
New Insights on the Zika Virus Arrival in the Americas and Spatiotemporal Reconstruction of the Epidemic Dynamics in Brazil

 , , , , , , ,
, , , , , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Phylogenetic and Phylogeographic Analyses
3. Results
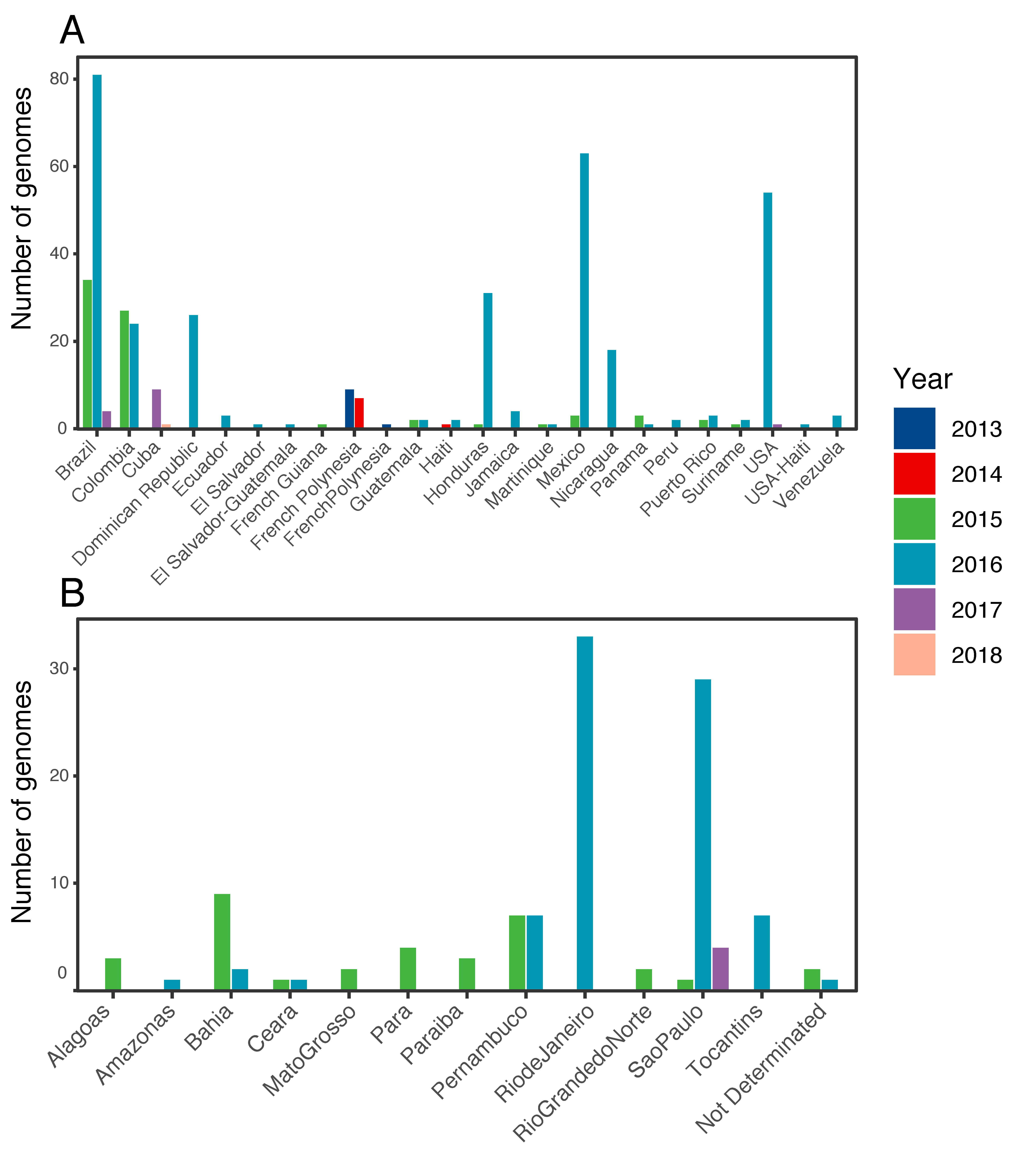
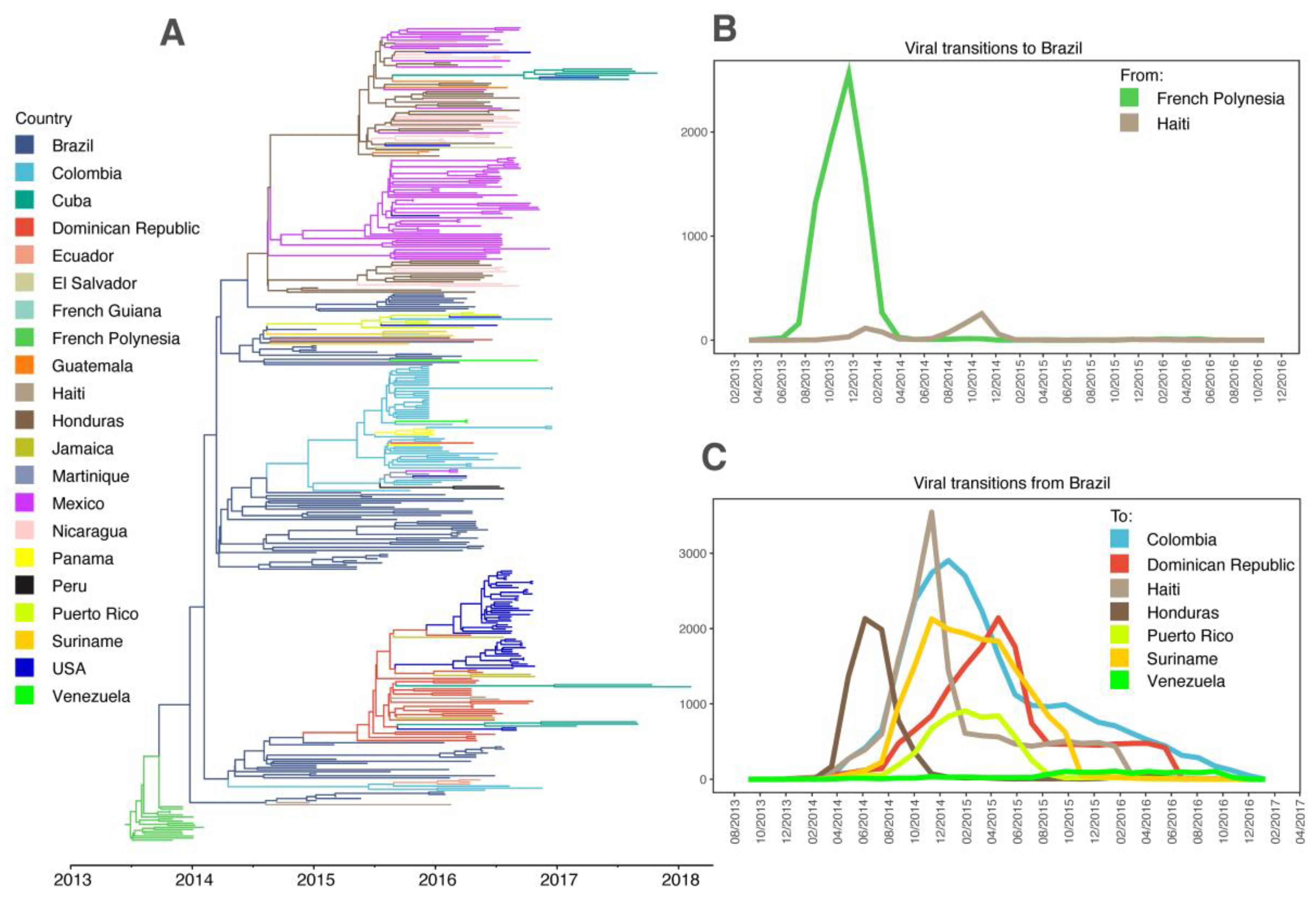
3.1. ZIKV in the Americas
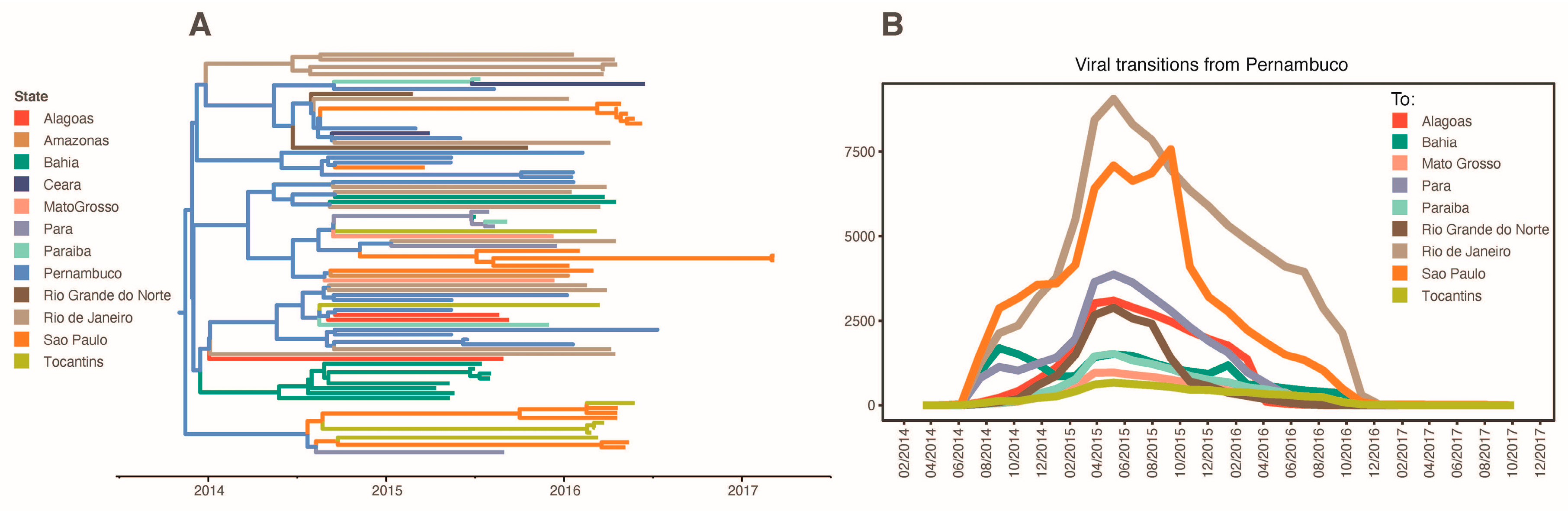
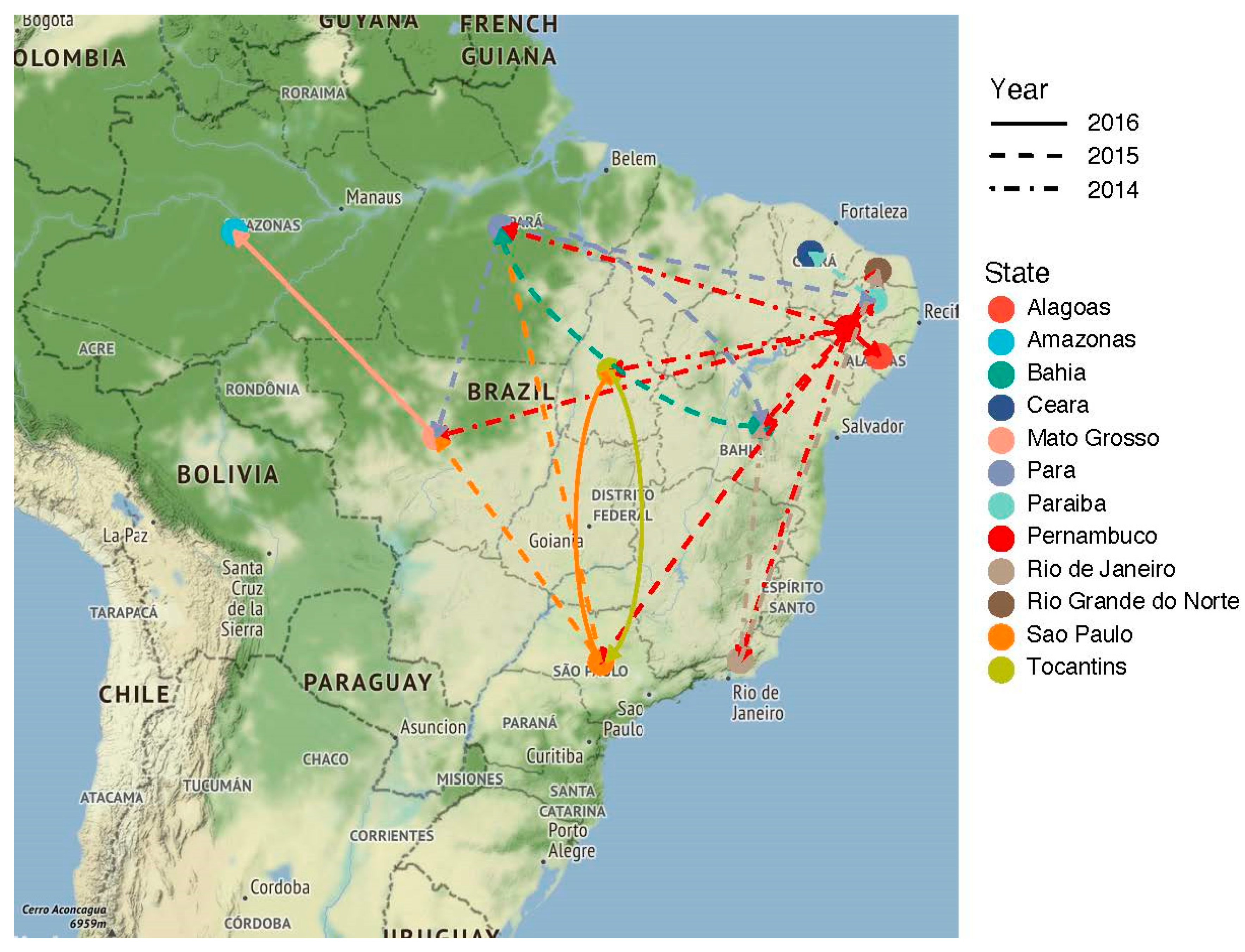
3.2. The ZIKV Dissemination across the Brazilian States
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Macnamara, F.N. Zika virus: A report on three cases of human infection during an epidemic of jaundice in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 1954, 48, 139–145. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C. Zika virus outbreak on Yap Island, federated states of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Oehler, E.; Watrin, L.; Larre, P.; Leparc-Goffart, I.; Lastere, S.; Valour, F.; Baudouin, L.; Mallet, H.P.; Musso, D.; Ghawche, F. Zika