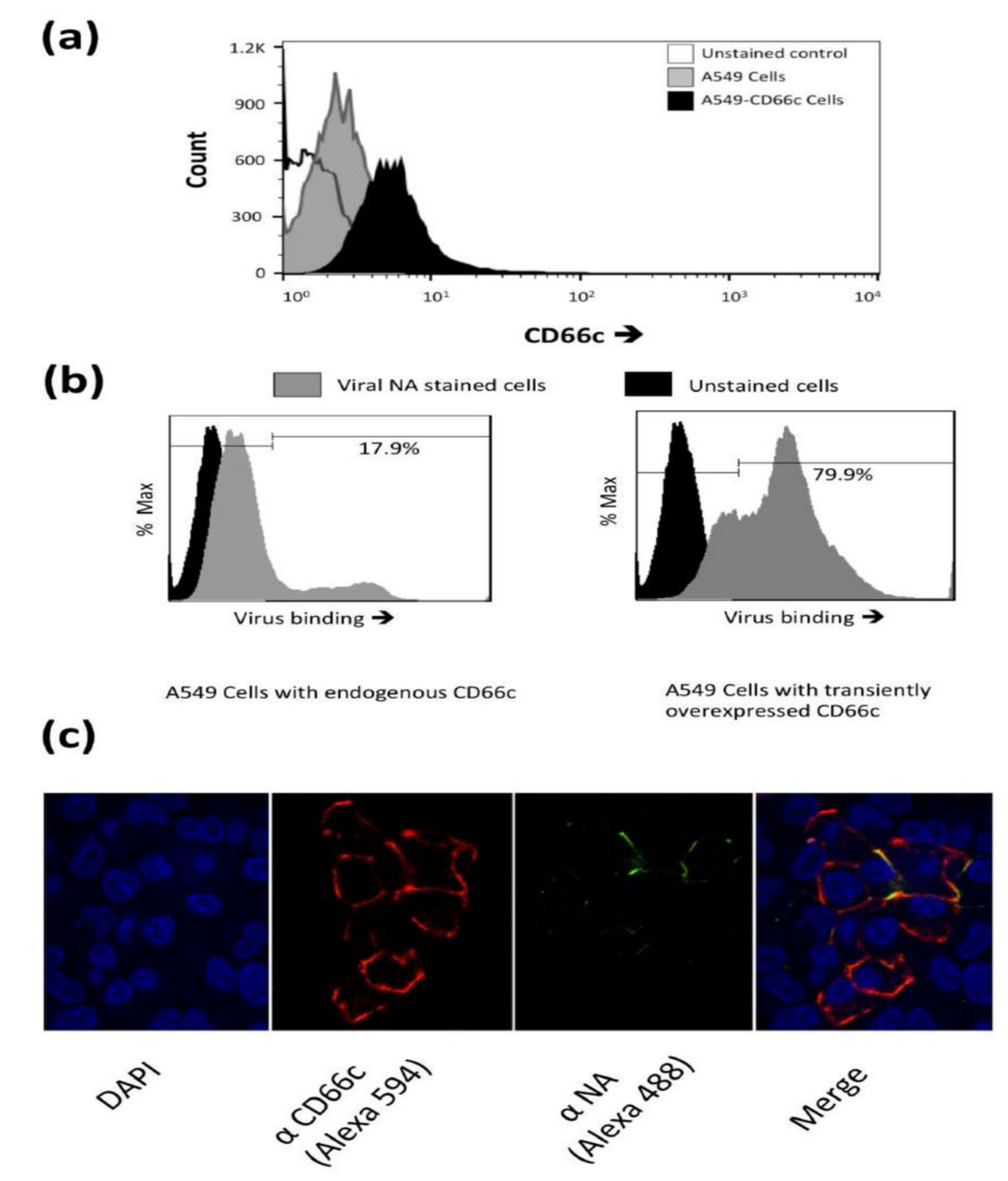
Figure 1.
(a): Flow cytometry analysis of surface expression of CD66c in A549 cells. Cell population transiently overexpressing CD66c shows signal for higher expression of CD66c on the cell surface (black) as compared to A549 cells (grey), unstained control cells (white). (b): Flow cytometry analysis of virus binding on A549 cells or CD66c overexpressing A549 cells. 5 multiplicity of infection (MOI) of PR8 virus binding signal corresponding to stained NA protein (Alexa-488) of the virus on host cell surface. Signal of unstained cells (black), viral NA (grey) corresponding to the virus binding on cell surface. PR8 virus binding is increased from ~18% (left panel) to >80% of CD66c overexpressing A549 cell population (right panel). (c): To a monolayer of A549 cells, binding of 5 multiplicity of infection (moi) of PR8 virus was observed under confocal microscope. Panels from left to right shows cell nuclei stained with DAPI (blue), CD66c (red), Viral NA (green) at the cell periphery. Colocalization (yellow) of NA protein of PR8 virus (green) with host membrane protein CD66c (red) at the periphery of cells.
Figure 1.
(a): Flow cytometry analysis of surface expression of CD66c in A549 cells. Cell population transiently overexpressing CD66c shows signal for higher expression of CD66c on the cell surface (black) as compared to A549 cells (grey), unstained control cells (white). (b): Flow cytometry analysis of virus binding on A549 cells or CD66c overexpressing A549 cells. 5 multiplicity of infection (MOI) of PR8 virus binding signal corresponding to stained NA protein (Alexa-488) of the virus on host cell surface. Signal of unstained cells (black), viral NA (grey) corresponding to the virus binding on cell surface. PR8 virus binding is increased from ~18% (left panel) to >80% of CD66c overexpressing A549 cell population (right panel). (c): To a monolayer of A549 cells, binding of 5 multiplicity of infection (moi) of PR8 virus was observed under confocal microscope. Panels from left to right shows cell nuclei stained with DAPI (blue), CD66c (red), Viral NA (green) at the cell periphery. Colocalization (yellow) of NA protein of PR8 virus (green) with host membrane protein CD66c (red) at the periphery of cells.
![Viruses 13 00726 g001]()
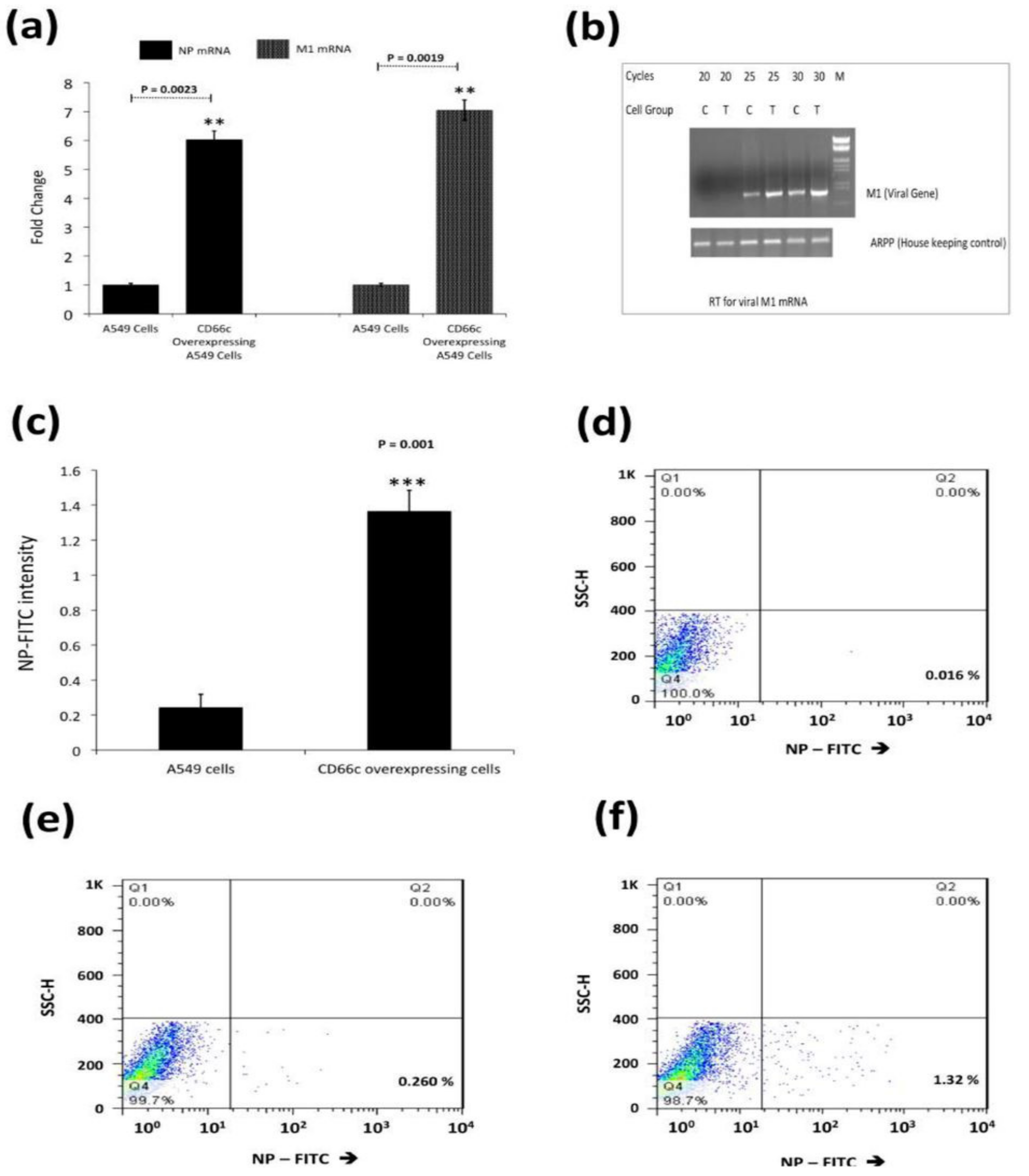
Figure 2.
