
Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Challenges with Malacoherpesviruses
2.2. Sample Collection, DNA, RNA Extraction, Library Preparation and Sequencing
2.3. Reconstruction of Malacoherpesvirus Genomes
2.4. Annotation of the Viral Genomes and Definition of a New Gene Nomenclature
2.5. Evaluation of Viral Transcription by RNA-seq
2.6. Transcript Diversity and ADAR Hyper-Editing Analysis
2.7. Single Nucleotide Polymorphism Analysis
2.8. Statistical Analysis
3. Results
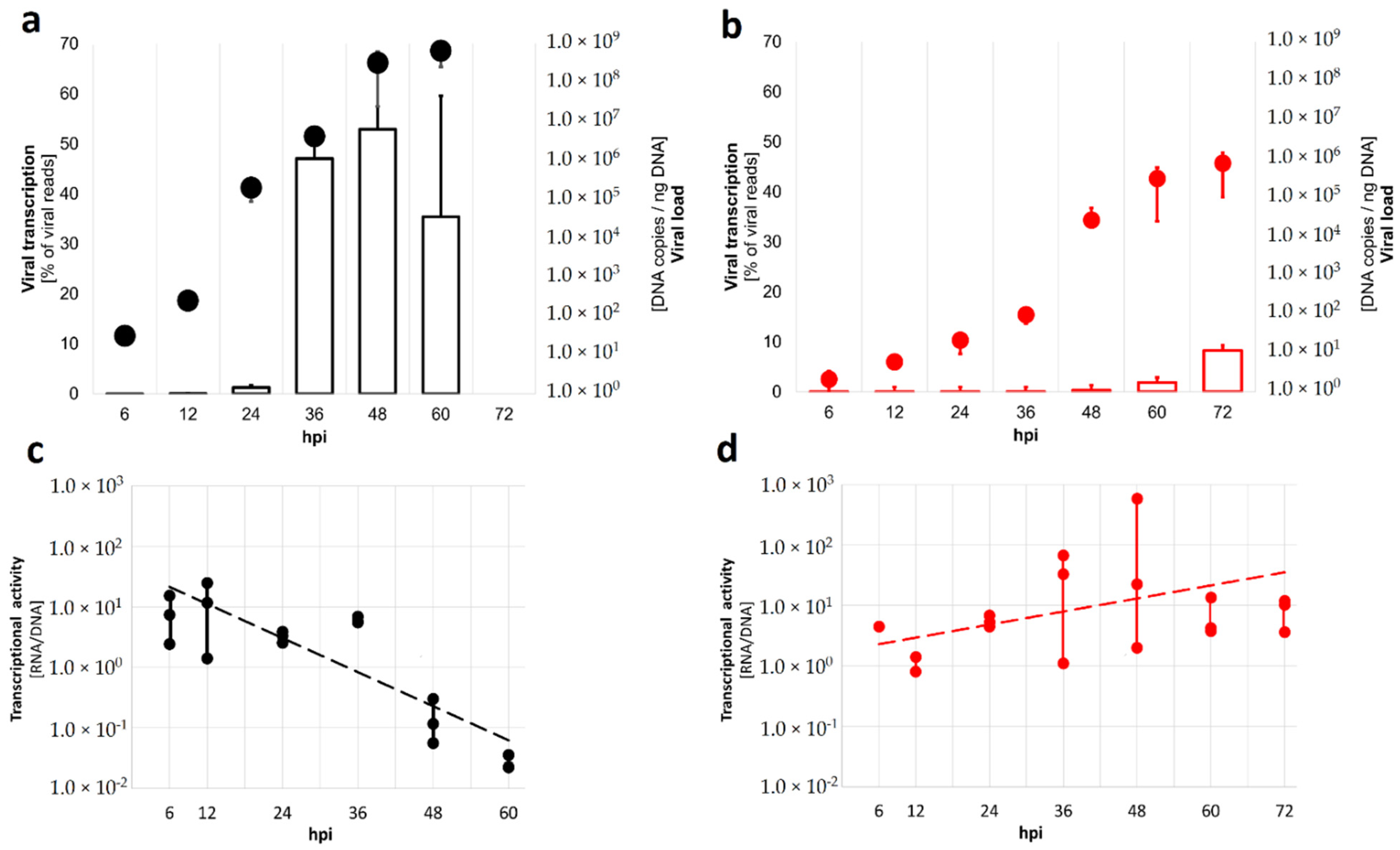
3.1. Virus Replication and Transcription Dynamics
3.2. Annotation of Malacoherpesvirus Genomes and Transcriptional Arrays
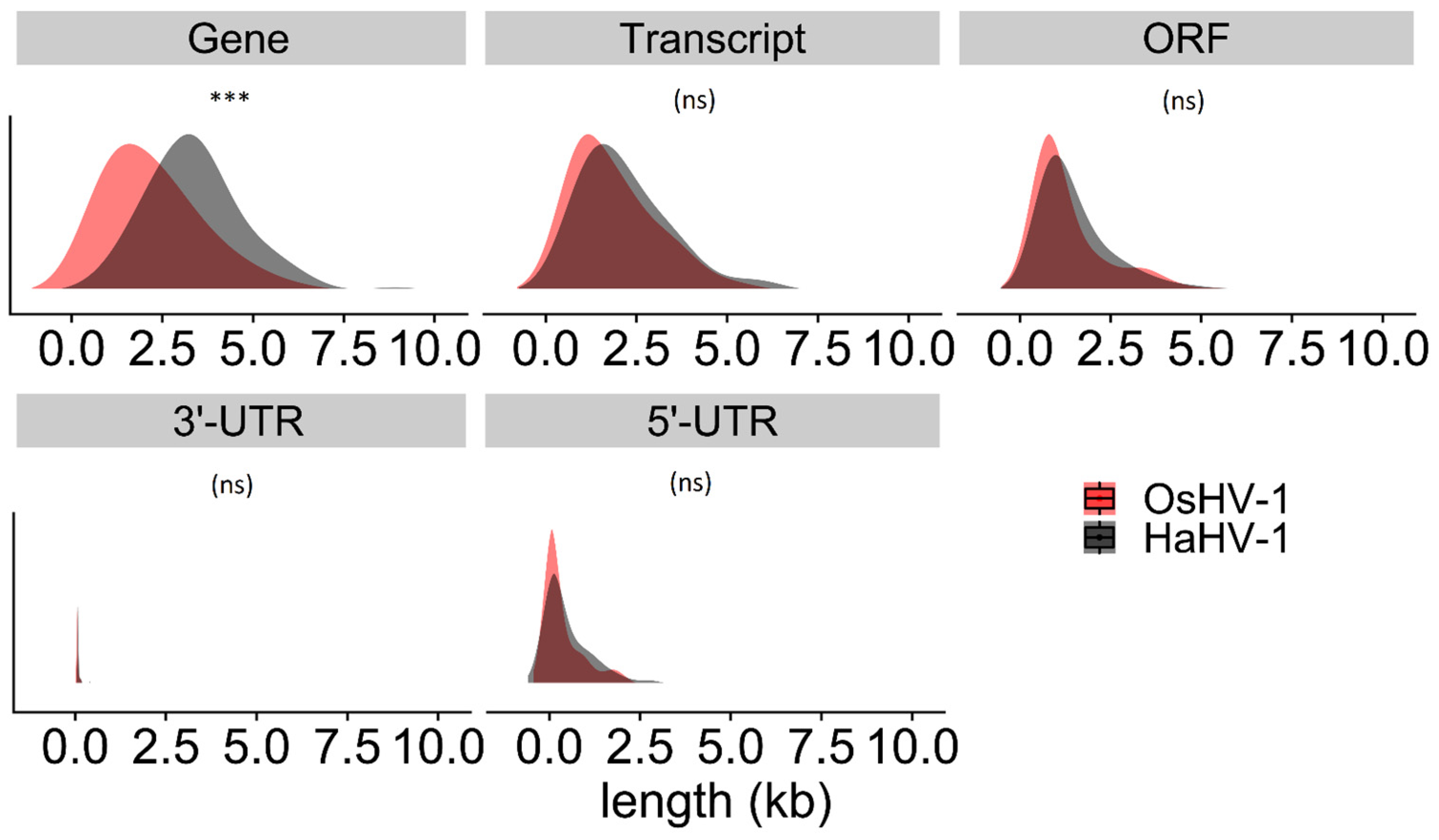
3.3. Gene Lengths Differ between HaHV-1 and OsHV-1
3.4. Transcript Diversity Differs between Malacoherpesviruses
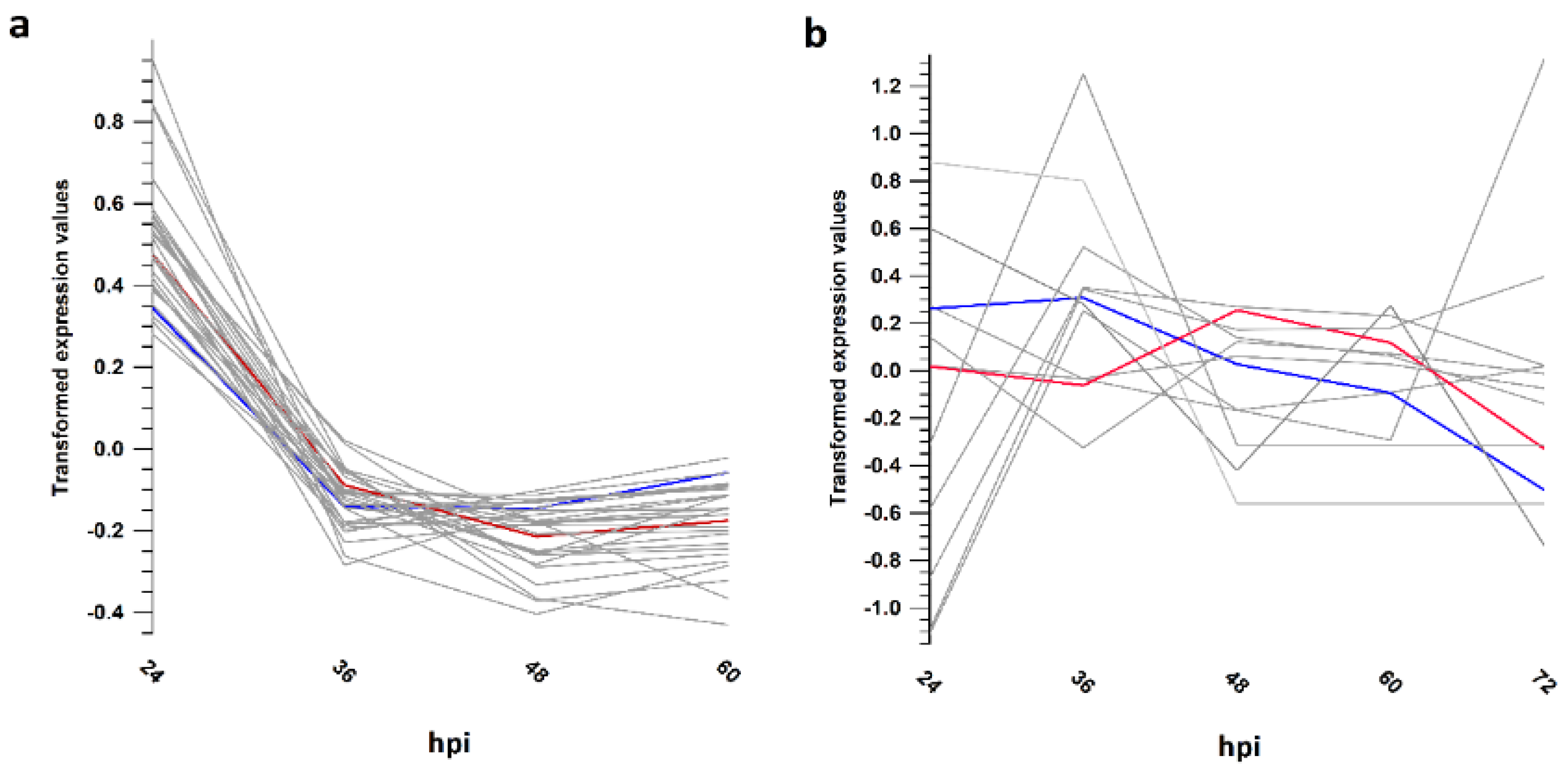
3.5. Gene Expression Trends Supported Different Transcriptional Strategies
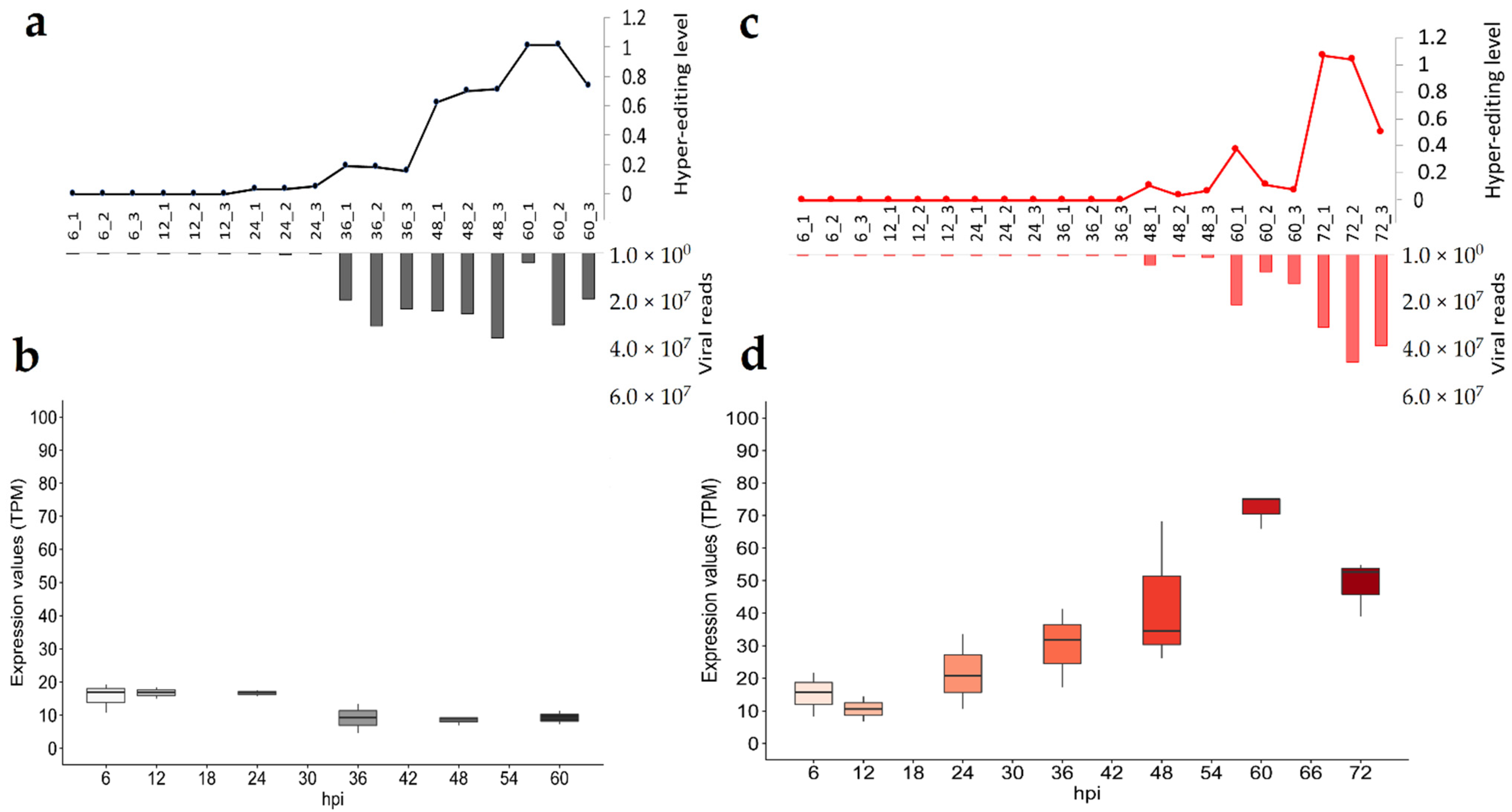
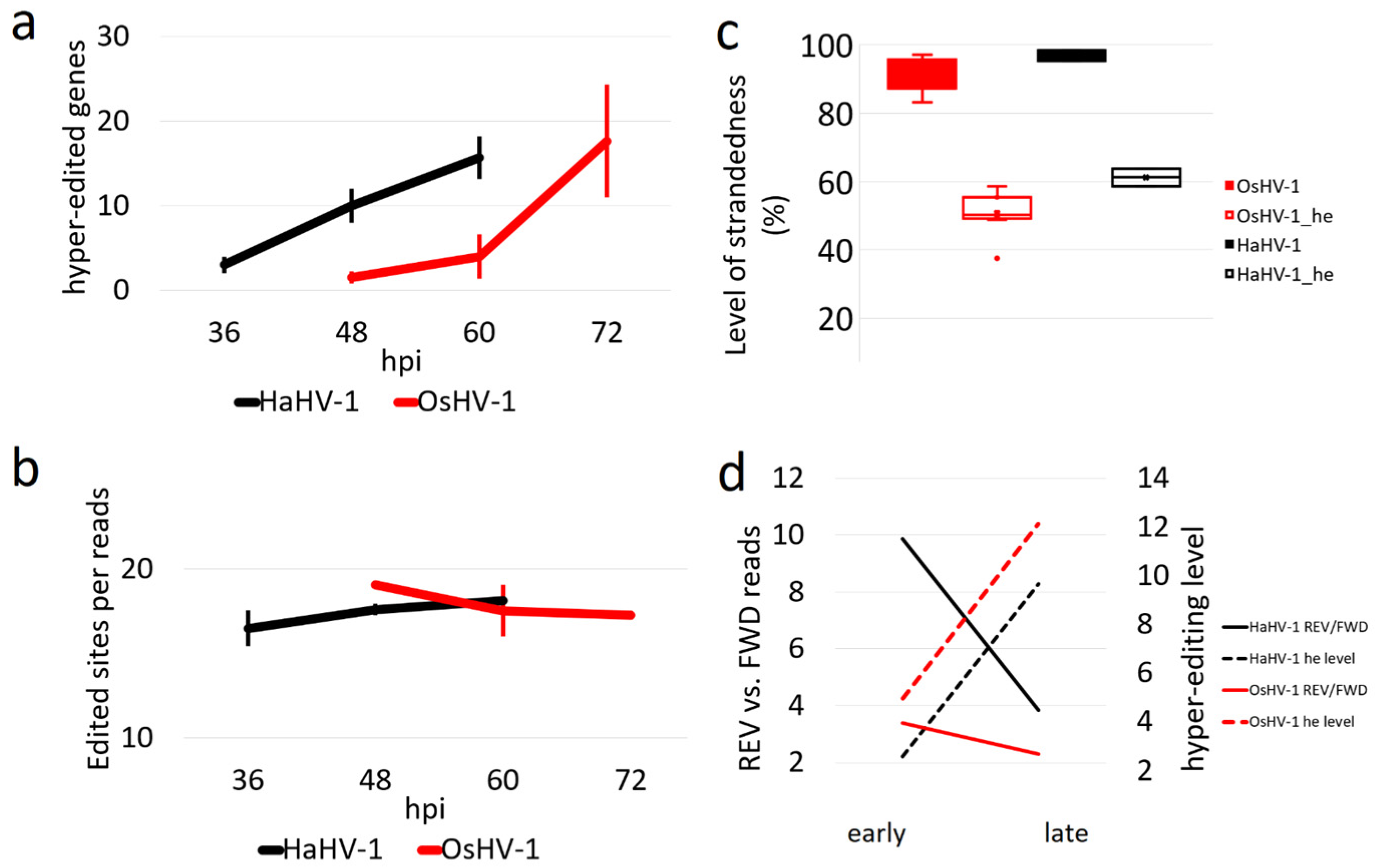
3.6. Tracing the Impact of ADAR1 Editing on Viral Transcripts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Marine Viruses—Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Faillace, C.A.; Lorusso, N.S.; Duffy, S. Overlooking the Smallest Matter: Viruses Impact Biological Invasions. Ecol. Lett. 2017, 20, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V. Viruses and Mobile Elements as Drivers of Evolutionary Transitions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150442. [Google Scholar] [CrossRef]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The Order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Chang, P.H.; Kuo, S.T.; Lai, S.H.; Yang, H.S.; Ting, Y.Y.; Hsu, C.L.; Chen, H.C. Herpes-like Virus Infection Causing Mortality of Cultured Abalone Haliotis Diversicolor Supertexta in Taiwan. Dis. Aquat. Organ. 