virus infection complicated by Guillain-Barre syndrome–case report, French Polynesia, December 2013. Eurosurveillance 2014, 19, 20720. [Google Scholar] [CrossRef] [Green Version]
- Pyke, A.T.; Daly, M.T.; Cameron, J.N.; Moore, P.R.; Taylor, C.T.; Hewitson, G.R.; Humphreys, J.L.; Gair, R. Imported Zika virus infection from the Cook Islands into Australia, 2014. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef]
- Tognarelli, J.; Ulloa, S.; Villagra, E.; Lagos, J.; Aguayo, C.; Fasce, R.; Parra, B.; Mora, J.; Becerra, N.; Lagos, N. A report on the outbreak of Zika virus on Easter Island, South Pacific, 2014. Arch. Virol. 2016, 161, 665–668. [Google Scholar] [CrossRef] [Green Version]
- Dupont-Rouzeyrol, M.; O’Connor, O.; Calvez, E.; Daures, M.; John, M.; Grangeon, J.-P.; Gourinat, A.-C. Co-infection with Zika and dengue viruses in 2 patients, New Caledonia, 2014. Emerg. Infect. Dis. 2015, 21, 381. [Google Scholar] [CrossRef] [Green Version]
- Wæhre, T.; Maagard, A.; Tappe, D.; Cadar, D.; Schmidt-Chanasit, J. Zika virus infection after travel to Tahiti, December 2013. Emerg. Infect. Dis. 2014, 20, 1412. [Google Scholar] [CrossRef]
- Faria, N.R.; da Silva Azevedo, R.D.S.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, J.H.-O.; Eldholm, V.; Seligman, S.J.; Lundkvist, Å.; Falconar, A.K.; Gaunt, M.W.; Musso, D.; Nougairède, A.; Charrel, R.; Gould, E.A. How did Zika virus emerge in the Pacific Islands and Latin America? MBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Sun, K.; Chinazzi, M.; Piontti, A.P.; Dean, N.E.; Rojas, D.P.; Merler, S.; Mistry, D.; Poletti, P.; Rossi, L. Spread of Zika virus in the Americas. Proc. Natl. Acad. Sci. USA 2017, 114, E4334–E4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Quick, J.; Claro, I.M.; Theze, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.G.; Hill, S.C.; Black, A.; da Costa, A.C. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Massad, E.; Burattini, M.N.; Khan, K.; Struchiner, C.J.; Coutinho, F.A.B.; Wilder-Smith, A. On the origin and timing of Zika virus introduction in Brazil. Epidemiol. Infect. 2017, 145, 2303–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, G.S.; Bandeira, A.C.; Sardi, S.I. Zika virus outbreak, bahia, brazil. Emerg. Infect. Dis. 2015, 21, 1885. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; de Melo, V.C.A.; Mosimann, A.L.P.; dos Santos, G.I.V.; dos Santos, C.N.D.; Luz, K. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Qu, J.; Baniecki, M.L.; Gladden-Young, A. Zika virus evolution and spread in the Americas. Nature 2017, 546, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Thézé, J.; Li, T.; Du Plessis, L.; Bouquet, J.; Kraemer, M.U.G.; Somasekar, S.; Yu, G.; de Cesare, M.; Balmaseda, A.; Kuan, G. Genomic epidemiology reconstructs the introduction and spread of Zika virus in Central America and Mexico. Cell Host Microbe 2018, 23, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Ebranati, E.; Veo, C.; Carta, V.; Percivalle, E.; Rovida, F.; Frati, E.R.; Amendola, A.; Ciccozzi, M.; Tanzi, E.; Galli, M. Time-scaled phylogeography of complete Zika virus genomes using discrete and continuous space diffusion models. Infect. Genet. Evol. 2019, 73, 33–43. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Kraemer, M.U.G.; Dudas, G.; Tan, A.L.; Gangavarapu, K.; Wiley, M.R.; White, S.; Thézé, J.; Magnani, D.M. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 2017, 546, 401–405. [Google Scholar] [CrossRef]
- World Health Organization. WHO Statement on the First Meeting of the International Health Regulations (2005) (IHR 2005) Emergency Committee on Zika Virus and Observed Increase in Neurological Disorders and Neonatal Malformations; 2016. Available online: http://www.who.int/mediacentre/news/statements/2016/1st-emergency-committee-zika/en (accessed on 21 December 2020).