(a): Real time quantification of NP and M1 mRNA in viral infected A549 cells 8 h post infection (8 h.p.i) infected cells. Figure shows higher mRNA levels of NP (left two bars) and M1 (right two bars) in cells overexpressing CD66c. Each bar represents mean of five independent experimental readings. (b): Semi quantification of viral M1 by RT-PCR. For the above-mentioned reaction condition (8 h.p.i, and 1) viral M1 mRNA was also measured semi-quantitatively. Before each lane A549 control cells is denoted as ‘’C’’, or A549 cells having overexpressed CD66c with ‘‘T’’. Cycles are the number of PCR cycles. (c): Flow cytometric analysis for virus load in A549 infected cell: The bar diagram shows rise in viral NP in CD66c overexpressing A549 cells (right) than that in A549 cells (left). Each bar represents mean values of at least three independent experiments ± SD. Statistical significance was assessed by student’s t-test, (**) for p ≤ 0.01 and (***) for p ≤ 0.001. (d): Representative FACS snapshot of unstained A549 cells showing no signal corresponding to NP-FITC in lower-right quadrant; cells with some signal in (e); cells overexpressing CD66c with increased NP-FITC signal in lower-right quadrant (f).
Figure 2.
(a): Real time quantification of NP and M1 mRNA in viral infected A549 cells 8 h post infection (8 h.p.i) infected cells. Figure shows higher mRNA levels of NP (left two bars) and M1 (right two bars) in cells overexpressing CD66c. Each bar represents mean of five independent experimental readings. (b): Semi quantification of viral M1 by RT-PCR. For the above-mentioned reaction condition (8 h.p.i, and 1) viral M1 mRNA was also measured semi-quantitatively. Before each lane A549 control cells is denoted as ‘’C’’, or A549 cells having overexpressed CD66c with ‘‘T’’. Cycles are the number of PCR cycles. (c): Flow cytometric analysis for virus load in A549 infected cell: The bar diagram shows rise in viral NP in CD66c overexpressing A549 cells (right) than that in A549 cells (left). Each bar represents mean values of at least three independent experiments ± SD. Statistical significance was assessed by student’s t-test, (**) for p ≤ 0.01 and (***) for p ≤ 0.001. (d): Representative FACS snapshot of unstained A549 cells showing no signal corresponding to NP-FITC in lower-right quadrant; cells with some signal in (e); cells overexpressing CD66c with increased NP-FITC signal in lower-right quadrant (f).
![Viruses 13 00726 g002]()
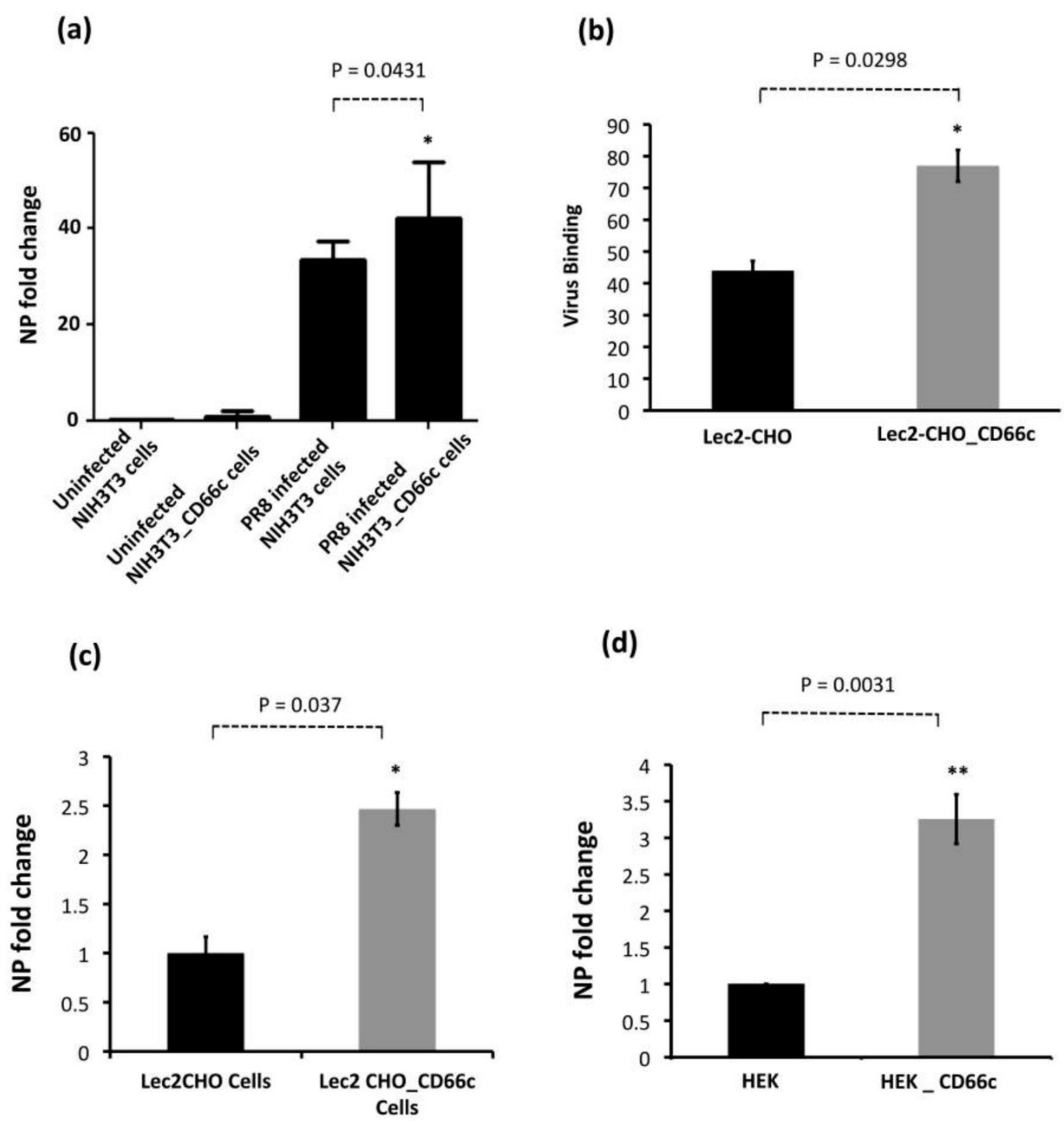
Figure 3.
Demonstration of rise in viral uptake in CD66c overexpressing NIH 3T3 and Lec2-CHO cell lines. (a): The level of infection as probed by the NP mRNA fold-change. The bar in the right end shows increased influenza infection in CD66c transfected NIH3T3 cell lines. (b): Lec2 CHO-CD66c is CD66c overexpressing Lec2 CHO cell lines. The right bar shows greater virus binding on the surface of cells overexpressing CD66c. (c): An increased level of viral NP mRNA in Lec2 CHO-CD66c cells (right bar) as compared to Lec2 CHO cells (left bar) suggesting increased virus entry. (d): Levels of mRNA corresponding to lower infection level in HEK cells (left bar) and an increase in infection in CD66c overexpressing HEK cells (right bar). Statistical significance was assessed by student’s t-test, (*) for p ≤ 0.05 and (**) for p ≤ 0.01.
Figure 3.
Demonstration of rise in viral uptake in CD66c overexpressing NIH 3T3 and Lec2-CHO cell lines. (a): The level of infection as probed by the NP mRNA fold-change. The bar in the right end shows increased influenza infection in CD66c transfected NIH3T3 cell lines. (b): Lec2 CHO-CD66c is CD66c overexpressing Lec2 CHO cell lines. The right bar shows greater virus binding on the surface of cells overexpressing CD66c. (c): An increased level of viral NP mRNA in Lec2 CHO-CD66c cells (right bar) as compared to Lec2 CHO cells (left bar) suggesting increased virus entry. (d): Levels of mRNA corresponding to lower infection level in HEK cells (left bar) and an increase in infection in CD66c overexpressing HEK cells (right bar). Statistical significance was assessed by student’s t-test, (*) for p ≤ 0.05 and (**) for p ≤ 0.01.