2005, 65, 23–27. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J.; Trus, B.L.; Cheng, N.; Steven, A.C.; Watson, M.S.; Cunningham, C.; Le Deuff, R.-M.; Renault, T. A Novel Class of Herpesvirus with Bivalve Hosts. J. Gen. Virol. 2005, 86, 41–53. [Google Scholar] [CrossRef]
- Bai, C.; Gao, W.; Wang, C.; Yu, T.; Zhang, T.; Qiu, Z.; Wang, Q.; Huang, J. Identification and Characterization of Ostreid Herpesvirus 1 Associated with Massive Mortalities of Scapharca Broughtonii Broodstocks in China. Dis. Aquat. Organ. 2016, 118, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Mushegian, A.; Karin, E.L.; Pupko, T. Sequence Analysis of Malacoherpesvirus Proteins: Pan-Herpesvirus Capsid Module and Replication Enzymes with an Ancient Connection to “Megavirales”. Virology 2018, 513, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Moreau, K.; Segarra, A.; Tourbiez, D.; Travers, M.-A.; Rubinsztein, D.C.; Renault, T. Autophagy Plays an Important Role in Protecting Pacific Oysters from OsHV-1 and Vibrio aestuarianus Infections. Autophagy 2015, 11, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.; Faury, N.; Barbosa-Solomieu, V.; Moreau, K. Suppression Substractive Hybridisation (SSH) and Real Time PCR Reveal Differential Gene Expression in the Pacific Cupped Oyster, Crassostrea Gigas, Challenged with Ostreid Herpesvirus 1. Dev. Comp. Immunol. 2011, 35, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.-M.; Rosani, U.; Xin, L.-S.; Li, G.-Y.; Li, C.; Wang, Q.-C.; Wang, C.-M. Dual Transcriptomic Analysis of Ostreid Herpesvirus 1 Infected Scapharca broughtonii with an Emphasis on Viral Anti-Apoptosis Activities and Host Oxidative Bursts. Fish Shellfish Immunol. 2018, 82, 554–654. [Google Scholar] [CrossRef]
