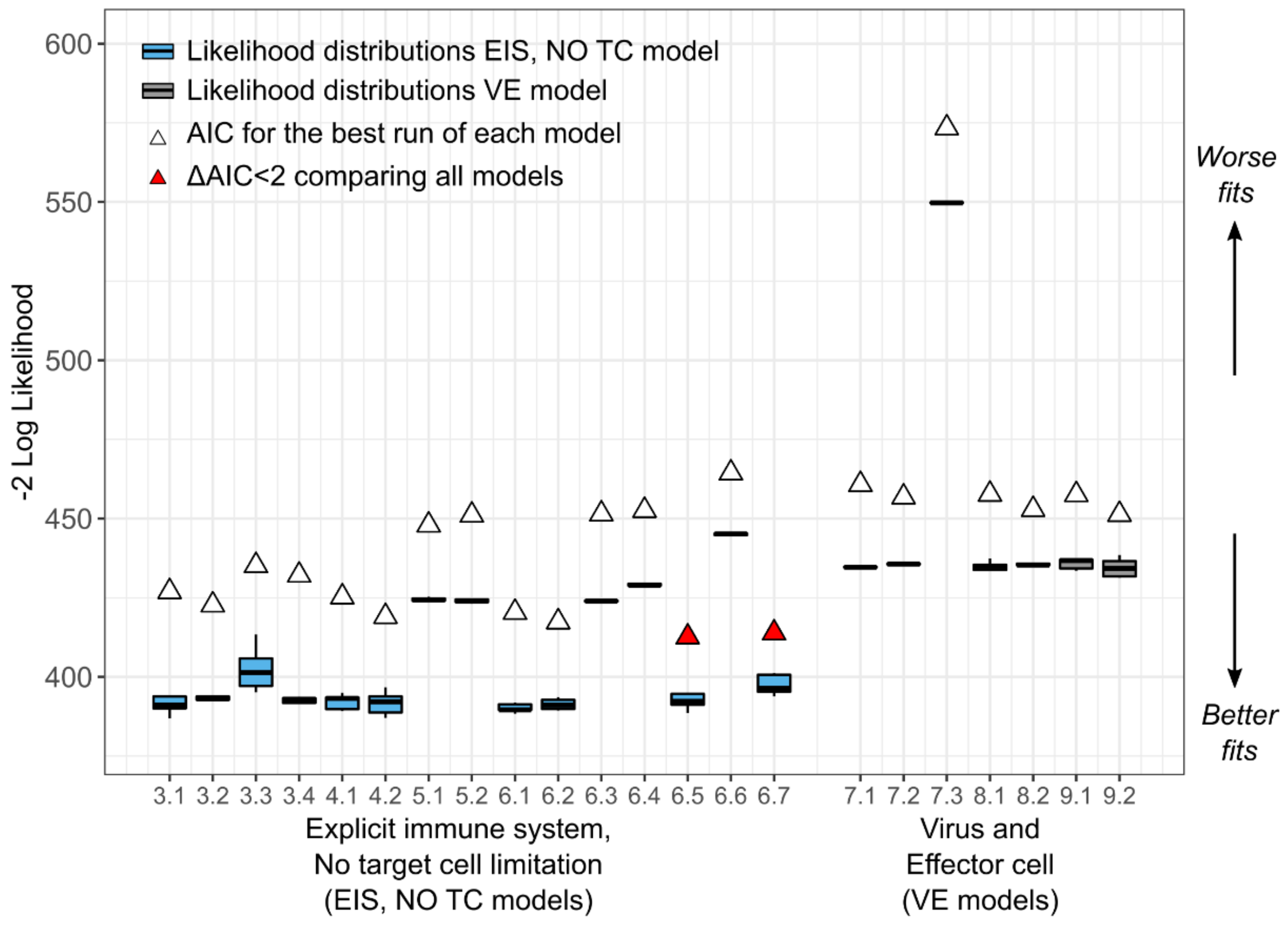
To determine the most parsimonious model, we calculated the log-likelihood () for all five assessments for each of the 38 models. Then, we computed the Akaike Information Criterion (AIC) for the assessment with the highest , where with being the number of parameters estimated. Then, we defined the delta score , where is the particular AIC for a model, and is the minimum AIC from all the models compared. We assumed two models had similar support from the data if the delta scores comparing them was less than two, i.e., .
