
Genomic Characterization of Diverse Bat Coronavirus HKU10 in Hipposideros Bats

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling
2.3. RNA Extraction, PCR Screening and Sequencing
2.4. Sequencing of Full-Length Genomes
2.5. Genome Analysis
2.6. Virus Isolation
2.7. Estimation of Divergence Time
2.8. Cell Lines
2.9. BtCoV HKU10 Spike-Mediated Pseudovirus Cell Tropism Screening
2.10. Nucleotide Sequence Accession Numbers
3. Results
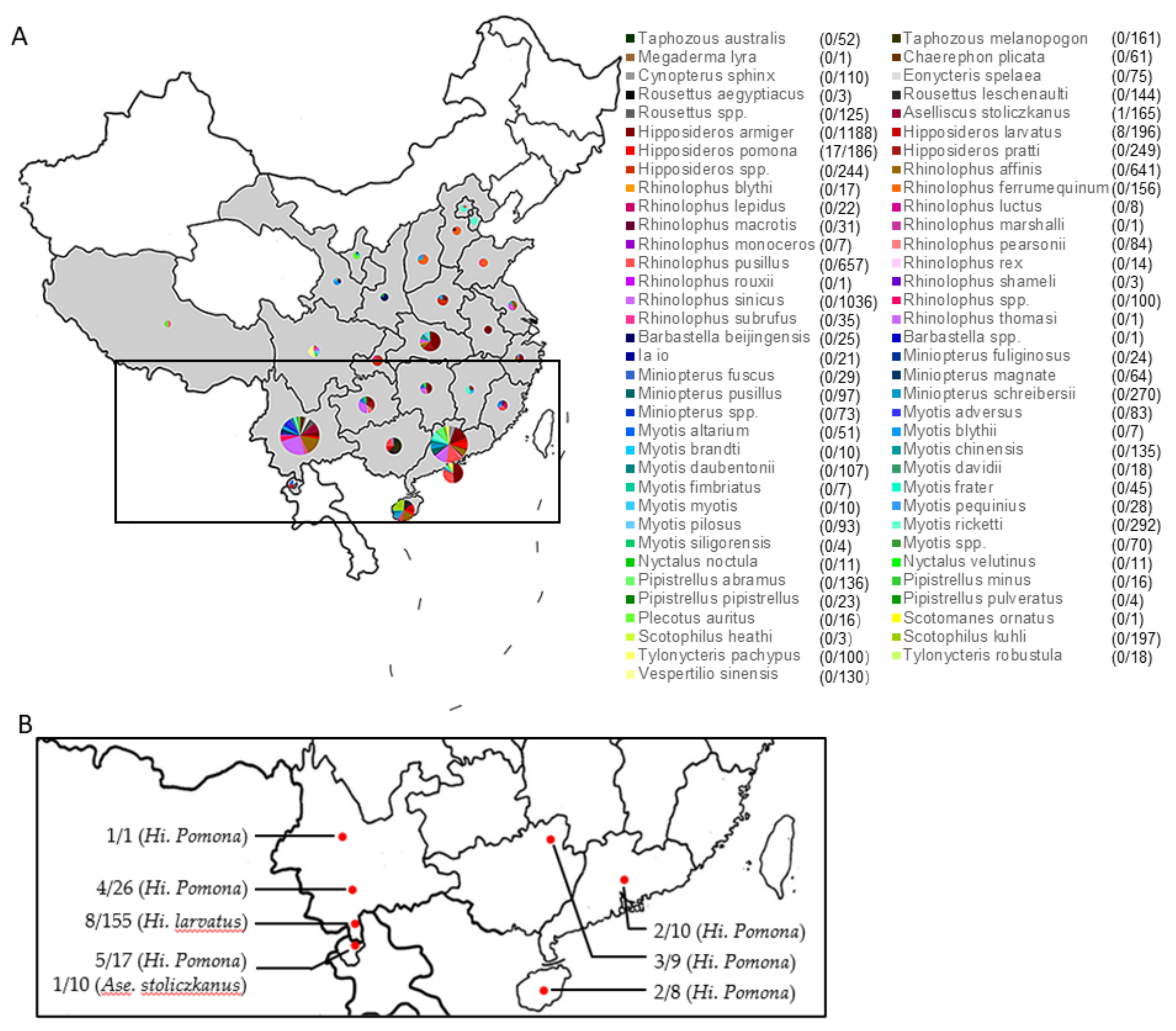
3.1. Prevalence of BtCoV HKU10
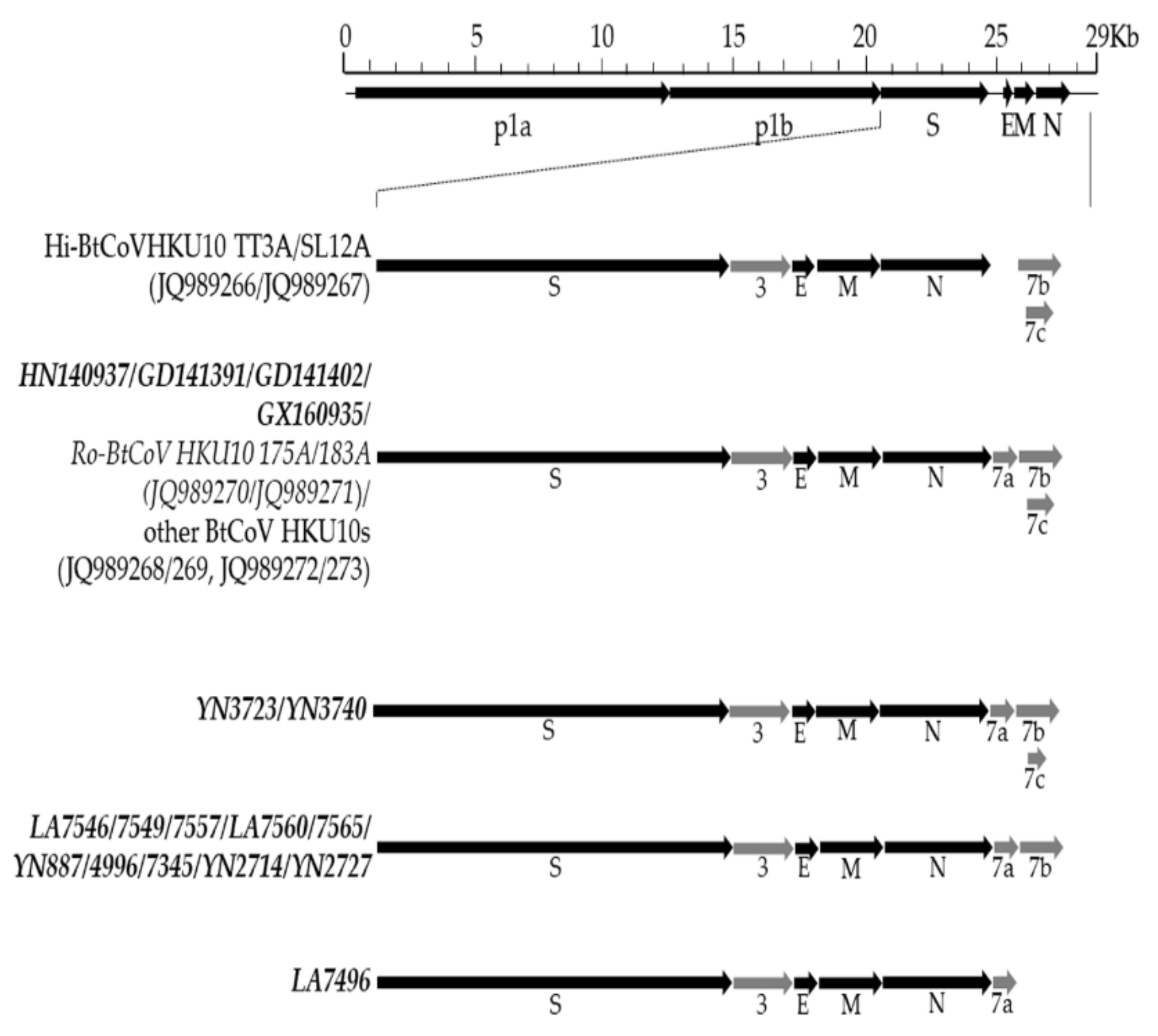
3.2. Genomic Characterization of Different BtCoV HKU10 Strains
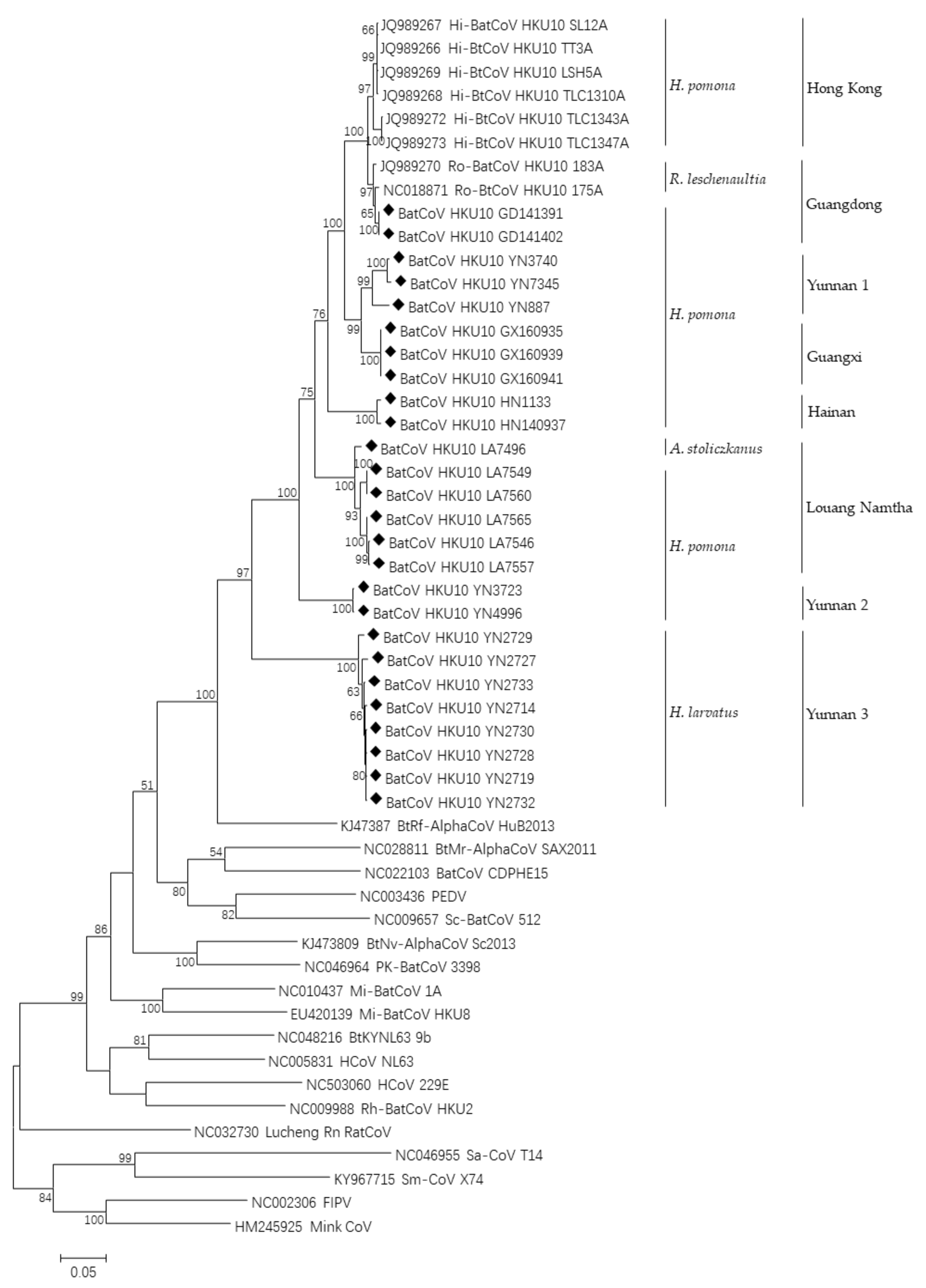
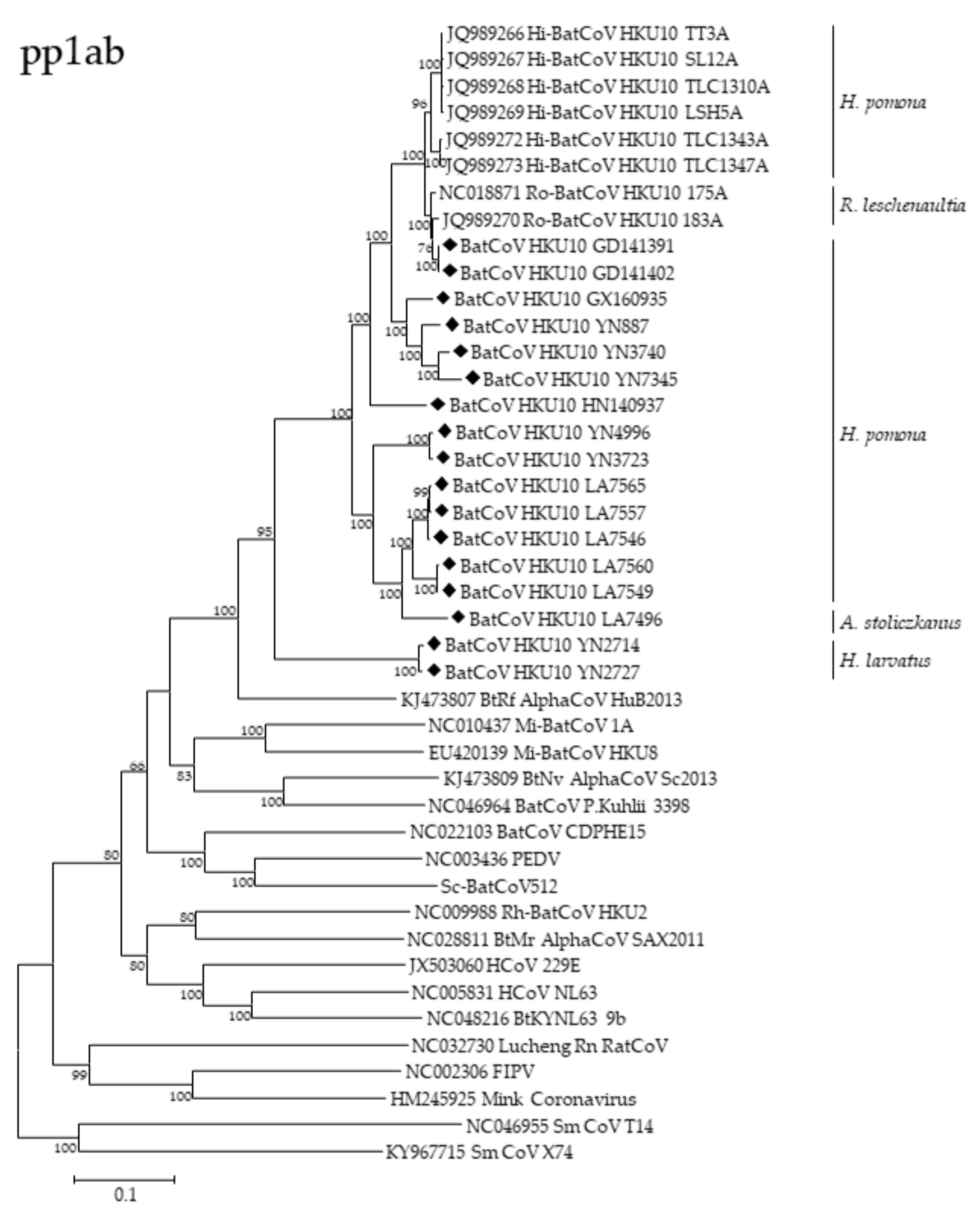
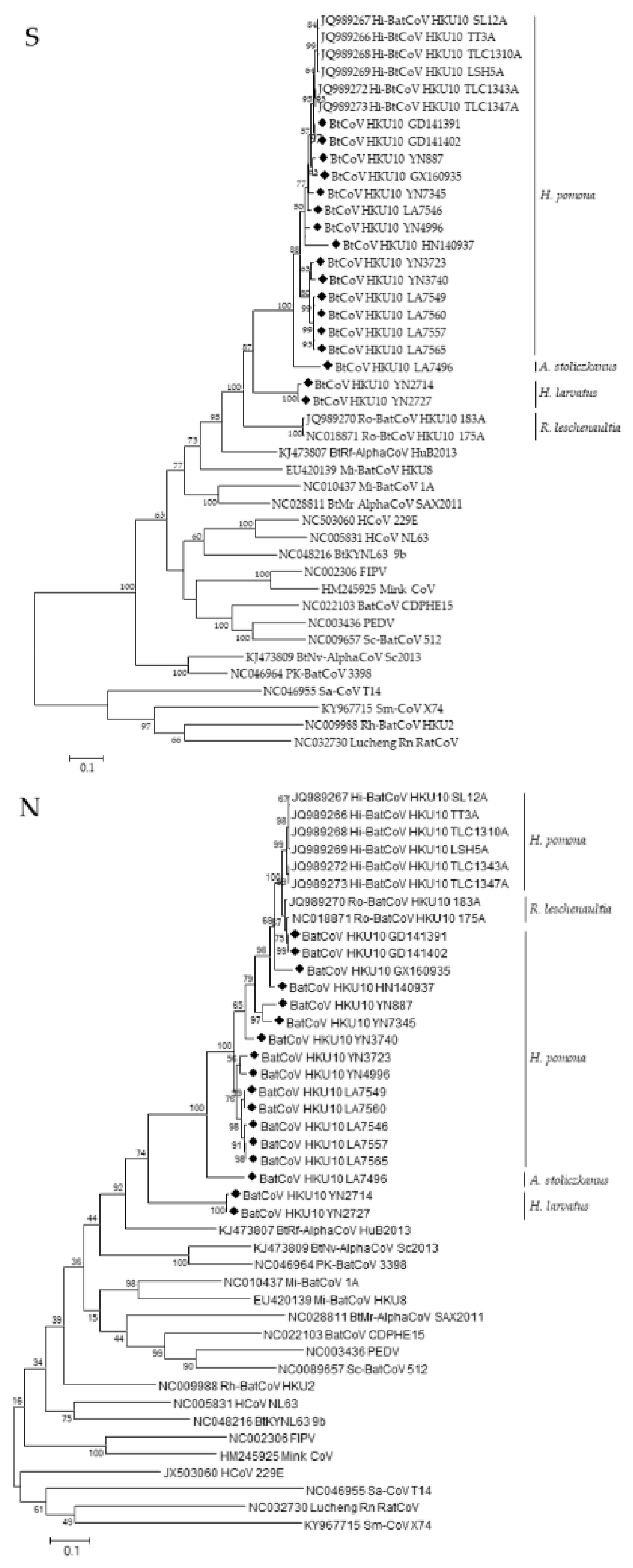
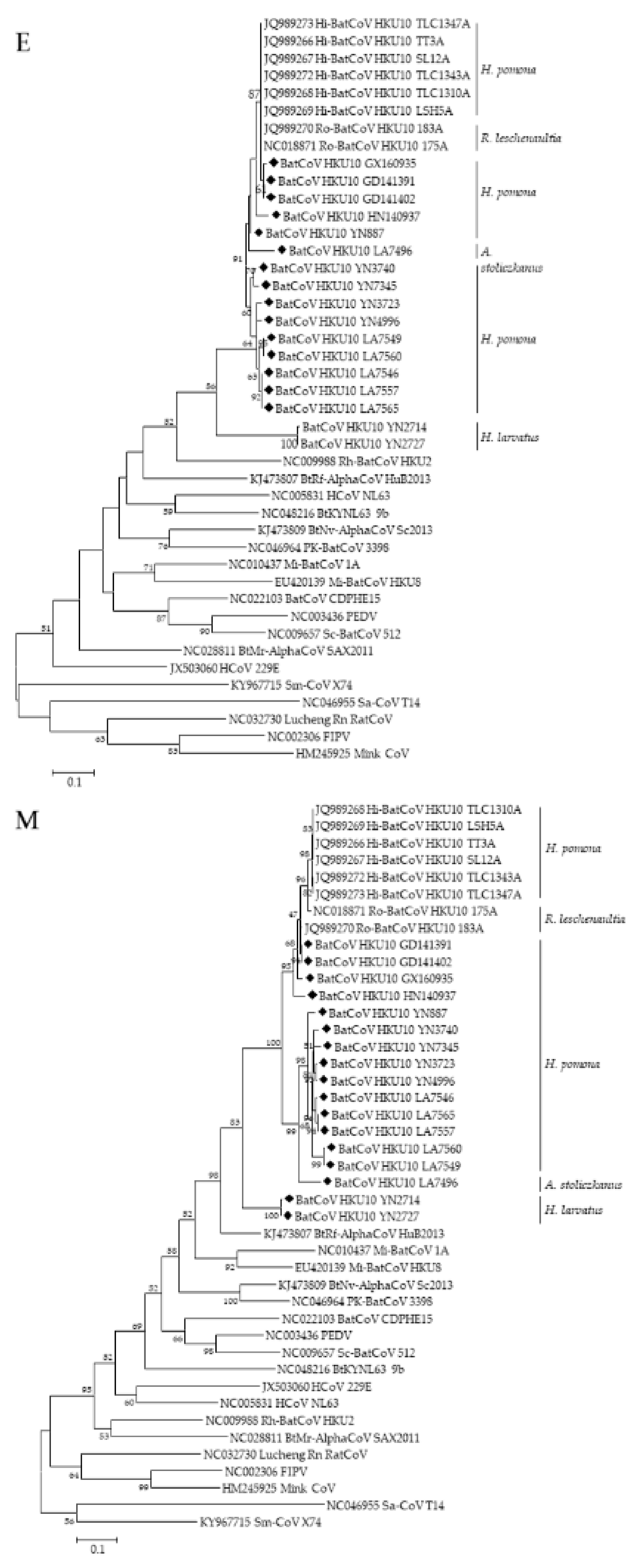
3.3. Phylogenetic Analyses of Polyprotein 1ab and Structural Proteins
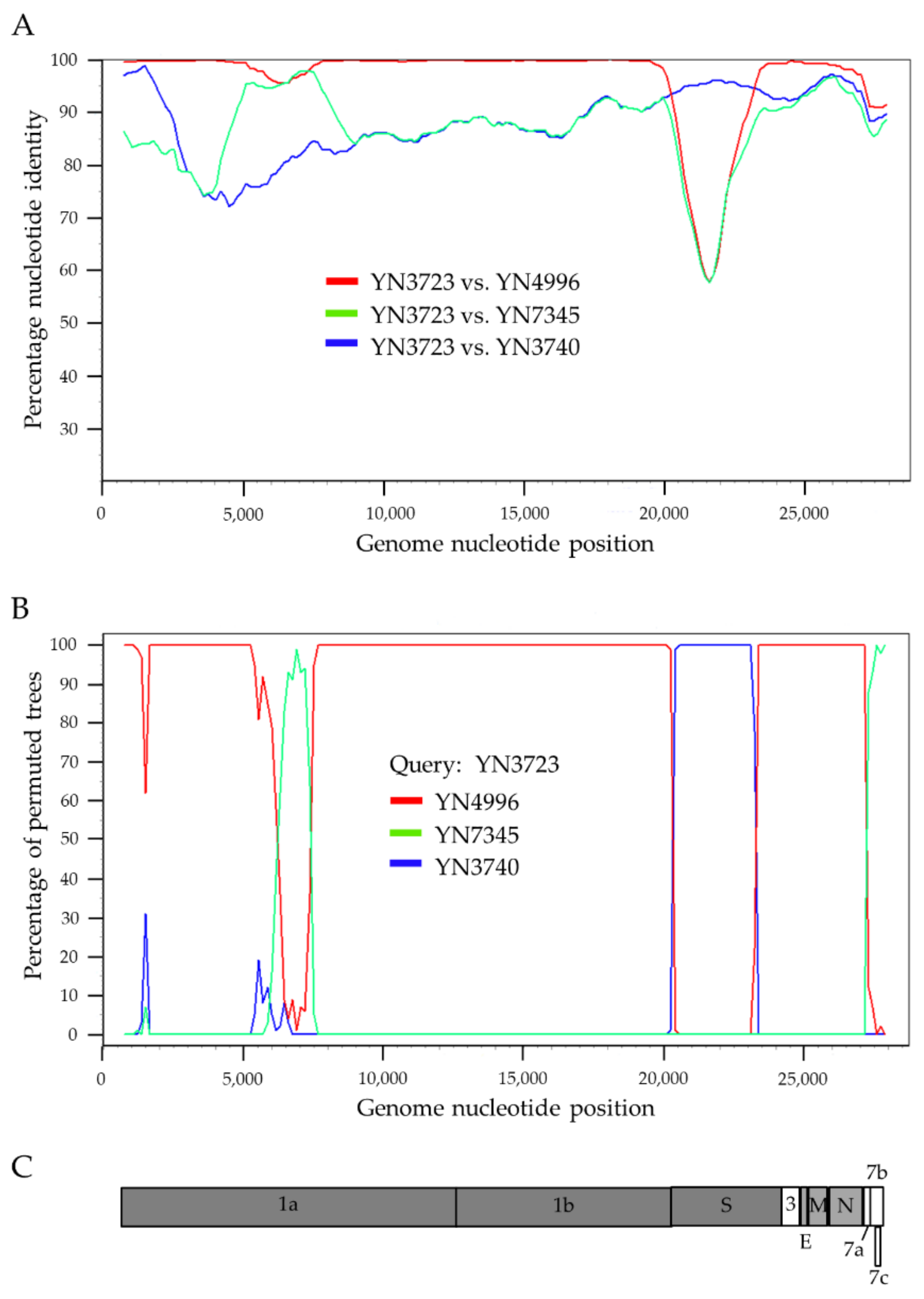
3.4. Recombination Analysis
3.5. Estmation of Synonymous and Nonsynonymous Substitution Rates
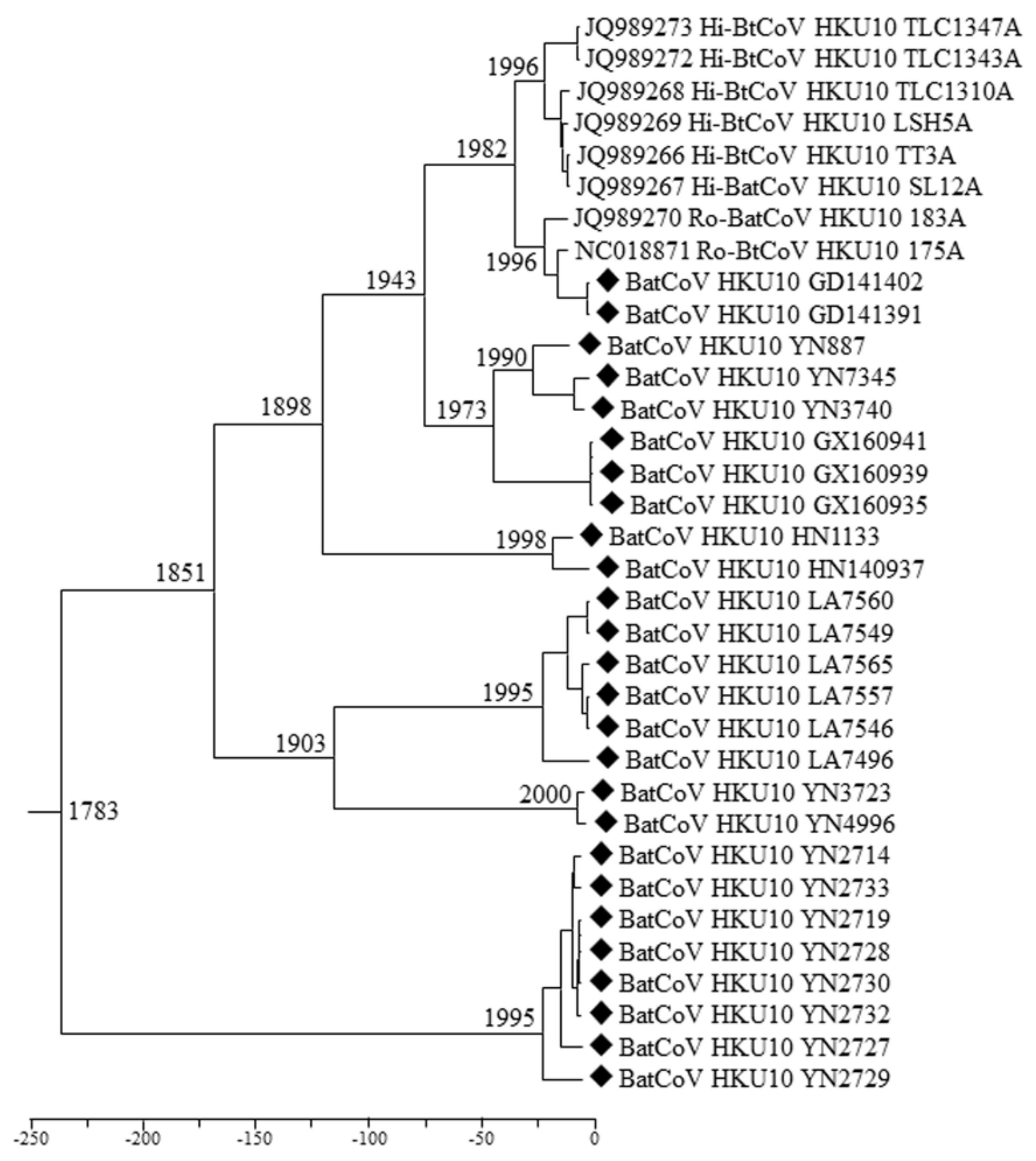
3.6. Estimation of Divergence Date
3.7. BtCoV HKU10 Spike-Mediated Pseudovirus Entry
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knipe, D.M.; Howley, P.M.; Cohen, J.I.; Griffin, D.E.; Lamb, R.A.; Martin, M.A.; Racaniello, V.R.; Roizman, B. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2013; Volume 1. [Google Scholar]
- International Committee on Taxonomy of Viruses. ICTV Master Species List 2018a v1. 2018. Available online: http://www.ictvonline.org/ (accessed on 7 April 2019).