- Rosani, U.; Varotto, L.; Domeneghetti, S.; Arcangeli, G.; Pallavicini, A.; Venier, P. Dual Analysis of Host and Pathogen Transcriptomes in Ostreid Herpesvirus 1-Positive Crassostrea gigas. Environ. Microbiol. 2015, 17, 4200–4212. [Google Scholar] [CrossRef]
- de Lorgeril, J.; Lucasson, A.; Petton, B.; Toulza, E.; Montagnani, C.; Clerissi, C.; Vidal-Dupiol, J.; Chaparro, C.; Galinier, R.; Escoubas, J.-M.; et al. Immune-Suppression by OsHV-1 Viral Infection Causes Fatal Bacteraemia in Pacific Oysters. Nat. Commun. 2018, 9, 4215. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.-M.; Rosani, U.; Li, Y.-N.; Zhang, S.-M.; Xin, L.-S.; Wang, C.-M. RNA-Seq of HaHV-1-Infected Abalones Reveals a Common Transcriptional Signature of Malacoherpesviruses. Sci. Rep. 2019, 9, 938. [Google Scholar] [CrossRef] [Green Version]
- Balázs, Z.; Tombácz, D.; Szűcs, A.; Snyder, M.; Boldogkői, Z. Long-Read Sequencing of the Human Cytomegalovirus Transcriptome with the Pacific Biosciences RSII Platform. Sci. Data 2017, 4, 170194. [Google Scholar] [CrossRef]
- Harel, N.; Meir, M.; Gophna, U.; Stern, A. Direct Sequencing of RNA with MinION Nanopore: Detecting Mutations Based on Associations. Nucleic Acids Res. 2019, 47, e148. [Google Scholar] [CrossRef] [Green Version]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-Read Sequencing—A Powerful Tool in Viral Transcriptome Research. Trends Microbiol. 2019, 27, 578–592. [Google Scholar] [CrossRef] [Green Version]