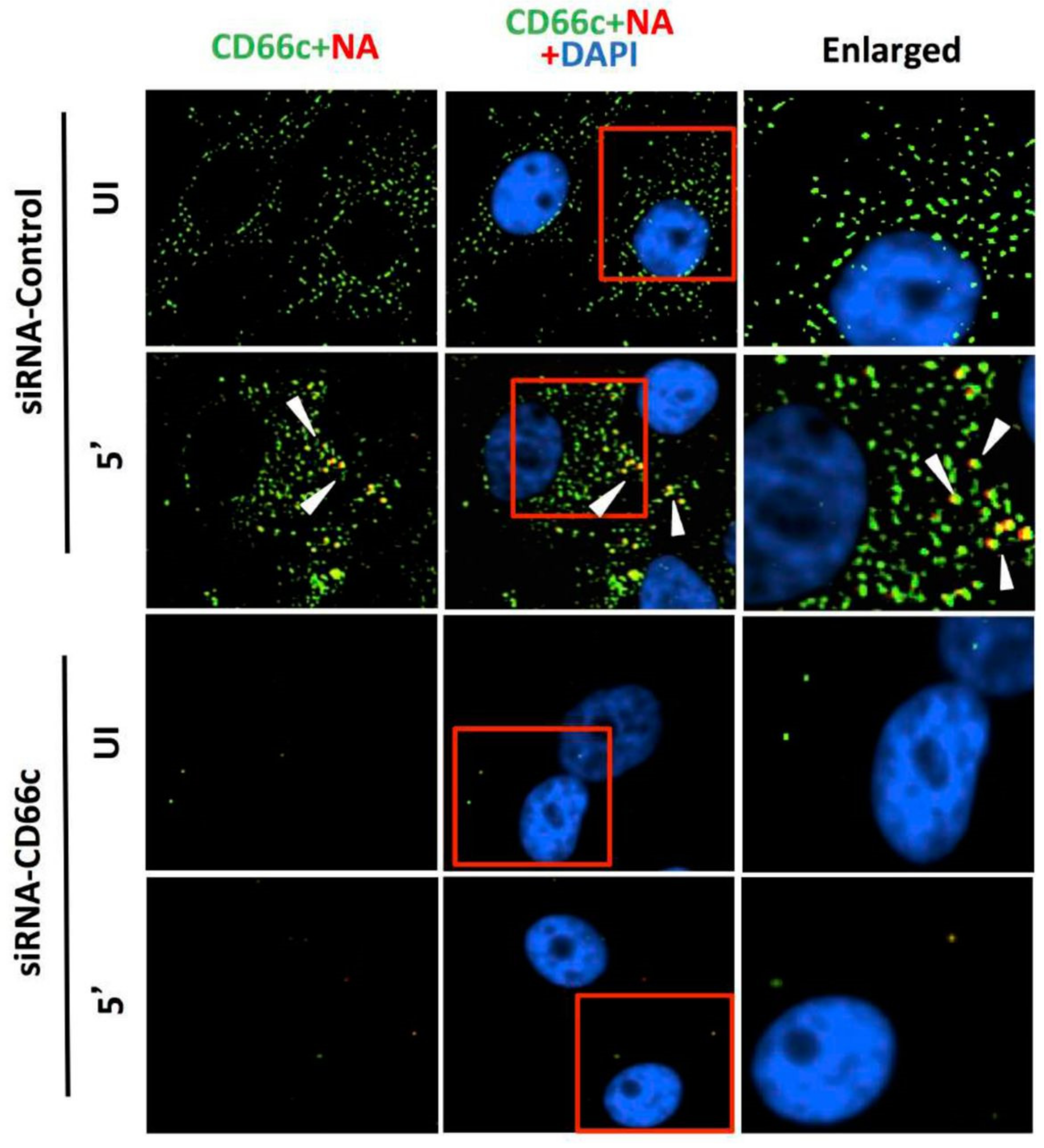
Figure 4.
Knockdown of CD66c shows inhibition of virus binding on cell surface under fluorescent microscope. Here, UI denotes uninfected cells; 5′, Cells after 5 min of virus binding to them. Figure shows that A549 cells treated with siRNA control (negative) do not inhibit expression level of CD66c (green) and therefore binding of 5 multiplicity of infection (MOI) of PR8 viruses on cell surface can be seen. Figure shows colocalization between NA (red) and CD66c (green) in merged view (yellow) (second panel from top, pointed with white arrow). Amount of colocalization between NA and CD66c and absence of NA (red color) in control siRNA treated cells signifies possible colocalization between CD66c and NA at the host cell surface. The lowest two panels show diminished green signals in cells treated with CD66c siRNA suggesting a poor expression of CD66c protein (green). Consequently, virus binding on siRNA-CD66c treated cells is not seen, as evident by the absence of any green (CD66c) or red (NA) signal (bottom panel).
Figure 4.
Knockdown of CD66c shows inhibition of virus binding on cell surface under fluorescent microscope. Here, UI denotes uninfected cells; 5′, Cells after 5 min of virus binding to them. Figure shows that A549 cells treated with siRNA control (negative) do not inhibit expression level of CD66c (green) and therefore binding of 5 multiplicity of infection (MOI) of PR8 viruses on cell surface can be seen. Figure shows colocalization between NA (red) and CD66c (green) in merged view (yellow) (second panel from top, pointed with white arrow). Amount of colocalization between NA and CD66c and absence of NA (red color) in control siRNA treated cells signifies possible colocalization between CD66c and NA at the host cell surface. The lowest two panels show diminished green signals in cells treated with CD66c siRNA suggesting a poor expression of CD66c protein (green). Consequently, virus binding on siRNA-CD66c treated cells is not seen, as evident by the absence of any green (CD66c) or red (NA) signal (bottom panel).
![Viruses 13 00726 g004]()
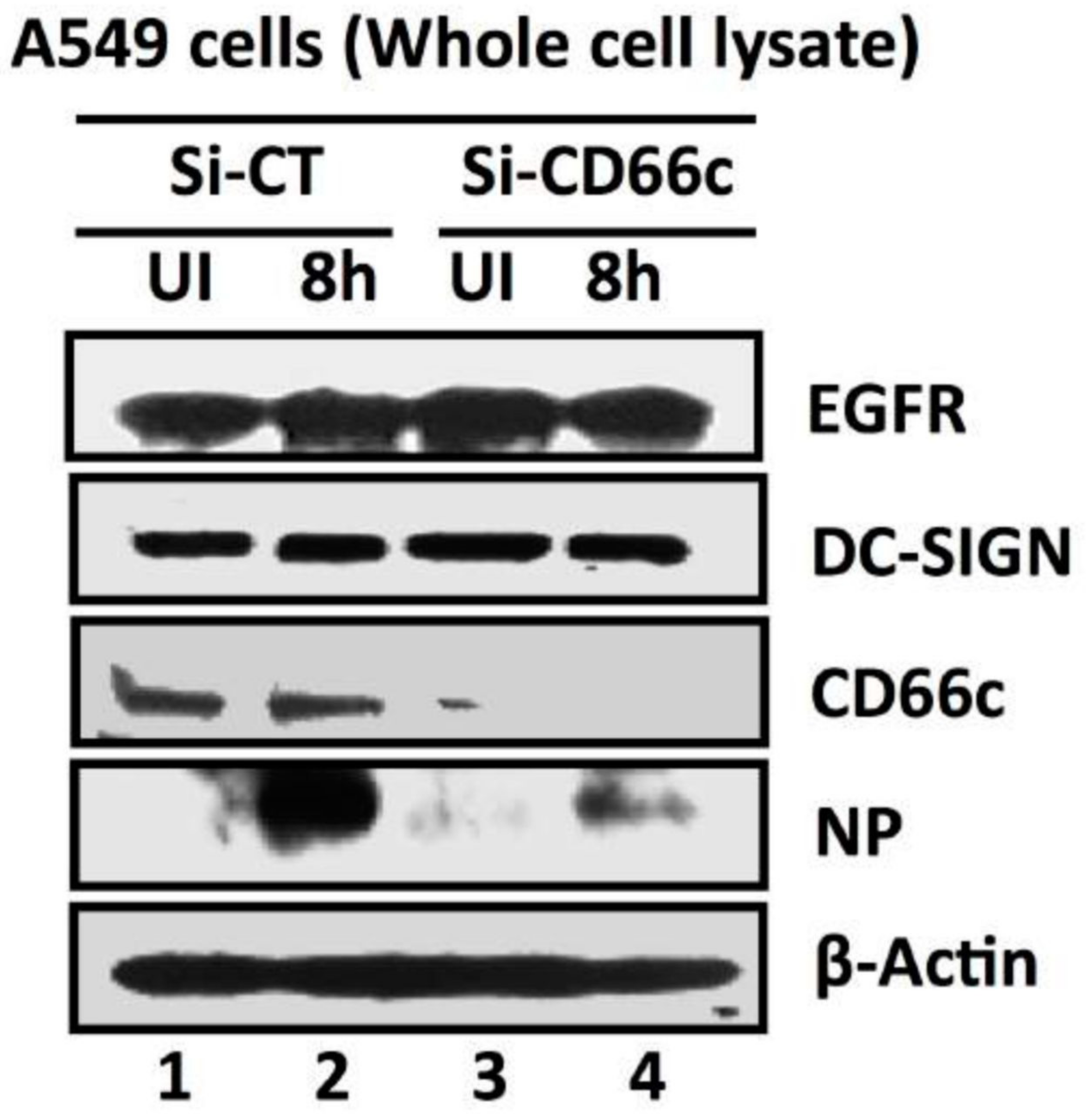
Figure 5.
siRNA-mediated knockdown of CD66c shows inhibition of virus entry into lung-cells infected with 1 MOI of PR8 virus, through western blot analysis. Here, Si-CT denotes control siRNA treated cells; Si-CD66c are cells treated with CD66c siRNA. 8h are cells harvested after 8 h of infection (one life cycle of IAV), UI are uninfected cells. A549 cells treated with siRNA show complete knock down of the receptor molecule CD66c (third panel from top) while control siRNA treated cells do not show any reduction in expression of CD66c (left two wells of third panel). The fourth panel from top shows level of A/PR8/34 influenza virus entry in cells determined by expression levels of viral NP protein. The second well from left shows significant expression level of viral NP protein in cells treated with control siRNA, eight hours after infection. In contrast, the right-most well shows a marked reduction of influenza A virus (IAV) entry in cells treated with CD66c siRNA, as determined by low expression level of viral protein NP eight hours after infection. Conclusively, virus entry was inhibited in absence of CD66c (CD66c siRNA treated cells). CD66c siRNA treated cells do not show any noticeable change in expression levels of EGRF (topmost panel) and DC-SIGN (second panel from top). For loading control β-Actin was probed (the bottom panel).