- Miller, W.A.; Koev, G. Synthesis of subgenomic RNAs by positive-strand RNA viruses. Virology 2000, 273, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [PubMed] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.J.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zeng, L.P.; Yang, X.L.; Ge, X.Y.; Zhang, W.; Li, B.; Xie, J.Z.; Shen, X.R.; Zhang, Y.Z.; Wang, N.; et al. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS Pathog. 2017, 13, e1006698. [Google Scholar] [CrossRef]
- Luo, C.-M.; Wang, N.; Yang, X.-L.; Liu, H.-Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.-B.; Zhu, G.-J.; et al. Discovery of novel bat coronaviruses in south China that use the same receptor as MERS coronavirus. J. Virol. 2018, 92, e00116-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History. J. Virol. 2017, 91, e01953-16. [Google Scholar] [CrossRef] [Green Version]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Lam, C.S.; Ahmed, S.; Chen, H.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: Implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 8638–8650. [Google Scholar]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, M.M.C. Recombination in large RNA viruses: Coronaviruses. Semin. Virol. 1996, 7, 381–388. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.X.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Gunther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Li, W.D.; Shi, Z.L.; Yu, M.; Ren, W.Z.; Smith, C.; Epstein, J.H.; Wang, H.Z.; Crameri, G.; Hu, Z.H.; Zhang, H.J.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.; Zhang, W.; Zhu, Y.; Zhang, Y.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.F.; Tian, X.Y.; Qin, P.; Wang, B.; Zhao, P.W.; Yang, Y.L.; Wang, L.X.; Wang, D.D.; Song, Y.H.; Zhang, X.B.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef]