- Depledge, D.P.; Mohr, I.; Wilson, A.C. Going the Distance: Optimizing RNA-Seq Strategies for Transcriptomic Analysis of Complex Viral Genomes. J. Virol. 2019, 93, e01342-18. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Moldován, N.; Balázs, Z.; Gulyás, G.; Csabai, Z.; Boldogkői, M.; Snyder, M.; Boldogkői, Z. Multiple Long-Read Sequencing Survey of Herpes Simplex Virus Dynamic Transcriptome. Front. Genet. 2019, 10, 834. [Google Scholar] [CrossRef]
- Kakuk, B.; Tombácz, D.; Balázs, Z.; Moldován, N.; Csabai, Z.; Torma, G.; Megyeri, K.; Snyder, M.; Boldogkői, Z. Combined Nanopore and Single-Molecule Real-Time Sequencing Survey of Human Betaherpesvirus 5 Transcriptome. Sci. Rep. 2021, 11, 14487. [Google Scholar] [CrossRef]
- Schlub, T.E.; Holmes, E.C. Properties and Abundance of Overlapping Genes in Viruses. Virus Evol. 2020, 6, veaa009. [Google Scholar] [CrossRef] [Green Version]
- Wight, M.; Werner, A. The Functions of Natural Antisense Transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [CrossRef]
- Lamers, M.M.; van den Hoogen, B.G.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef] [Green Version]
- Piontkivska, H.; Frederick, M.; Miyamoto, M.M.; Wayne, M.L. RNA Editing by the Host ADAR System Affects the Molecular Evolution of the Zika Virus. Ecol. Evol. 2017, 7, 4475–4485. [Google Scholar] [CrossRef] [Green Version]
- Rosani, U.; Bai, C.-M.; Maso, L.; Shapiro, M.; Abbadi, M.; Domeneghetti, S.; Wang, C.-M.; Cendron, L.; MacCarthy, T.; Venier, P. A-to-I Editing of Malacoherpesviridae RNAs Supports the Antiviral Role of ADAR1 in Mollusks. BMC Evol. Biol. 2019, 19, 149. [Google Scholar] [CrossRef]