Figure 5.
siRNA-mediated knockdown of CD66c shows inhibition of virus entry into lung-cells infected with 1 MOI of PR8 virus, through western blot analysis. Here, Si-CT denotes control siRNA treated cells; Si-CD66c are cells treated with CD66c siRNA. 8h are cells harvested after 8 h of infection (one life cycle of IAV), UI are uninfected cells. A549 cells treated with siRNA show complete knock down of the receptor molecule CD66c (third panel from top) while control siRNA treated cells do not show any reduction in expression of CD66c (left two wells of third panel). The fourth panel from top shows level of A/PR8/34 influenza virus entry in cells determined by expression levels of viral NP protein. The second well from left shows significant expression level of viral NP protein in cells treated with control siRNA, eight hours after infection. In contrast, the right-most well shows a marked reduction of influenza A virus (IAV) entry in cells treated with CD66c siRNA, as determined by low expression level of viral protein NP eight hours after infection. Conclusively, virus entry was inhibited in absence of CD66c (CD66c siRNA treated cells). CD66c siRNA treated cells do not show any noticeable change in expression levels of EGRF (topmost panel) and DC-SIGN (second panel from top). For loading control β-Actin was probed (the bottom panel).
![Viruses 13 00726 g005]()
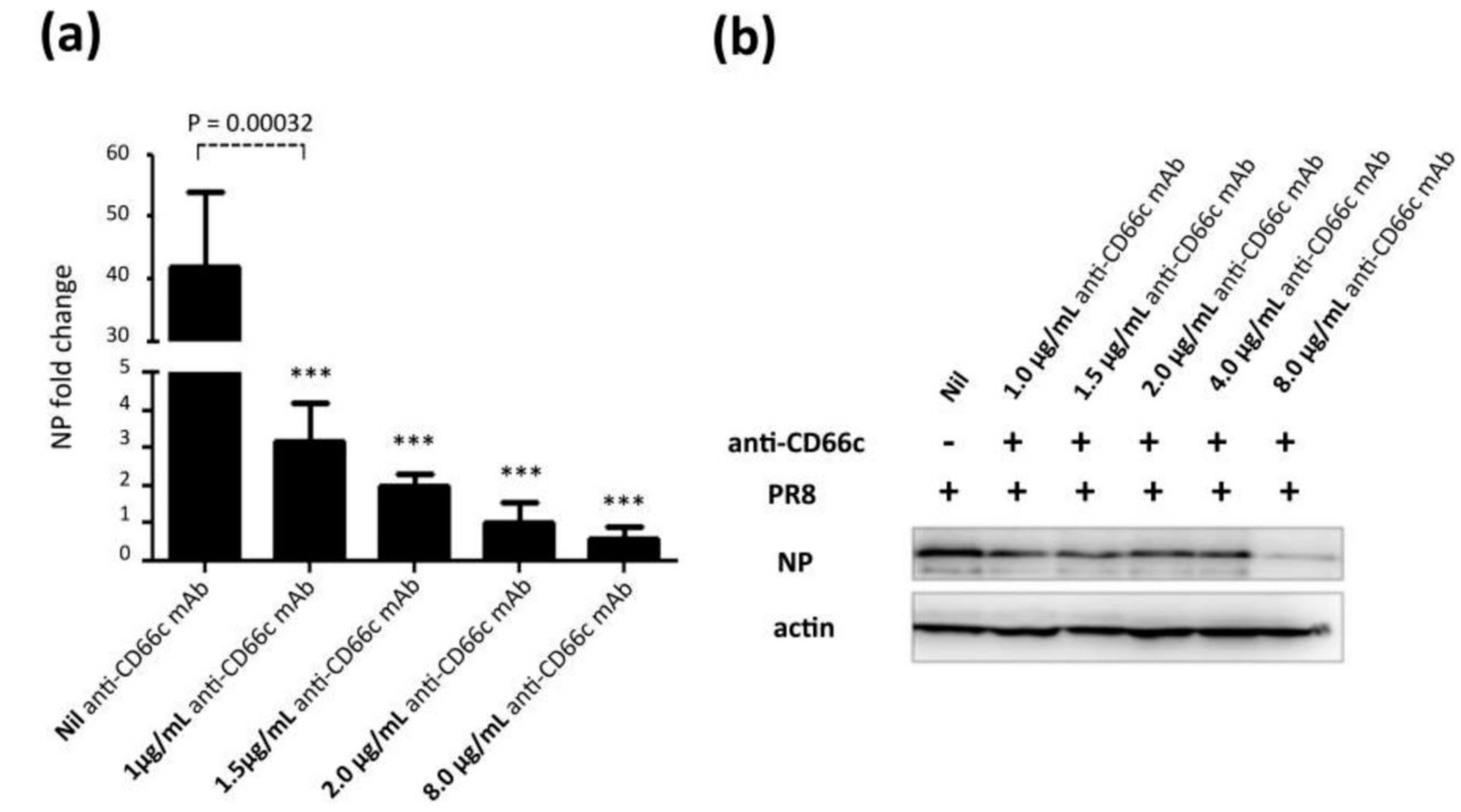
Figure 6.
Antibody mediated receptor blockade experiments in CD66c overexpressing cells. (a): The figure shows level of A/PR8/34 influenza virus entry in a monolayer of NIH3T3-CD66c cells when treated with mAb anti-CD66c prior to infection. From left to right, the first bar in the figure represents levels of viral NP mRNA (a measure of virus entry) in untreated NIH3T3-CD66c. Bars second to fifth from left show levels of viral NP mRNA in virus-infected cells treated with mAb anti-CD66c at a concentration of 1.0 μg/mL, 1.5 μg/mL, 2.0 μg/mL and 8.0 μg/mL respectively. Conclusively, the data show a decrease in virus entry in cells treated with anti-CD66c in a dose dependent manner. Data represent mean values of at least three independent experiments ± SD. Statistical significance was assessed by student’s t-test (GraphPad), (*) for p ≤ 0.05, (**) for p ≤ 0.01 and (***) for p ≤ 0.001. (b): A western blot showing expression levels of viral NP protein in virus-infected NIH3T3-CD66c when treated with corresponding concentrations of mAb anti-CD66c 1.0 μg/mL, 1.5 μg/mL, 2.0 μg/mL, 4.0 μg/mL and 8.0 μg/mL, prior to viral infection. The inhibition of virus entry in anti-CD66c mAb treated cells was significant at a concentration of 8.0 μg/mL of CD66c.
Figure 6.
Antibody mediated receptor blockade experiments in CD66c overexpressing cells. (a): The figure shows level of A/PR8/34 influenza virus entry in a monolayer of NIH3T3-CD66c cells when treated with mAb anti-CD66c prior to infection. From left to right, the first bar in the figure represents levels of viral NP mRNA (a measure of virus entry) in untreated NIH3T3-CD66c. Bars second to fifth from left show levels of viral NP mRNA in virus-infected cells treated with mAb anti-CD66c at a concentration of 1.0 μg/mL, 1.5 μg/mL, 2.0 μg/mL and 8.0 μg/mL respectively. Conclusively, the data show a decrease in virus entry in cells treated with anti-CD66c in a dose dependent manner. Data represent mean values of at least three independent experiments ± SD. Statistical significance was assessed by student’s t-test (GraphPad), (*) for p ≤ 0.05, (**) for p ≤ 0.01 and (***) for p ≤ 0.001. (b): A western blot showing expression levels of viral NP protein in virus-infected NIH3T3-CD66c when treated with corresponding concentrations of mAb anti-CD66c 1.0 μg/mL, 1.5 μg/mL, 2.0 μg/mL, 4.0 μg/mL and 8.0 μg/mL, prior to viral infection. The inhibition of virus entry in anti-CD66c mAb treated cells was significant at a concentration of 8.0 μg/mL of CD66c.