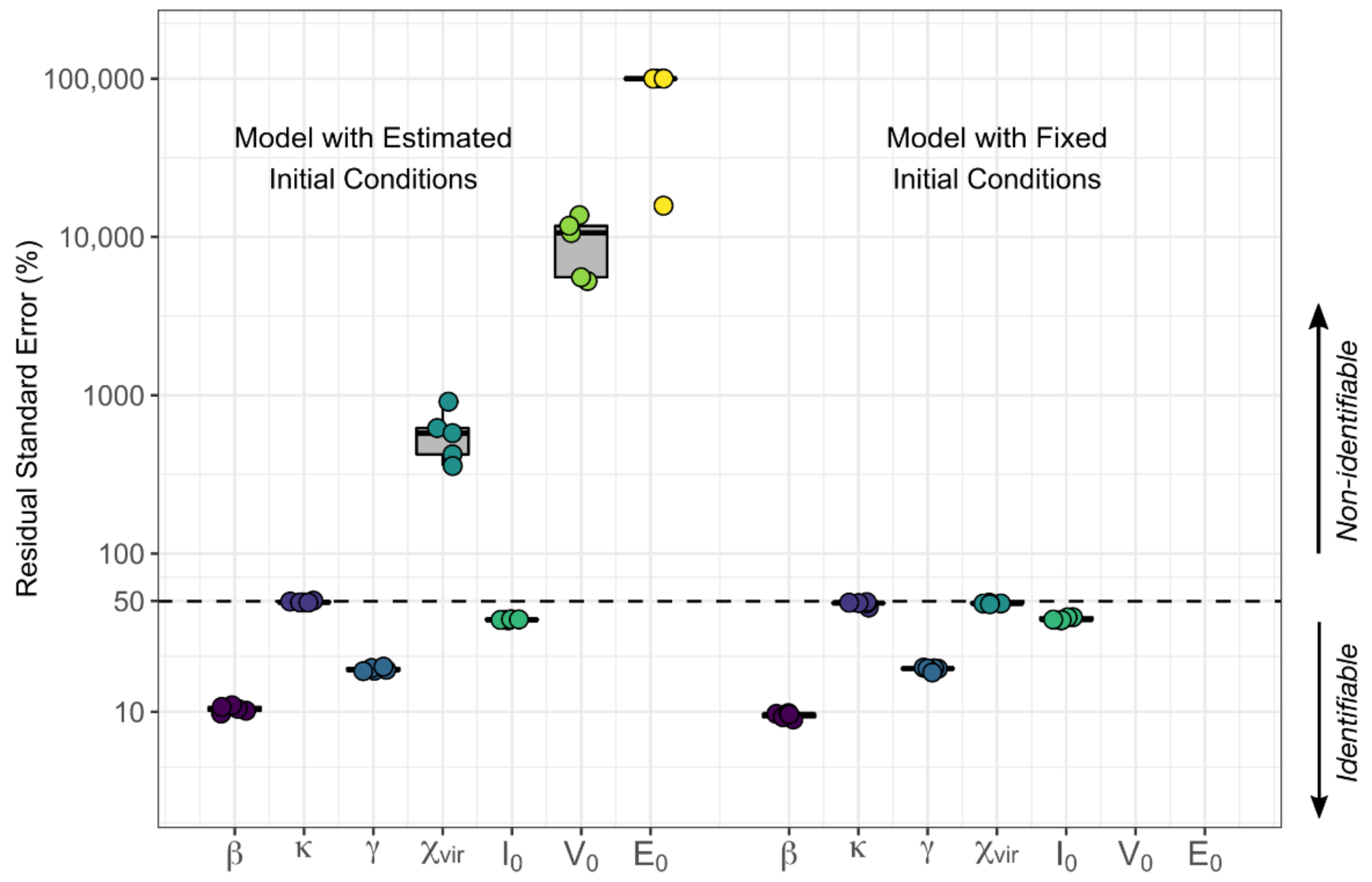
We analyzed the selection of our models further by assessing the relative standard error of the parameters for practical identifiability. During the estimation process, if a large change in a parameter causes no change in the likelihood, the data is not informing the value of the parameter under that specific structural model. The relative standard error (RSE) is a summary measure obtained during the parameter estimation process for each parameter and is small if the data provides adequate information to estimate the parameter. Specifically, Monolix calculates the Fisher Information Matrix (FIM) for each set of estimated parameters. In this matrix, the contribution of each parameter to the likelihood is indicated along the diagonal. The standard error (SE) vector is the square root of the diagonal of the inverse of the FIM. In that sense, the smaller the SE for a parameter, the more the data is informing that parameter. Finally, the RSE is the SE divided by the estimated parameter value such that the RSE is the uncertainty in estimation of a parameter normalized by its estimated value. When the SE of a parameter is greater than its estimated value (RSE > 100%), that parameter is generally regarded not to be practically identifiable [
24]. To be stringent, for those models with parameters with RSE percentages above 50%, we attempted to reduce the number of parameters while still maintaining some biological plausibility to which we could map the parameter values. We chose final models based on AIC, but also on identifiability and biological plausibility.