- Gong, L.; Li, J.; Zhou, Q.F.; Xu, Z.C.; Chen, L.; Zhang, Y.; Xue, C.Y.; Wen, Z.F.; Cao, Y.C. A New Bat-HKU2-like Coronavirus in Swine, China, 2017. Emerg. Infect. Dis 2017, 23, 1607–1609. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Y.; Wang, N.; Zhang, W.; Hu, B.; Li, B.; Zhang, Y.Z.; Zhou, J.H.; Luo, C.M.; Yang, X.L.; Wu, L.J.; et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 2016, 31, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Wacharapluesadee, S.; Duengkae, P.; Rodpan, A.; Kaewpom, T.; Maneeorn, P.; Kanchanasaka, B.; Yingsakmongkon, S.; Sittidetboripat, N.; Chareesaen, C.; Khlangsap, N.; et al. Diversity of coronavirus in bats from Eastern Thailand. Virol. J. 2015, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 48, 10–18. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Li, K.S.M.; Tsang, A.K.L.; Shek, C.T.; Wang, M.; Choi, G.K.Y.; Guo, R.T.; Wong, B.H.L.; Poon, R.W.S.; Lam, C.S.F.; et al. Recent Transmission of a Novel Alphacoronavirus, Bat Coronavirus HKU10, from Leschenault’s Rousettes to Pomona Leaf-Nosed Bats: First Evidence of Interspecies Transmission of Coronavirus between Bats of Different Suborders. J. Virol. 2012, 86, 11906–11918. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Luo, C.M.; Liu, H.Z.; Yang, X.L.; Hu, B.; Zhang, W.; Li, B.; Zhu, Y.; Zhu, G.J.; Shen, X.R.; et al. Characterization of a New Member of Alphacoronavirus with Unique Genomic Features in Rhinolophus Bats. Viruses 2019, 11, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg Infect Dis 2010, 16, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the Cytochrome-B Gene of Mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.; von Helversen, O. Cryptic diversity in European bats. Proc. R. Soc. B Biol. Sci. 2001, 268, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Leibowitz, J.L. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Res. 2015, 206, 120–133. [Google Scholar] [CrossRef]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Siltberg, J.; Liberles, D.A. A simple covarion-based approach to analyse nucleotide substitution rates. J. Evol. Biol. 2002, 15, 588–594. [Google Scholar] [CrossRef] [Green Version]
- Luna, L.K.D.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Crameri, G.; Todd, S.; Grimley, S.; McEachern, J.A.; Marsh, G.A.; Smith, C.; Tachedjian, M.; De Jong, C.; Virtue, E.R.; Yu, M.; et al. Establishment, immortalisation and characterisation of pteropid bat cell lines. PLoS ONE 2009, 4, e8266. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.Y.; Liu, C.; Wang, L.L.; Ma, C.Q.; Tang, J.; Baric, R.S.; Jiang, S.; Li, F. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc. Natl. Acad. Sci. USA 2014, 111, 12516–12521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.D.; Tu, C.C.; Zhang, G.W.; Wang, S.Y.; Zheng, K.; Lei, L.C.; Chen, Q.X.; Gao, Y.W.; Zhou, H.Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; Van der Hoek, L. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.H.; Smits, S.L.; Dekkers, D.H.W.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.; Chan, K.H.; Choi, G.K.; To, K.K.; Tse, H.; Cai, J.P.; Yeung, M.L.; Cheng, V.C.; Chen, H.; Che, X.Y.; et al. Differential cell line susceptibility to the emerging novel human betacoronavirus 2c EMC/2012: Implications for disease pathogenesis and clinical manifestation. J. Infect. Dis. 2013, 207, 1743–1752. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Tang, J.; Ma, Y.M.; Liang, X.Y.; Yang, Y.; Peng, G.Q.; Qi, Q.Q.; Jiang, S.B.; Li, J.R.; Du, L.Y.; et al. Receptor Usage and Cell Entry of Porcine Epidemic Diarrhea Coronavirus. J. Virol. 2015, 89, 6121–6125. [Google Scholar] [CrossRef] [Green Version]
- Li, W.T.; Hulswit, R.J.G.; Widjaja, I.; Raj, V.S.; McBride, R.; Peng, W.J.; Widagdo, W.; Tortorici, M.A.; van Dieren, B.; Lang, Y.; et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. USA 2017, 114, E8508–E8517. [Google Scholar] [CrossRef] [Green Version]
- Hulswit, R.J.G.; Lang, Y.F.; Bakkers, M.J.G.; Li, W.T.; Li, Z.S.; Schouten, A.; Ophorst, B.; van Kuppeveld, F.J.M.; Boons, G.J.; Bosch, B.J.; et al. Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. Proc. Natl. Acad. Sci. USA 2019, 116, 2681–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.W.; Hu, Y.W.; Wang, Q.H.; Qi, J.X.; Gao, F.; Li, Y.; Zhang, Y.F.; Zhang, W.; Yuan, Y.; Bao, J.K.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Li, W.H.; Moore, M.J.; Choe, H.; Farzan, M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J. Biol. Chem. 2004, 279, 3197–3201. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, H.; Simmons, G.; Rennekamp, A.J.; Chaipan, C.; Gramberg, T.; Heck, E.; Geier, M.; Wegele, A.; Marzi, A.; Bates, P.; et al. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J. Virol. 2006, 80, 8639–8652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Shang, J.; Yang, Y.; Liu, C.; Wan, Y.S.; Geng, Q.B.; Wang, M.; Baric, R.; Li, F. Lysosomal Proteases Are a Determinant of Coronavirus Tropism. J. Virol. 