![Viruses 13 00726 g006]()
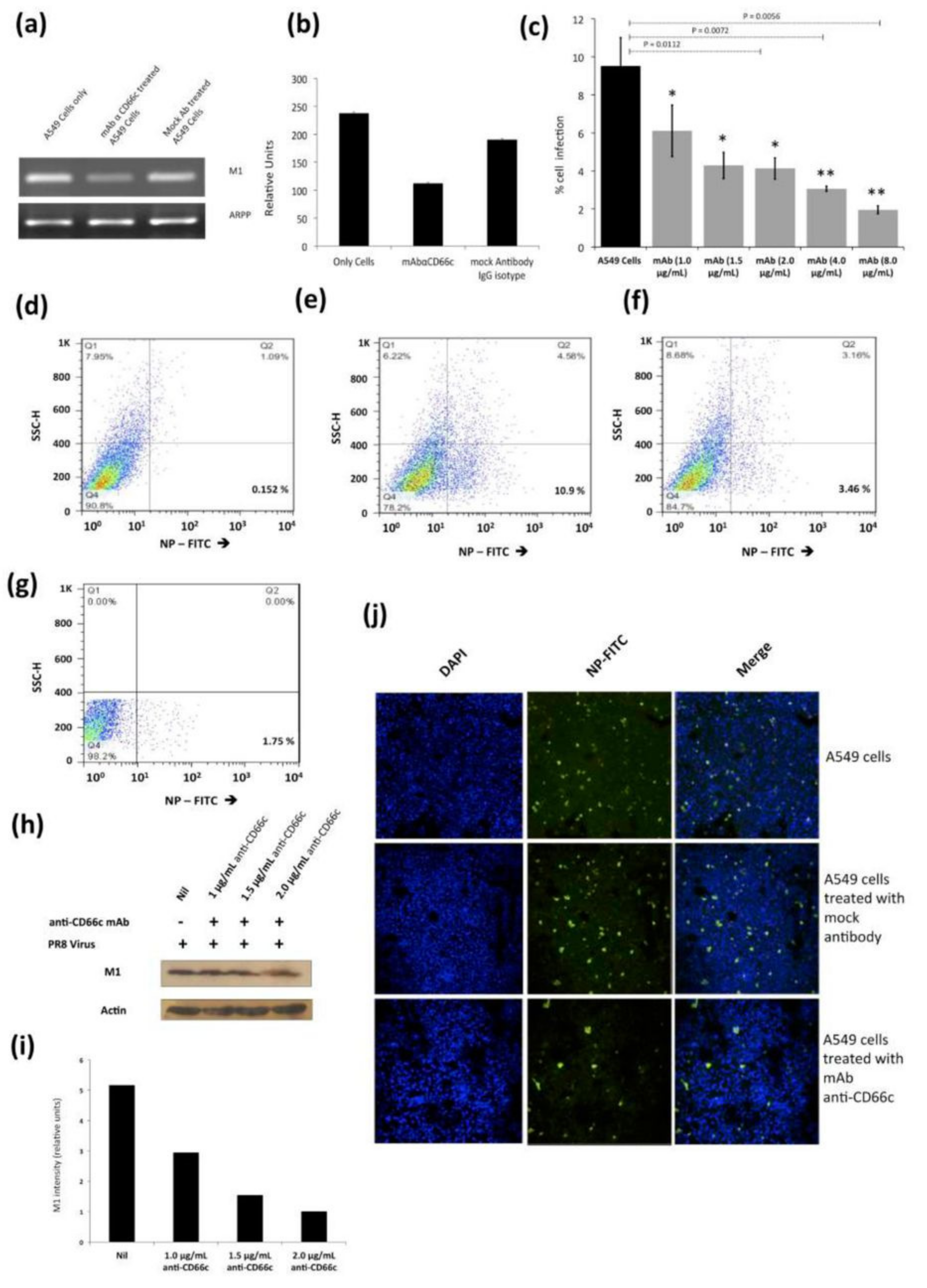
Figure 7.
Antibody mediated receptor blockade experiments in A549 cells expressing endogenous levels of CD66c, during A/PR8/34 influenza virus infection in lung A549 cells (24 HPI), when incubated with increasing concentrations of anti-CD66c mAb prior to infection. (a): Left well shows M1 mRNA level in A549 cells (left) in cells incubated with mAb anti-CD66c (middle) and the right well shows M1 mRNA level in cells incubated with mock antibody (IgG isotype antibody) prior to viral infection. (b): Densitometry analysis of image (a). The expression level of housekeeping gene acidic ribosomal phosphoprotein (ARPP) as experimental control. (c): A549 cells expressing viral NP protein (a measure of virus entry) after infection without any anti-CD66c treatment (black bar). Grey bars (from left to right) represent A549 cells treated with anti-CD66c mAb, prior to infection, at the concentrations of 1.0 μg/mL, 1.5 μg/mL, 2 μg/mL, 4 μg/mL or 8 μg/mL, respectively. Each bar shows the mean of three independent experimental readings (*, p < 0.05; **, p < 0.01). Figures (d–g) are representative snapshots of NP stained cells from flow cytometry. (d): Unstained A549 cells (e): A549 cells with NP-FITC signal in 10.9% cell population. (f): A549 cells incubated with 4.0 μg/mL of mAb anti-CD66c with relatively reduced signal, in 3.46% cell population. (g): A549 cells incubated with 8.0 μg/mL of mAb anti-CD66c. (h): Western blot analysis of infection in A549 cells incubated with anti-CD66c mAb at a concentration of 1.0 μg/mL, 1.5 μg/mL or 2.0 μg/mL, prior to infection by the virus. (i): Densitometry analysis of (h). (j): Immunofluorescent assay (IFA) and confocal microscopic analysis of A/PR8/34 virus-infected A549 cells at 24 HPI, staining viral NP (green). Uppermost panel shows infection level in A549 cells, the middle panel shows infection in cells incubated with mock antibody (IgG Isotype control) and the bottom panel shows reduced infection in cells incubated with 4 μg/mL anti-CD66c mAb before viral infection.
Figure 7.
Antibody mediated receptor blockade experiments in A549 cells expressing endogenous levels of CD66c, during A/PR8/34 influenza virus infection in lung A549 cells (24 HPI), when incubated with increasing concentrations of anti-CD66c mAb prior to infection. (a): Left well shows M1 mRNA level in A549 cells (left) in cells incubated with mAb anti-CD66c (middle) and the right well shows M1 mRNA level in cells incubated with mock antibody (IgG isotype antibody) prior to viral infection. (b): Densitometry analysis of image (a). The expression level of housekeeping gene acidic ribosomal phosphoprotein (ARPP) as experimental control. (c): A549 cells expressing viral NP protein (a measure of virus entry) after infection without any anti-CD66c treatment (black bar). Grey bars (from left to right) represent A549 cells treated with anti-CD66c mAb, prior to infection, at the concentrations of 1.0 μg/mL, 1.5 μg/mL, 2 μg/mL, 4 μg/mL or 8 μg/mL, respectively. Each bar shows the mean of three independent experimental readings (*, p < 0.05; **, p < 0.01). Figures (d–g) are representative snapshots of NP stained cells from flow cytometry. (d): Unstained A549 cells (e): A549 cells with NP-FITC signal in 10.9% cell population. (f): A549 cells incubated with 4.0 μg/mL of mAb anti-CD66c with relatively reduced signal, in 3.46% cell population. (g): A549 cells incubated with 8.0 μg/mL of mAb anti-CD66c. (h): Western blot analysis of infection in A549 cells incubated with anti-CD66c mAb at a concentration of 1.0 μg/mL, 1.5 μg/mL or 2.0 μg/mL, prior to infection by the virus. (i): Densitometry analysis of (h). (j): Immunofluorescent assay (IFA) and confocal microscopic analysis of A/PR8/34 virus-infected A549 cells at 24 HPI, staining viral NP (green). Uppermost panel shows infection level in A549 cells, the middle panel shows infection in cells incubated with mock antibody (IgG Isotype control) and the bottom panel shows reduced infection in cells incubated with 4 μg/mL anti-CD66c mAb before viral infection.