- Bai, C.-M.; Xin, L.-S.; Rosani, U.; Wu, B.; Wang, Q.-C.; Duan, X.-K.; Liu, Z.-H.; Wang, C.-M. Chromosomal-Level Assembly of the Blood Clam, Scapharca (Anadara) Broughtonii, Using Long Sequence Reads and Hi-C. GigaScience 2019, 8, giz067. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Burioli, E.A.V.; Prearo, M.; Houssin, M. Complete Genome Sequence of Ostreid Herpesvirus Type 1 µVar Isolated during Mortality Events in the Pacific Oyster Crassostrea gigas in France and Ireland. Virology 2017, 509, 239–251. [Google Scholar] [CrossRef]
- Abbadi, M.; Zamperin, G.; Gastaldelli, M.; Pascoli, F.; Rosani, U.; Milani, A.; Schivo, A.; Rossetti, E.; Turolla, E.; Gennari, L.; et al. Identification of a Newly Described OsHV-1 µvar from the North Adriatic Sea (Italy). J. Gen. Virol. 2018, 99, 693. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinforma. Oxf. Engl. 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A Genome-Wide Map of Hyper-Edited RNA Reveals Numerous New Sites. Nat. Commun. 2014, 5, 4726. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Poggiali, D.; Whitaker, K.; Marshall, T.R.; van Langen, J.; Kievit, R.A. Raincloud Plots: A Multi-Platform Tool for Robust Data Visualization. Wellcome Open Res. 2021, 4, 63. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 9 November 2020).
- Rosani, U.; Venier, P. Oyster RNA-Seq Data Support the Development of Malacoherpesviridae Genomics. Front. Microbiol. 2017, 8, 1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Žugich, J.; Goodrum, F. Transcriptome-Wide Characterization of Human Cytomegalovirus in Natural Infection and Experimental Latency. Proc. Natl. Acad. Sci. USA 2017, 114, E10586–E10595. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Jouaux, A.; Ford, S.E.; Lelong, C.; Sourdaine, P.; Mathieu, M.; Guo, X. Transcriptome Analysis Reveals Strong and Complex Antiviral Response in a Mollusc. Fish Shellfish Immunol. 2015, 46, 131–144. [Google Scholar] [CrossRef]