4. Discussion
Despite the development of effective antiviral therapies such as ganciclovir, CMV continues to cause substantial morbidity after HCT, and viral resistance may develop over time on current therapies [
2,
19]. Safer, more convenient, yet potent treatments are needed. Intrahost mathematical modeling could be an important tool for understanding the dynamics of CMV virus-host interactions and has the potential to improve the clinical trials process through simulation [
3,
4,
5,
13,
15]. A natural history mathematical model of CMV would allow us to perturb the model with proposed antiviral therapies and would provide a baseline for understanding required potencies and optimal dosing intervals for eliminating virus. Additionally, a baseline, quantitative understanding of the degree of immune control required for controlling virus might aid the development of a therapeutic CMV vaccine given after HCT.
Historically, mathematical modeling of CMV has been limited by the lack of availability of quantitative viral loads from untreated episodes of CMV viremia. Despite this, several groups have made important observations through modeling and other quantitative analysis of this pathogen [
11,
12,
13,
14,
15,
29,
30,
31]. However, because of this lack of data, in vivo estimation of basic model parameter values has proven difficult. In vitro estimates are unlikely to be reliable given the differences between in vitro and in vivo systems [
9]. In addition, whereas viral loads from other chronic viral infections such as HIV and hepatitis C tend to both grow, plateau, and respond to antiviral therapies in stereotypic patterns, in immunocompromised hosts, such as HCT recipients, viral dynamic patterns vary with the immunologic status of the host [
13,
32,
33].
Because we were able to obtain viral loads from frozen blood samples collected from HCT recipients from the placebo group in the historic randomized controlled trial evaluating ganciclovir for the early treatment of CMV after transplant, we have developed a natural history mathematical model for CMV after HCT [
16,
17]. We followed a systematic model selection procedure exploring four mechanistic ODE models with several parameterizations for a total of 38 competing models. We compared models mostly based on the Akaike Information Criteria, but in addition, we also considered the identifiability of their parameters and biological plausibility. From the competing models, we have identified two that fit the viral load data well and from which we can identify the model parameters.
In the process of fitting these models, we discovered that the data supported the inclusion of an explicit, dynamic immune system in the best models whereas a dynamic target cell compartment was not needed to recapitulate the observed data well. Rather assuming a constant, large pool of susceptible cells, whose number was unaffected by infection, was sufficient for model fitting, consistent with the fact that CMV can infect many tissue and cell types throughout the body. Given that we were fitting only to viral load data, we were limited in the number of parameters that could be identified independently. Thus, we formed some composite parameters, which limits some parameters’ independent interpretability somewhat.
For the best model with an explicit, dynamic immune system compartment and without target cell limitation (
EIS,
no TC, Equation (6), model 6.7), we estimated three parameters:
,
, and
.
is a composite parameter that describes viral infectivity, viral productivity, and the constant supply of target cells that CMV infects and thus reflects the overall viral growth rate.
contains elements of both the killing rate of immune effector cells and the proliferation rate of those cells in response to virus and thus may be a marker of the adaptive immune response. Therefore, parameters
and
may be useful in comparing participants to each other, but the actual parameter estimates may be difficult to interpret. On the other hand,
represents a measurable rate, the clearance rate of CMV viral particles from the blood. From the best fit of this model, we estimated that the CMV genome clearance rate in plasma has a median of 0.27 per day, equivalent to a half-life of 2.6 days, which is slow. For comparison, the clearance rates of HIV and hepatitis C viruses have been estimated to be 23 per day and 8 per day, respectively [
34]. Hepatitis B, a DNA virus, has complex clearance dynamics and two forms of viral DNA such that estimating the clearance rate is difficult, but the median half-life has been estimated to range from 9 to 21 h, also significantly faster than CMV [
35]. The model clearance rate estimate is distinct from our viral kinetics calculation of clearance slope (equivalent to a half-life of 2.5 days) and a prior estimate made by Emery and colleagues in which the slope of viral decline in bone marrow transplant receiving ganciclovir was calculated to be equivalent to a half-life of 1.5 days. First, Emery et al. estimated this rate during treatment rather than during natural immune clearance [
9]. Second, the downslope of viral load during therapy may reflect the death rate of infected cells or the removal rate of viral particles from the blood, but this cannot be disentangled without either additional knowledge of the biological system or a mechanistic model or both. Interestingly, despite this difference in calculation methods, the viral particle removal rate and the calculated viral decline kinetic from our data set in those clearing virus spontaneously was substantially slower than those receiving ganciclovir in the Emery et al. study [
9]. In another study from this group, patients with HIV starting antiretroviral therapy that included a protease inhibitor and with CMV viremia were not given specific anti-CMV therapy and were followed by CMV PCR. The median time to viral clearance was 13.5 weeks (range 5–40 weeks). Granted, we cannot calculate a reliable clearance slope from this data because the median sampling interval was nine weeks, but this study further supports the slow natural clearance rate of CMV [
36].