![Viruses 13 00726 g007]()
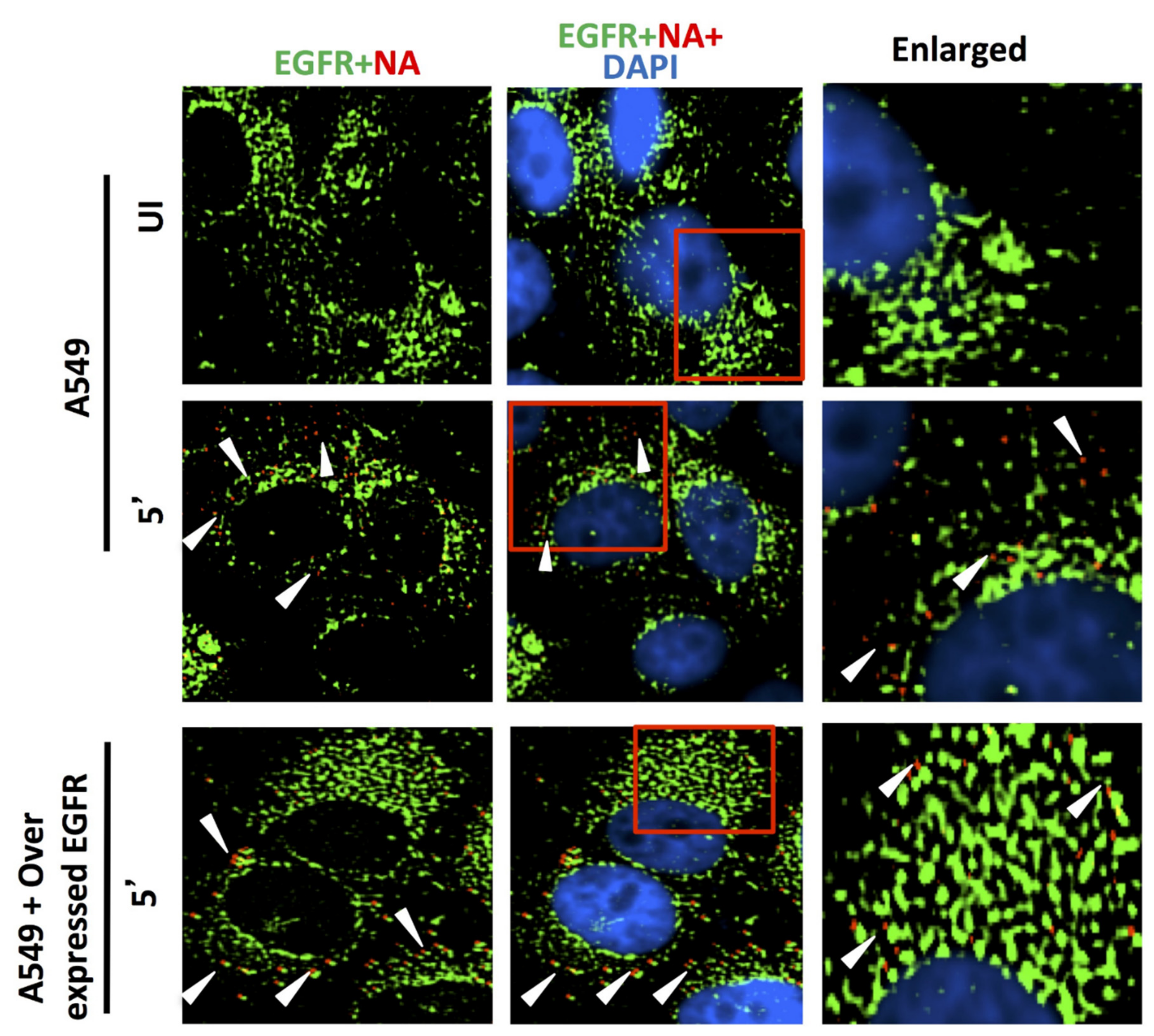
Figure 8.
Comparing the ability of host membrane proteins CD66c, EGFR and DC-SIGN respectively in IAV binding on A549 cells through fluorescent microscopy. UI denotes uninfected cells; 5′, Cells after 5 min of virus binding. A549 cells were cultured either with endogenous level of expression or overexpression of receptor candidates EGFR, CD66c or DC-SIGN. Cells were either uninfected or infected with 5 MOI of A/PR8/34 influenza virus for 5 min. The secondary antibody Alexa 488 (green) probed host EGFR and Alexa 594 (red) probed influenza NA; cell nuclei are stained with DAPI (blue). No trace of virus binding in uninfected cells (top panel); modest virus binding on cell surface is observed after 5 min of incubation influenza NA (red). Only few virus particles are colocalizing with EGFR (middle panel, yellow spots, white arrow). In EGFR overexpressing A549 cells there is neither corresponding increase in virus binding nor in colocalization of viral NA with EGFR. Rather cells overexpressing EGFR show similar virus binding pattern as cells with endogenous level of EGFR (bottom panel). Altogether, virus binding is not significantly increased with overexpression of EGFR, suggesting its poor binding ability.
Figure 8.
Comparing the ability of host membrane proteins CD66c, EGFR and DC-SIGN respectively in IAV binding on A549 cells through fluorescent microscopy. UI denotes uninfected cells; 5′, Cells after 5 min of virus binding. A549 cells were cultured either with endogenous level of expression or overexpression of receptor candidates EGFR, CD66c or DC-SIGN. Cells were either uninfected or infected with 5 MOI of A/PR8/34 influenza virus for 5 min. The secondary antibody Alexa 488 (green) probed host EGFR and Alexa 594 (red) probed influenza NA; cell nuclei are stained with DAPI (blue). No trace of virus binding in uninfected cells (top panel); modest virus binding on cell surface is observed after 5 min of incubation influenza NA (red). Only few virus particles are colocalizing with EGFR (middle panel, yellow spots, white arrow). In EGFR overexpressing A549 cells there is neither corresponding increase in virus binding nor in colocalization of viral NA with EGFR. Rather cells overexpressing EGFR show similar virus binding pattern as cells with endogenous level of EGFR (bottom panel). Altogether, virus binding is not significantly increased with overexpression of EGFR, suggesting its poor binding ability.
![Viruses 13 00726 g008]()
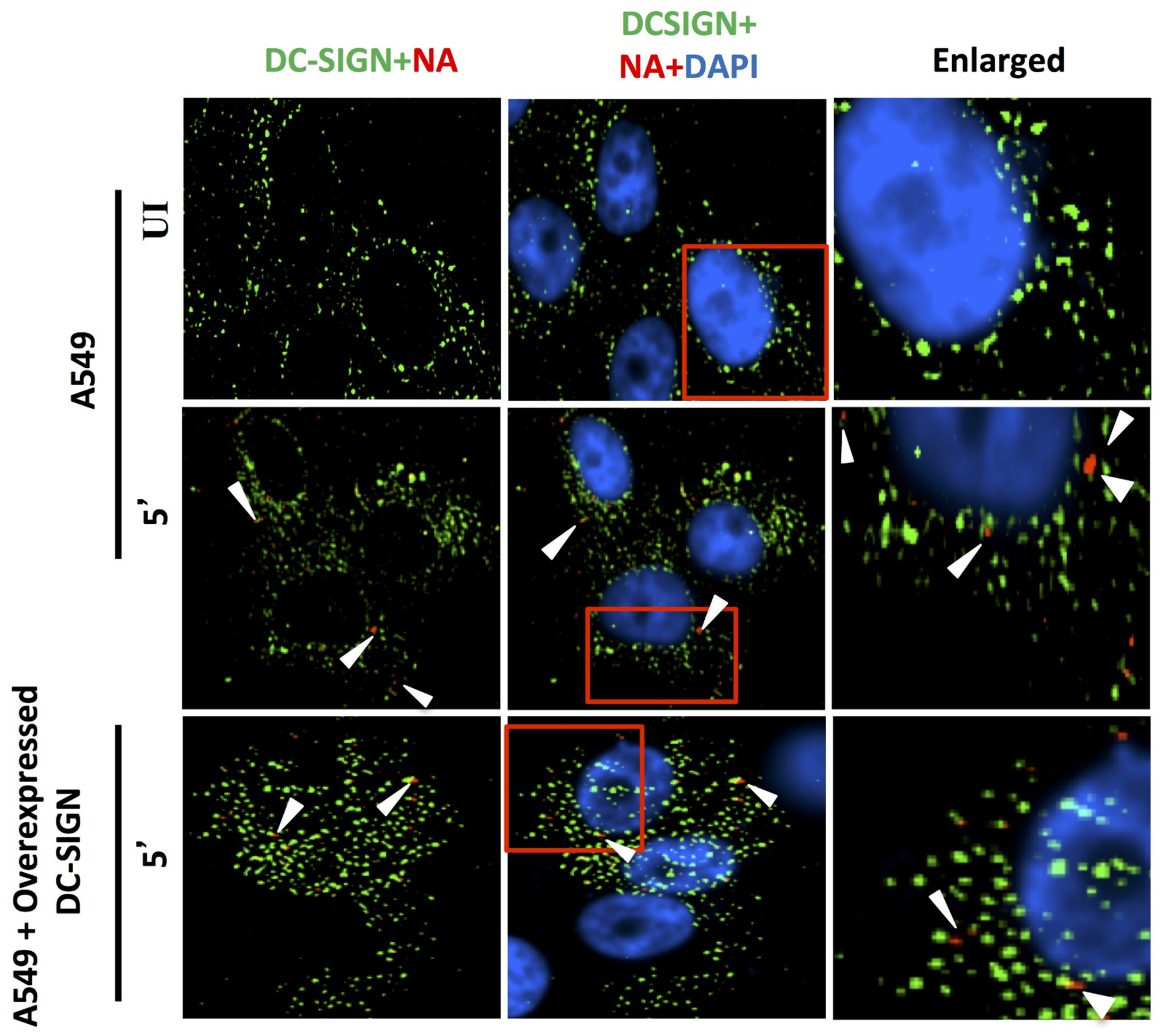
Figure 9.
DC-SIGN (green) and influenza NA (red) after 5 min of virus binding. No trace of virus binding in uninfected cells (upper panel). Few co-localization spots (yellow spots, middle panel) in cells with endogenous level of DC-SIGN; in cells overexpressing DC-SIGN (green) there is no consequent increase in virus binding (bottom panel, white arrow) as compared to cells with endogenous level of DC-SIGN (middle panel, white arrow). Altogether, virus binding is not significantly increased with overexpression of DC-SIGN.