- Rivas, H.G.; Schmaling, S.K.; Gaglia, M.M. Shutoff of Host Gene Expression in Influenza A Virus and Herpesviruses: Similar Mechanisms and Common Themes. Viruses 2016, 8, 102. [Google Scholar] [CrossRef]
- Smiley, J.R. Herpes Simplex Virus Virion Host Shutoff Protein: Immune Evasion Mediated by a Viral RNase? J. Virol. 2004, 78, 1063–1068. [Google Scholar] [CrossRef] [Green Version]
- Savin, K.W.; Cocks, B.G.; Wong, F.; Sawbridge, T.; Cogan, N.; Savage, D.; Warner, S. A Neurotropic Herpesvirus Infecting the Gastropod, Abalone, Shares Ancestry with Oyster Herpesvirus and a Herpesvirus Associated with the Amphioxus Genome. Virol. J. 2010, 7, 308. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Hardy-Smith, P.; Handlinger, J. Ganglioneuritis Causing High Mortalities in Farmed Australian Abalone (Haliotis laevigata and Haliotis rubra). Aust. Vet. J. 2007, 85, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Schikorski, D.; Faury, N.; Pepin, J.F.; Saulnier, D.; Tourbiez, D.; Renault, T. Experimental Ostreid Herpesvirus 1 Infection of the Pacific Oyster Crassostrea Gigas: Kinetics of Virus DNA Detection by q-PCR in Seawater and in Oyster Samples. Virus Res. 2011, 155, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, C.V.; Woodcock, D.J.; Lam, C.; Garcia-Albornoz, M.; Adamson, A.; Ashall, L.; Rowe, W.; Downton, P.; Schmidt, L.; West, S.; et al. Temperature Regulates NF-ΚB Dynamics and Function through Timing of A20 Transcription. Proc. Natl. Acad. Sci. USA 2018, 