2018, 92, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSP | Putative Functional Domain(s) | YN2714 | LA7496 | YN887 | GD141391 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino Acids Position in ORF1ab | Predicted Size (aa of Protein) | C-End Predicted Cleavage Site | Amino Acids Position in ORF1ab | Predicted Size (aa of Protein) | C-End Predicted Cleavage Site | Amino Acids Position in ORF1ab | Predicted Size (aa of Protein) | C-End Predicted Cleavage Site | Amino Acids Position in ORF1ab | Predicted Size (aa of Protein) | C-End Predicted Cleavage Site | ||
| NSP1 | Unknown | M1-A195 | 195 | NA|GP | M1-A195 | 195 | VA|KP | M1-A195 | 195 | VA|KP | M1-A195 | 195 | VA|KV |
| NSP2 | Unknown | G196-G888 | 693 | TG|GG | K196-G888 | 693 | RG|SG | K196-G888 | 693 | RG|SG | K196-G888 | 693 | RG|SG |
| NSP3 | ADRP, PL2 pro | G889-G2463 | 1575 | CG|SG | S889-G2529 | 1641 | CG|SG | S889-G2523 | 1635 | CG|SG | S889-G2518 | 1630 | CG|SG |
| NSP4 | Hydrophobid domain | S2464-Q2941 | 478 | LQ|AG | S2530-Q3007 | 478 | LQ|SG | S2524-Q3001 | 478 | LQ|SG | S2519-Q2996 | 478 | LQ|SG |
| NSP5 | 3CL pro | A2942-Q3243 | 302 | LQ|ST | S3008-Q3309 | 302 | LQ|ST | S3002-Q3303 | 302 | LQ|SN | S2997-Q3298 | 302 | LQ|SN |
| NSP6 | Hydrophobid domain | S3244-Q3519 | 276 | VQ|SK | S3310-Q3585 | 276 | VQ|SK | S3304-Q3579 | 276 | VQ|SK | S3299-Q3574 | 276 | VQ|SK |
| NSP7 | Replicase | S3520-Q3602 | 83 | LQ|SV | S3586-Q3668 | 83 | LQ|SV | S3580-Q3662 | 83 | LQ|SV | S3575-Q3657 | 83 | LQ|SV |
| NSP8 | Replicase | S3603-Q3797 | 195 | LQ|NN | S3669-Q3863 | 195 | LQ|NN | S3663-Q3857 | 195 | LQ|NN | S3658-Q3852 | 195 | LQ|NN |
| NSP9 | Replicase | N3798-Q3905 | 108 | LQ|AG | N3864-Q3971 | 108 | LQ|AG | N3858-Q3965 | 108 | LQ|AG | N3853-Q3960 | 108 | LQ|AG |