Figure 9.
DC-SIGN (green) and influenza NA (red) after 5 min of virus binding. No trace of virus binding in uninfected cells (upper panel). Few co-localization spots (yellow spots, middle panel) in cells with endogenous level of DC-SIGN; in cells overexpressing DC-SIGN (green) there is no consequent increase in virus binding (bottom panel, white arrow) as compared to cells with endogenous level of DC-SIGN (middle panel, white arrow). Altogether, virus binding is not significantly increased with overexpression of DC-SIGN.
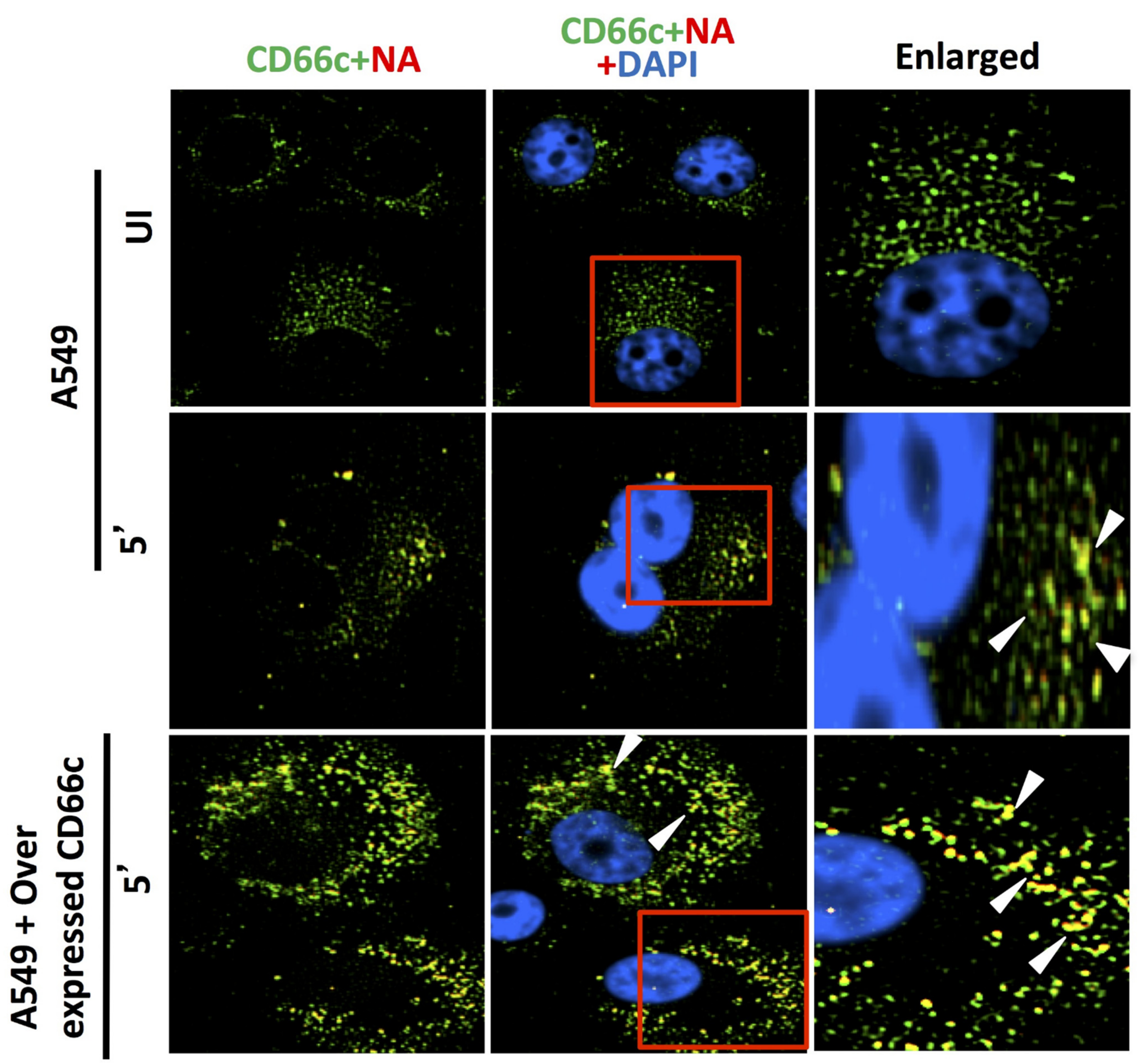
Figure 10.
Cells stained for endogenous expression level of host CD66c (green) and influenza NA (red) after 5 min (5′) showed significant virus binding on cell surface as probed by influenza neuraminidase NA (red) (middle panel). Cells also showed colocalization (yellow spots) between endogenous levels of receptor CD66c (green) with influenza neuraminidase NA (red). Unbound influenza NA (red) is not easily identified as most are seen merged (as yellow) with CD66c. No trace of virus binding in uninfected cells (UI, upper panel). Cells overexpressing receptor CD66c shows significant increase in virus binding and co-localization of CD66c with NA (yellow dots) on the cell surface (bottom panel) when compared to cells with endogenous level of CD66c (middle panel). CD66c (green) mainly at the periphery of cells interacting with significant number of NA (yellow after merge, white arrow). Altogether, virus binding with CD66c is prominent and significantly increased with overexpression, highlighting the strong binding ability of receptor CD66c as compared to EGFR and DC-SIGN.
Figure 10.
Cells stained for endogenous expression level of host CD66c (green) and influenza NA (red) after 5 min (5′) showed significant virus binding on cell surface as probed by influenza neuraminidase NA (red) (middle panel). Cells also showed colocalization (yellow spots) between endogenous levels of receptor CD66c (green) with influenza neuraminidase NA (red). Unbound influenza NA (red) is not easily identified as most are seen merged (as yellow) with CD66c. No trace of virus binding in uninfected cells (UI, upper panel). Cells overexpressing receptor CD66c shows significant increase in virus binding and co-localization of CD66c with NA (yellow dots) on the cell surface (bottom panel) when compared to cells with endogenous level of CD66c (middle panel). CD66c (green) mainly at the periphery of cells interacting with significant number of NA (yellow after merge, white arrow). Altogether, virus binding with CD66c is prominent and significantly increased with overexpression, highlighting the strong binding ability of receptor CD66c as compared to EGFR and DC-SIGN.
![Viruses 13 00726 g010]()
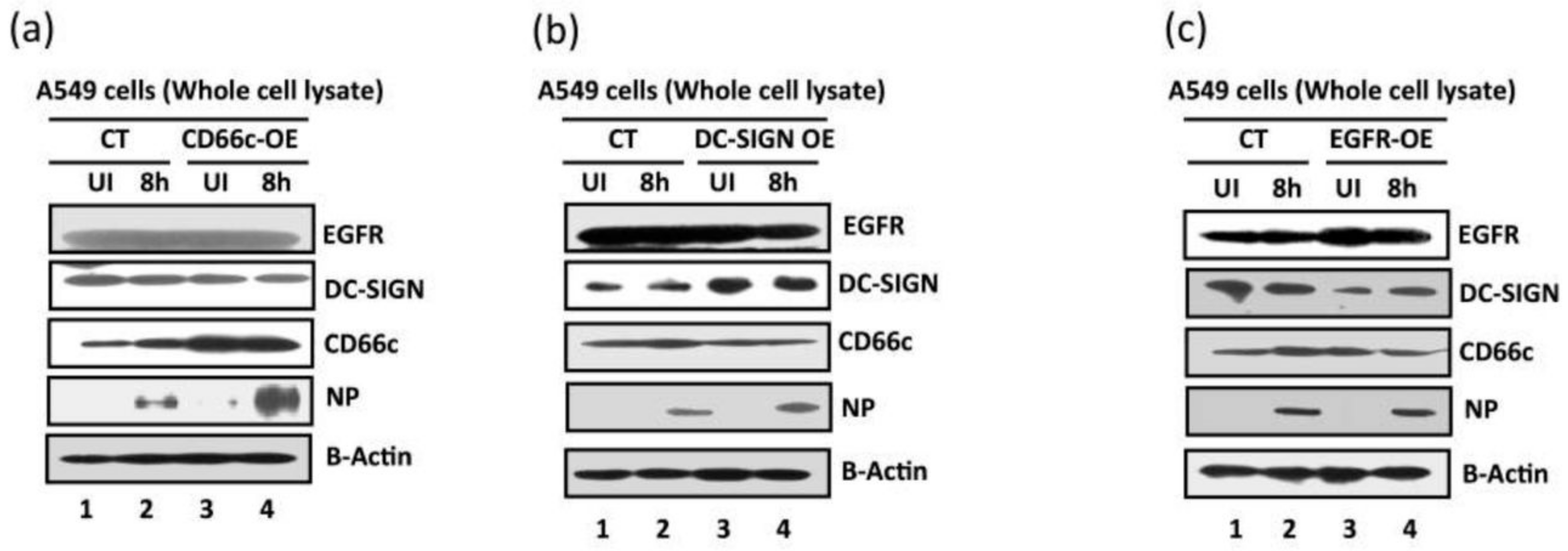
Figure 11.