115, E5243–E5249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansarah-Sobrinho, C.; Nelson, S.; Jost, C.A.; Whitehead, S.S.; Pierson, T.C. Temperature-Dependent Production of Pseudoinfectious Dengue Reporter Virus Particles by Complementation. Virology 2008, 381, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Bookelaar, B.; Lynch, S.A.; Culloty, S.C. Host Plasticity Supports Spread of an Aquaculture Introduced Virus to an Ecosystem Engineer. Parasit. Vectors 2020, 13, 498. [Google Scholar] [CrossRef]
- Delisle, L.; Pauletto, M.; Vidal-Dupiol, J.; Petton, B.; Bargelloni, L.; Montagnani, C.; Pernet, F.; Corporeau, C.; Fleury, E. High Temperature Induces Transcriptomic Changes in Crassostrea gigas That Hinder Progress of Ostreid Herpesvirus (OsHV-1) and Promote Survival. J. Exp. Biol. 2020, 223, jeb226233. [Google Scholar] [CrossRef]
- Boldogköi, Z. Transcriptional Interference Networks Coordinate the Expression of Functionally Related Genes Clustered in the Same Genomic Loci. Front. Genet. 2012, 3, 122. [Google Scholar] [CrossRef] [Green Version]
- Rosani, U.; Abbadi, M.; Green, T.; Bai, C.-M.; Turolla, E.; Arcangeli, G.; Wegner, K.M.; Venier, P. Parallel Analysis of MiRNAs and MRNAs Suggests Distinct Regulatory Networks in Crassostrea gigas Infected by Ostreid Herpesvirus 1. BMC Genom. 2020, 21, 620. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E.; Rovnak, J.; Badani, H.; Cohrs, R.J. A Comparison of Herpes Simplex Virus Type 1 and Varicella-Zoster Virus Latency and Reactivation. J. Gen. Virol. 2015, 96, 1581–1602. [Google Scholar] [CrossRef]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 Latent Transcription and Non-Coding RNA: A Critical Retrospective. J. Neuroimmunol. 2017, 308, 65–101. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Sample | Technology | Seq. Depth | Total ccs or Reads | No. of FLNC-ccs | No. of FLNC |