Of note, the size of the CMV DNA genome, which is considerably larger than the RNA genome of either HIV or hepatitis C, may also play a role in the slow clearance of CMV. In addition, whereas the model has allowed us to estimate this rate, a more accurate estimation could be obtained via plasmapheresis experiments as were performed in HIV and hepatitis C [
34]. Not only will this parameter value be helpful to us in modeling antiviral therapy in the future but also suggests that the ability of the immune system to clear viral DNA particles from the blood after HCT is limited. In addition, in this best-fitting version of Equation (6), the death rate of immune effector cells (
) was zero. Granted the model is fit only over a period of 100 days, so biologically, it is unrealistic to conclude that these cells are immortal. However, this finding suggests that the CMV-specific cells are long-lived and may represent memory cells. The literature supports this notion with reports that CD4 and CD8 T cells are likely the most important cells for controlling CMV infection after transplant [
2,
18,
19,
21,
22].
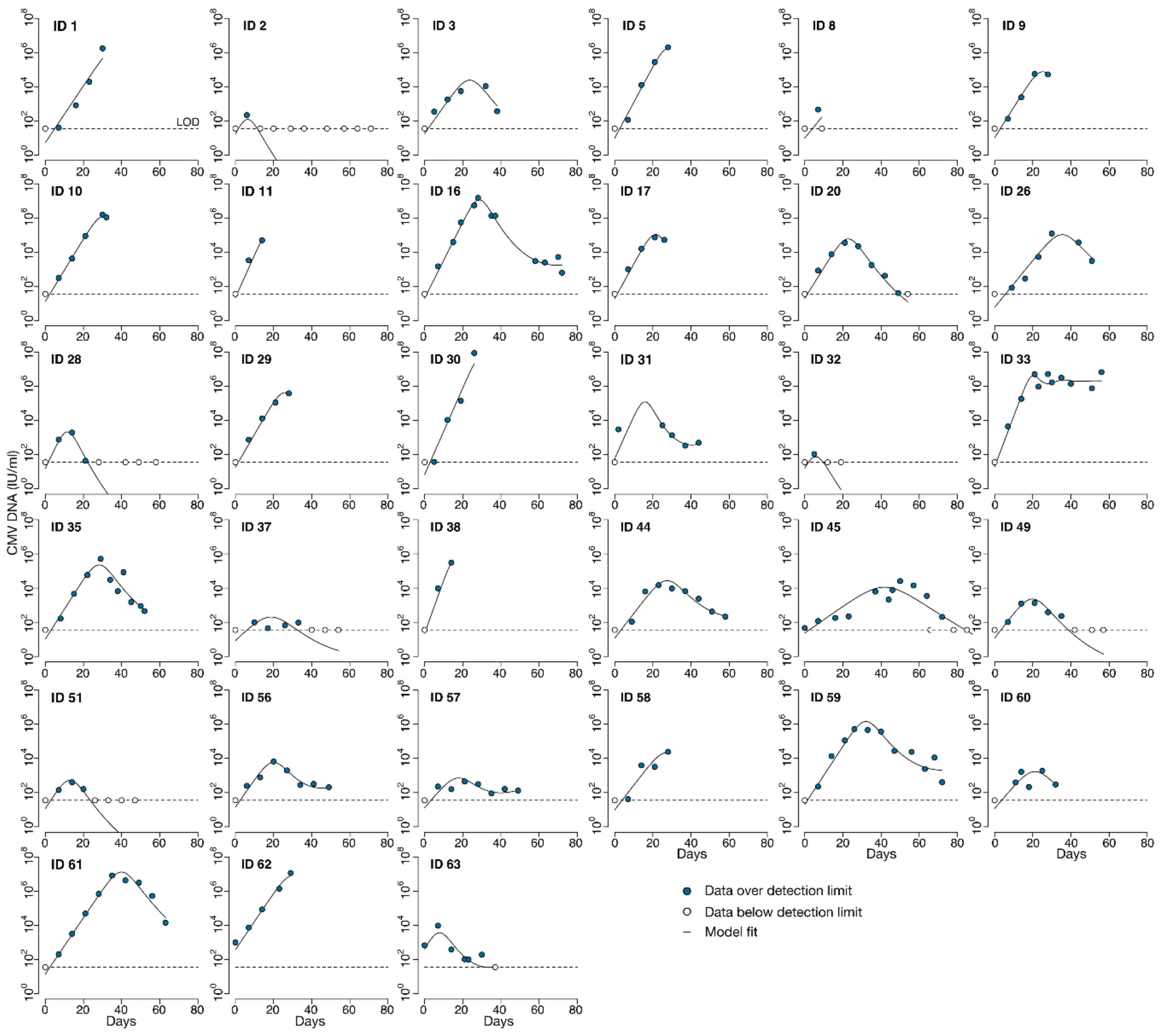
Our preferred model, the best-fitting version of the semi-mechanistic model that tracks only CMV viral load and CMV-specific effector cells (VE model 9.2), recapitulates the data well and contains only identifiable parameters. However, in terms of AIC, the EIS, no TC model performs better than the VE model. Whereas we can learn from both models, we chose to validate the VE model against the clinical trial risk factors and outcomes because it captures not only the viral episodes that end in complete viral clearance but also the episodes that plateau or increase after initial decrease. Additionally, the model appeared to be more stable and less sensitive to initial parameter conditions with low variability between the five assessments of each version of the model.
From the
VE model, we estimated the viral turnover rate (
) as 0.39 per day, equivalent to a doubling time of 1.8 days. For HIV, the doubling time has been estimated at 1.1 days [
37]. In this regard, CMV appears to replicate more slowly than HIV but surprisingly quickly for a virus usually considered to be slow in vitro [
9]. Emery and colleagues found a similar median viral doubling time of 1.3 days in bone marrow transplant recipients albeit with a direct approach rather than with a mechanistic model [
9]. In our viral kinetics calculations, we found the median doubling times to be 1.4 and 1.8 days depending on the calculation method (from start of estimated detectable DNAemia to first measured viral load versus to peak). However, the calculation of slope from data sampled relatively sparsely during the expansion phase, as in our data and in the CMV literature, is problematic [