| NSP10 | RNA synthesis protein | A3906-Q4041 | 136 | VQ|AF | A3972-Q4108 | 137 | MQ|AF | A3966-Q4102 | 137 | MQ|AF | A3961-Q4097 | 137 | MQ|AF |
| NSP11 | Unknown (short peptide at the end of ORF1a) | A4042-N4058 | 17 | A4109-N4125 | 17 | A4103-N4119 | 17 | A4098-N4114 | 17 | ||||
| NSP12 | RdRp | A4042-Q4968 | 927 | LQ|AA | A4109-Q5035 | 927 | LQ|SA | A4103-Q5029 | 927 | LQ|SA | A4098-Q5024 | 927 | LQ|SA |
| NSP13 | Hel, NTPase | A4969-Q5565 | 597 | LQ|AG | S5036-Q5632 | 597 | LQ|AG | S5030-Q5626 | 597 | LQ|AG | S5025-Q5621 | 597 | LQ|AG |
| NSP14 | ExoN, NMT | A5566-Q6083 | 518 | LQ|SL | A5633-Q6150 | 518 | LQ|SL | A5627-Q6144 | 518 | LQ|SL | A5622-Q6139 | 518 | LQ|SL |
| NSP15 | NeudoU | S6084-Q6422 | 339 | LQ|SA | S6151-Q6487 | 337 | LQ|SA | S6145-Q6483 | 339 | LQ|SA | S6140-Q6476 | 337 | LQ|SA |
| NSP16 | 2′-O-MT | S6423-K6724 | 302 | S6488-C6799 | 312 | S6484-K6785 | 302 | S6477-H6783 | 307 | ||||
| Genes | Ka/Ks Ratio | ||||
|---|---|---|---|---|---|
| BtCoV HKU10 | BtCoV HKU10 | BtCoV HKU10 | BtCoV HKU10 | BtCoV HKU10 | |
| (H. pomona, 20 Strains) | (A. stoliczkanus, 1 Strain) | (H. lavatus, 2 Strains) | (R. leschenaultia, 2 Strains) | (All, 25 Strains) | |
| NSP1 | 0.354 | 0.214 | 0.237 | 0.150 | 0.297 |
| NSP2 | 0.309 | 0.131 | 0.391 | 0.171 | 0.403 |
| NSP3 | 0.359 | 0.190 | 0.303 | 0.139 | 0.370 |
| NSP4 | 0.189 | 0.104 | 0.225 | 0.059 | 0.274 |
| NSP5 | 0.176 | 0.033 | 0.205 | 0.003 | 0.221 |
| NSP6 | 0.317 | 0.167 | 0.097 | 0.053 | 0.477 |
| NSP7 | 0.265 | Ka = 0, Ks = 0.035 | Ka = 0, Ks = 0 | Ka = 0, Ks = 0 | 0.166 |
| NSP8 | 0.174 | 0.500 | 0.010 | 0.012 | 0.287 |