Comparative measurement of viral infection in cells overexpressing CD66c, EGFR and DC-SIGN respectively. CT, control A549 cells (untransfected) with endogenous levels of protein expression; CD66c-OE is A549 cells overexpressing CD66c; DC-SIGN-OE, A549 cells overexpressing DC-SIGN; EGFR-OE, A549 cells overexpressing EGFR; UI, uninfected cell groups; 8 h, cells harvested after 8 h of A/PR8/34 influenza virus infection. (a) The A549 cells overexpressing CD66c (CD66c-OE) demonstrate significantly increased virus entry as against A549 cells with endogenous levels of CD66c (CT) when determined by the expression levels of viral protein NP (fourth panel from top). Top two panels in figure also show that overexpression of CD66c has not affected expression levels of EGFR and DC-SIGN, and their expression levels remained same as in untransfected A549 cells (top two panels). β-Actin is loading control. (b): cells overexpressing DC-SIGN (DC-SIGN-OE) showed a slight increase in virus entry into cells as against A549 cells with endogenous levels of DC-SIGN (CT), when determined by the level of viral NP protein (fourth panel from top). More importantly, overexpression of DC-SIGN has not increased expression levels of EGFR and CD66c, whose levels remained same as in untransfected A549 cells (top and third panel from top). β-Actin is loading control here. (c): The expression levels of viral NP protein (fourth panel from top) suggests that overexpression of EGFR in A549 cells (EGRF-OE) does not result in further increase in virus entry. The virus entry in these cells is same as in A549 cells with endogenous levels of EGFR (CT). Accordingly, overexpression of EGFR does not increase expression levels of DC-SIGN and CD66c (second and third panel from top). β-Actin is loading control.
Figure 11.
Comparative measurement of viral infection in cells overexpressing CD66c, EGFR and DC-SIGN respectively. CT, control A549 cells (untransfected) with endogenous levels of protein expression; CD66c-OE is A549 cells overexpressing CD66c; DC-SIGN-OE, A549 cells overexpressing DC-SIGN; EGFR-OE, A549 cells overexpressing EGFR; UI, uninfected cell groups; 8 h, cells harvested after 8 h of A/PR8/34 influenza virus infection. (a) The A549 cells overexpressing CD66c (CD66c-OE) demonstrate significantly increased virus entry as against A549 cells with endogenous levels of CD66c (CT) when determined by the expression levels of viral protein NP (fourth panel from top). Top two panels in figure also show that overexpression of CD66c has not affected expression levels of EGFR and DC-SIGN, and their expression levels remained same as in untransfected A549 cells (top two panels). β-Actin is loading control. (b): cells overexpressing DC-SIGN (DC-SIGN-OE) showed a slight increase in virus entry into cells as against A549 cells with endogenous levels of DC-SIGN (CT), when determined by the level of viral NP protein (fourth panel from top). More importantly, overexpression of DC-SIGN has not increased expression levels of EGFR and CD66c, whose levels remained same as in untransfected A549 cells (top and third panel from top). β-Actin is loading control here. (c): The expression levels of viral NP protein (fourth panel from top) suggests that overexpression of EGFR in A549 cells (EGRF-OE) does not result in further increase in virus entry. The virus entry in these cells is same as in A549 cells with endogenous levels of EGFR (CT). Accordingly, overexpression of EGFR does not increase expression levels of DC-SIGN and CD66c (second and third panel from top). β-Actin is loading control.
![Viruses 13 00726 g011]()
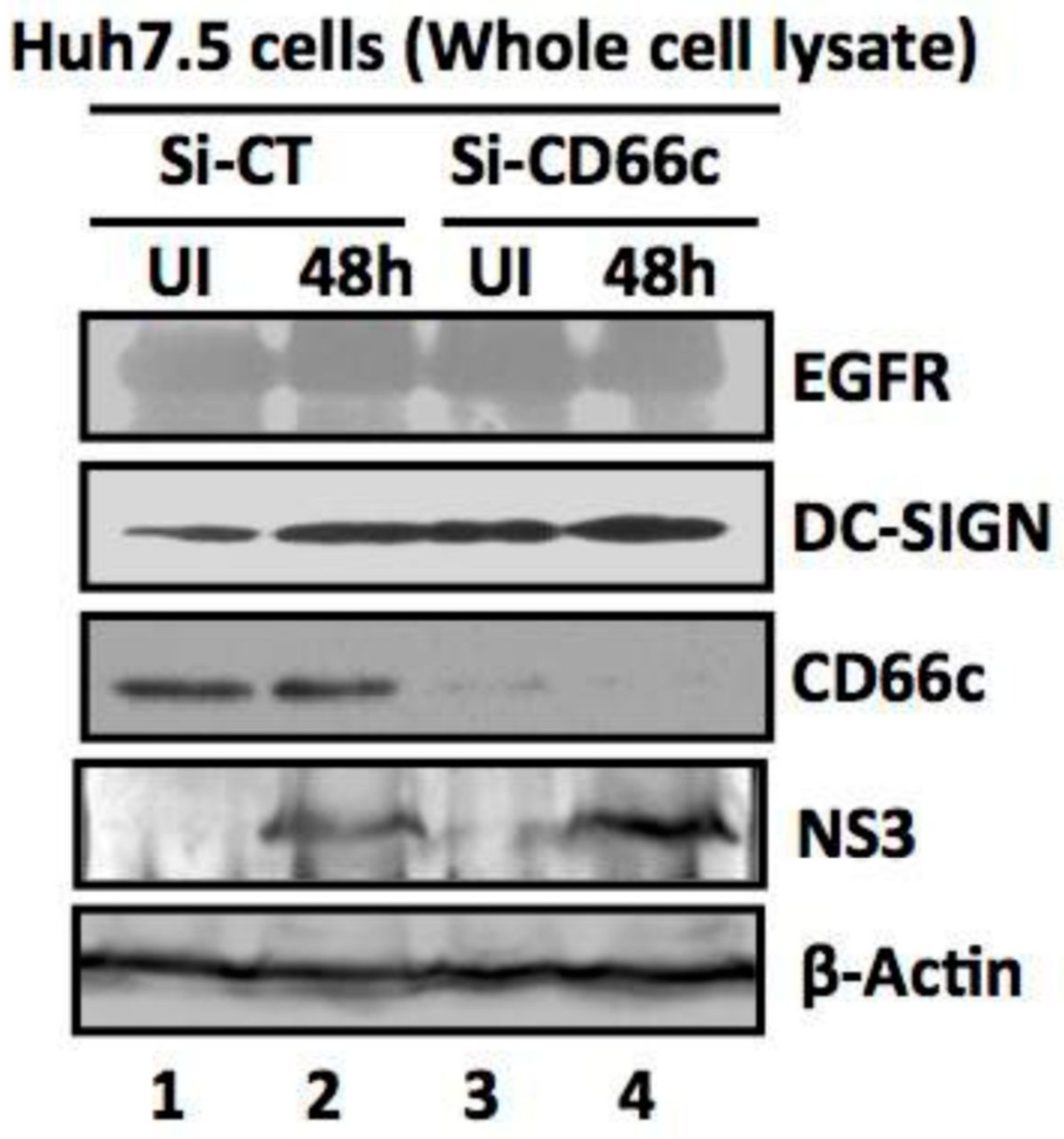
Figure 12.
siRNA knockdown of CD66c in Huh7.5 cells have not inhibited entry of another non-IAV virus (Hepatitis C virus, HCV). Here, Si-CT denotes control siRNA treated human hepatoma cells; Si-CD66c are human hepatoma cells treated with CD66c siRNA. Cells are either left uninfected (UI) or infected with 0.5 MOI of HCV for 48 h (48 h). Human hepatoma cells harvested 48 h post infection are subjected to immunoblotting. The figure shows that CD66c siRNA treated human hepatoma Huh7.5 cells show reduced expression of CD66c (third panel from top). However, this siRNA-mediated knockdown of CD66c in Huh7.5 cells does not inhibit HCV entry as determined by the expression level of HCV viral protein NS3 (fourth panel from top). Huh7.5 cells knocked down for CD66c expression do not show any inhibitory effect in the expression levels of EGFR and DC-SIGN (upper two panels). β-Actin is loading control.
Figure 12.
siRNA knockdown of CD66c in Huh7.5 cells have not inhibited entry of another non-IAV virus (Hepatitis C virus, HCV). Here, Si-CT denotes control siRNA treated human hepatoma cells; Si-CD66c are human hepatoma cells treated with CD66c siRNA. Cells are either left uninfected (UI) or infected with 0.5 MOI of HCV for 48 h (48 h). Human hepatoma cells harvested 48 h post infection are subjected to immunoblotting. The figure shows that CD66c siRNA treated human hepatoma Huh7.5 cells show reduced expression of CD66c (third panel from top). However, this siRNA-mediated knockdown of CD66c in Huh7.5 cells does not inhibit HCV entry as determined by the expression level of HCV viral protein NS3 (fourth panel from top). Huh7.5 cells knocked down for CD66c expression do not show any inhibitory effect in the expression levels of EGFR and DC-SIGN (upper two panels). β-Actin is loading control.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