9,
11,
27]. Because this mathematical model contains only one parameter for viral expansion,
, this rate should estimate the true viral expansion rate. Arguably, because the model interpolates between measured points accurately, we were able to calculate the rate of expansion more reliably with this version of the model. Additionally, consistent with the
EIS,
no TC model, we found that immune effector cells were long-lived with a median half-life of 50 days.
We validated the VE model against clinical data from the randomized trial and found that both the viral turnover rate and effector cell proliferation rate in response to virus correlated with important clinical features. Those with CMV-naïve HCT donors and acute graft-versus-host disease had higher viral turnover rate parameters (). Those who were diagnosed with tissue-invasive CMV disease during the clinical trial had slower proliferation of effector cells in response to virus (), suggesting that our model parameters have some clinical relevance.
Our study presents two intrahost models fit to long, untreated CMV reactivation episodes following HCT measured by quantitative CMV DNA PCR. In addition, we have expanded quantitative knowledge of intrahost virus-host interactions. However, there are some important limitations to this work. First, we have direct measurements only from the viral compartment and thus are limited in the number of parameters that we can identify independently. Next, our measurements from the viral compartment are measures of DNA rather than infectious virus. This limitation plagues our field, as we do not generally use viral culture clinically in humans due to problems with speed, reliability, quantification, and sensitivity. Especially in the setting of the SARS-CoV-2 pandemic, the persistence of viral genome (RNA) shedding in the absence of infectious virus has become evident [
38]. Addressing this limitation is a challenge for the field of intrahost modeling.
Moving forward, we can use our best, data-validated models to simulate the ranges of viral dynamics that we observed in the placebo group while modeling the effect of ganciclovir therapy in the ganciclovir arm of the randomized trial. This will allow us to estimate the efficacy of ganciclovir and propose optimal dosing strategies for ganciclovir and other CMV antivirals. Prior to this study, we would have been unable to distinguish natural immunity from the antiviral effect.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