- Teixeira, M.G.; da Conceição, N.; Costa, M.; de Oliveira, W.K.; Nunes, M.L.; Rodrigues, L.C. The epidemic of Zika virus–related microcephaly in Brazil: Detection, control, etiology, and future scenarios. Am. J. Public Health 2016, 106, 601–605. [Google Scholar] [CrossRef]
- Laguardia, J.; Domingues, C.M.A.; Carvalho, C.; Lauerman, C.R.; Macário, E.; Glatt, R. Sistema de informação de agravos de notificação em saúde (Sinan): Desafios no desenvolvimento de um sistema de informação em saúde. Epidemiol. Serviços Saúde 2004, 13, 135–146. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, B.; Rambaut, A.; Drummond, A.J. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 2006, 23, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian population dynamics inference: A coalescent-based model for multiple loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 gateway computing environments workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Oliveira, J.F.; Rodrigues, M.S.; Skalinski, L.M.; Santos, A.E.S.; Costa, L.C.; Cardim, L.L.; Paixão, E.S.; Costa, M.d.C.N.; Oliveira, W.K.; Barreto, M.L. Interdependence between confirmed and discarded cases of dengue, chikungunya and Zika viruses in Brazil: A multivariate time-series analysis. PLoS ONE 2020, 15, e0228347. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, W.K.; de França, G.V.A.; Carmo, E.H.; Duncan, B.B.; de Souza Kuchenbecker, R.; Schmidt, M.I. Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: A surveillance-based analysis. Lancet 2017, 390, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Campos, T.D.L.; Durães-Carvalho, R.; Rezende, A.M.; de Carvalho, O.V.; Kohl, A.; Wallau, G.L.; Pena, L.J. Revisiting key entry routes of human epidemic Arboviruses into the mainland Americas through large-scale Phylogenomics. Int. J. Genom. 2018, 2018, 6941735. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lormeau, V.-M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.-L.; Mallet, H.-P.; Sall, A.A.; Musso, D. Zika virus, French polynesia, South pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085. [Google Scholar] [CrossRef]
- Giovanetti, M.; Faria, N.R.; Lourenço, J.; de Jesus, J.G.; Xavier, J.; Claro, I.M.; Kraemer, M.U.G.; Fonseca, V.; Dellicour, S.; Thézé, J. Genomic and epidemiological surveillance of Zika virus in the Amazon region. Cell Rep. 2020, 30, 2275–2283. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Barcellos, C.; Brasil, P.; Cruz, O.G.; Honório, N.A.; Kuper, H.; Carvalho, M.S. The Zika virus epidemic in Brazil: From discovery to future implications. Int. J. Environ. Res. Public Health 2018, 15, 96. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, L.C.; Veiga, R.V.; Oliveira, J.F.; Rodrigues, M.S.; Andrade, R.F.S.; Paixão, E.S.; Teixeira, M.G.; Costa, M.d.C.N.; Cardim, L.L.; Carmo, E.H.; et al. New Insights on the Zika Virus Arrival in the Americas and Spatiotemporal Reconstruction of the Epidemic Dynamics in Brazil. Viruses 2021, 13, 12. https://doi.org/10.3390/v13010012
Costa LC, Veiga RV, Oliveira JF, Rodrigues MS, Andrade RFS, Paixão ES, Teixeira MG, Costa MdCN, Cardim LL, Carmo EH, et al. New Insights on the Zika Virus Arrival in the Americas and Spatiotemporal Reconstruction of the Epidemic Dynamics in Brazil. Viruses. 2021; 13(1):12. https://doi.org/10.3390/v13010012
Chicago/Turabian StyleCosta, Larissa Catharina, Rafael Valente Veiga, Juliane Fonseca Oliveira, Moreno S. Rodrigues, Roberto F. S. Andrade, Enny S. Paixão, Maria Glória Teixeira, Maria da Conceição N. Costa, Luciana L. Cardim, Eduardo H. Carmo, and et al. 2021. "New Insights on the Zika Virus Arrival in the Americas and Spatiotemporal Reconstruction of the Epidemic Dynamics in Brazil" Viruses 13, no. 1: 12. https://doi.org/10.3390/v13010012