|---|---|---|---|---|---|---|
| HaHV-1 | H-36P | PacBio | 20.0 Gb | 257,457 | 218,594 (50.4%) | 25,010 (15.8%) |
| H-48P | PacBio | 24.9 Gb | 310,674 | 252,845 (75.7%) | 25,774 (23.5%) | |
| H-36I | Illumina | 6.8 Gb | 45,723,904 (45%) | / | / | |
| H-48I | Illumina | 8.3 Gb | 55,518,872 (67%) | / | / | |
| OsHV-1 | O-60P | PacBio | 38.1 Gb | 550,552 | 383,717 (4.8%) | 17,074 (7%) |
| O-72P | PacBio | 45.9 Gb | 613,981 | 320,968 (11.7%) | 24,378 (8.5%) | |
| O-60I | Illumina | 14.4 Gb | 83,222,092 (2.8%) | / | / | |
| O-72I | Illumina | 11.6 Gb | 55,015,939 (9.9%) | / | / |
| Scheme ID | No. of Transcripts | Diversity Value | 5′-UTRs | 3′-UTRs |
|---|---|---|---|---|
| H-36P | 1216 (417) | 0.31 | 774 | 145 |
| H-48P | 1656 (636) | 0.27 | 1359 (+76%) | 194 (+34%) |
| O-60P | 142 | 0.12 | 417 | 119 |
| O-72P | 188 | 0.09 | 436 (+5%) | 139 (+17%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, C.-M.; Rosani, U.; Zhang, X.; Xin, L.-S.; Bortoletto, E.; Wegner, K.M.; Wang, C.-M. Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing. Viruses 2021, 13, 1971. https://doi.org/10.3390/v13101971
Bai C-M, Rosani U, Zhang X, Xin L-S, Bortoletto E, Wegner KM, Wang C-M. Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing. Viruses. 2021; 13(10):1971. https://doi.org/10.3390/v13101971
Chicago/Turabian StyleBai, Chang-Ming, Umberto Rosani, Xiang Zhang, Lu-Sheng Xin, Enrico Bortoletto, K. Mathias Wegner, and Chong-Ming Wang. 2021. "Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing" Viruses 13, no. 10: 1971. https://doi.org/10.3390/v13101971