| NSP9 | 0.198 | Ka = 0, Ks = 0 | 0.073 | 0.031 | 0.598 |
| NSP10 | 0.200 | Ka = 0, Ks = 0.032 | 0.120 | 0.056 | 0.138 |
| NSP11 | Ka = 0, Ks = 0.066 | Ka = 0, Ks = 0.131 | Ka = 0, Ks = 0.066 | Ka = 0, Ks = 0 | 1.176 |
| NSP12 | 0.123 | 0.053 | 0.092 | 0.012 | 0.067 |
| NSP13 | 0.112 | 0.161 | 0.087 | 0.008 | 0.0738 |
| NSP14 | 0.081 | 0.052 | 0.142 | 0.034 | 0.109 |
| NSP15 | 0.127 | 0.114 | 0.086 | 0.026 | 0.167 |
| NSP16 | 0.103 | 0.117 | 0.126 | 0.114 | 0.105 |
| S | 0.240 | 0.243 | 0.272 | 0.382 | 0.259 |
| ORF3a | 0.069 | 0.169 | Ka = 0, Ks = 0 | 0.051 | 0.274 |
| E | 0 | 0.204 | 0.227 | Ka = 0, Ks = 0 | 0.218 |
| M | 0.192 | 0.024 | 0.147 | 0.275 | 0.066 |
| N | 0.151 | 0.188 | 0.218 | 0.146 | 0.207 |
| ORF7a | 0.339 | 0.403 | 0.442 | 0.336 | 0.662 |
| ORF7b | 1.0368 | NA | 1.161 | 0.136 | 0.951 |
| ORF7c | 0.630 | NA | NA | 0.609 | 0.4433 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Luo, C.-M.; Yang, X.-L.; Liu, H.-Z.; Zhang, L.-B.; Zhang, W.; Li, B.; Zhu, Y.; Peng, C.; Shi, Z.-L.; et al. Genomic Characterization of Diverse Bat Coronavirus HKU10 in Hipposideros Bats. Viruses 2021, 13, 1962. https://doi.org/10.3390/v13101962
Wang N, Luo C-M, Yang X-L, Liu H-Z, Zhang L-B, Zhang W, Li B, Zhu Y, Peng C, Shi Z-L, et al. Genomic Characterization of Diverse Bat Coronavirus HKU10 in Hipposideros Bats. Viruses. 2021; 13(10):1962. https://doi.org/10.3390/v13101962
Chicago/Turabian StyleWang, Ning, Chu-Ming Luo, Xing-Lou Yang, Hai-Zhou Liu, Li-Biao Zhang, Wei Zhang, Bei Li, Yan Zhu, Cheng Peng, Zheng-Li Shi, and et al. 2021. "Genomic Characterization of Diverse Bat Coronavirus HKU10 in Hipposideros Bats" Viruses 13, no. 10: 1962. https://doi.org/10.3390/v13101962