Equine Herpesvirus Type 1 Modulates Cytokine and Chemokine Profiles of Mononuclear Cells for Efficient Dissemination to Target Organs

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
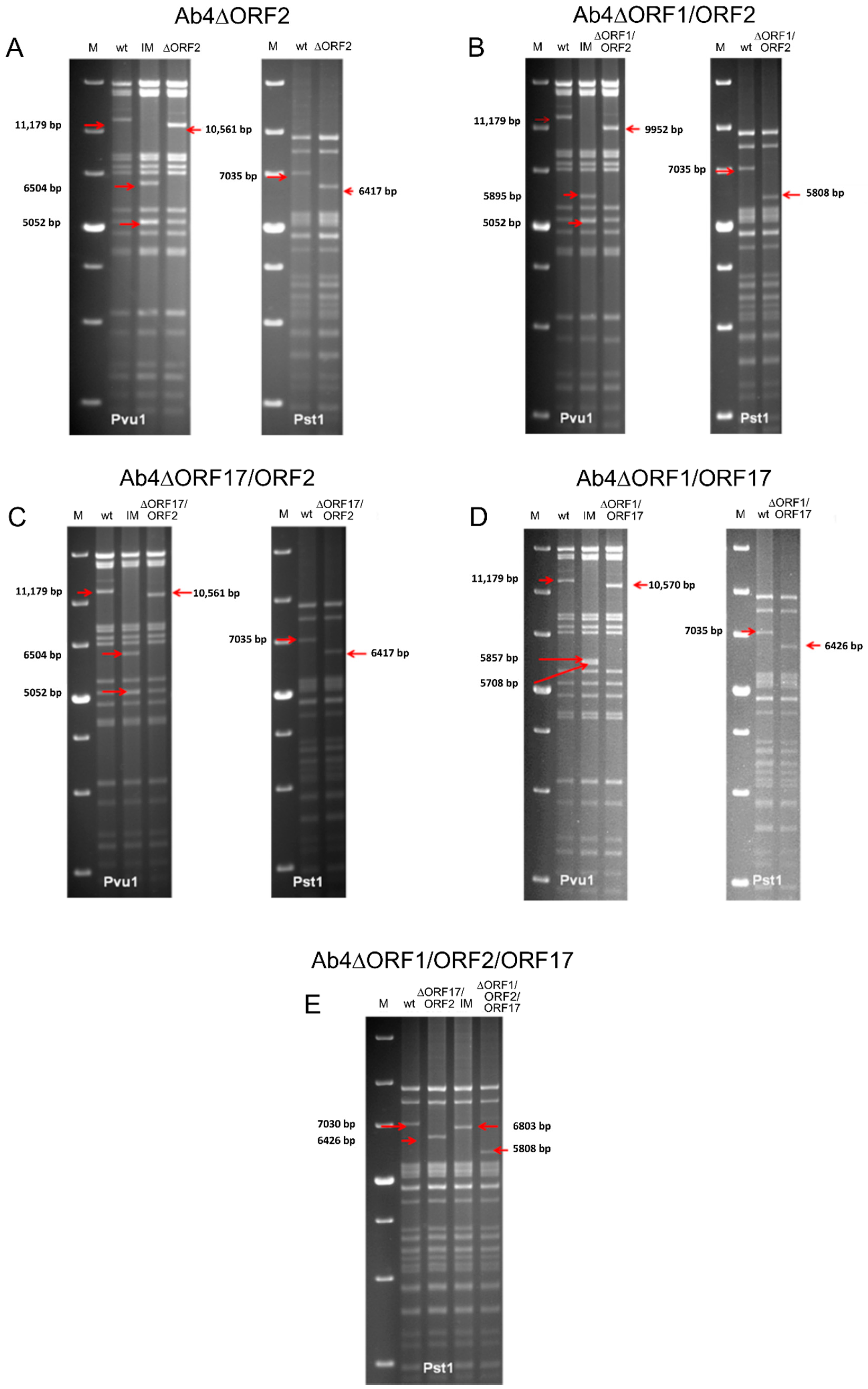
2.2. Engineering of EHV-1 BAC Mutants and Revertants
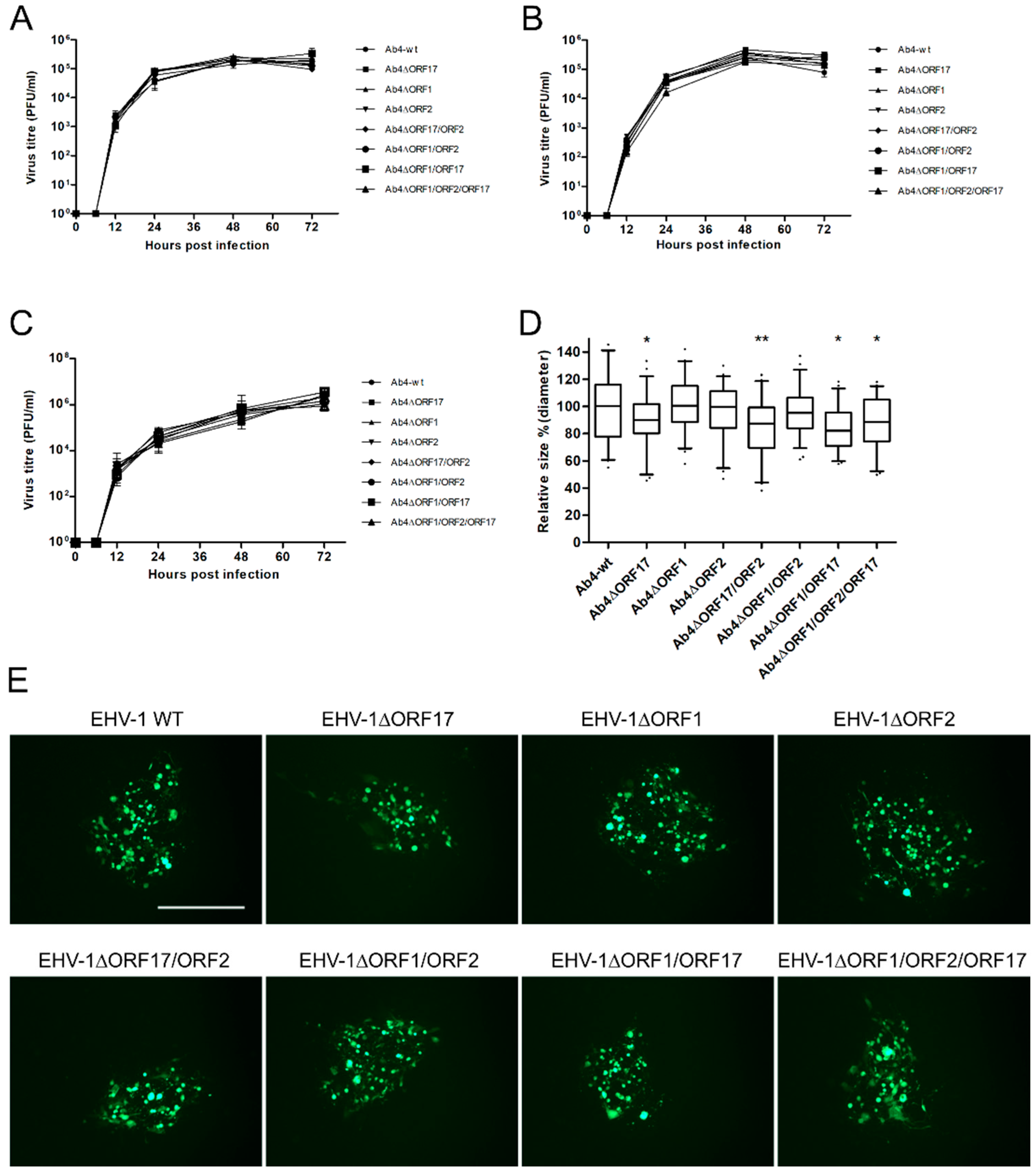
2.3. Growth Kinetics and Plaque Size Assay
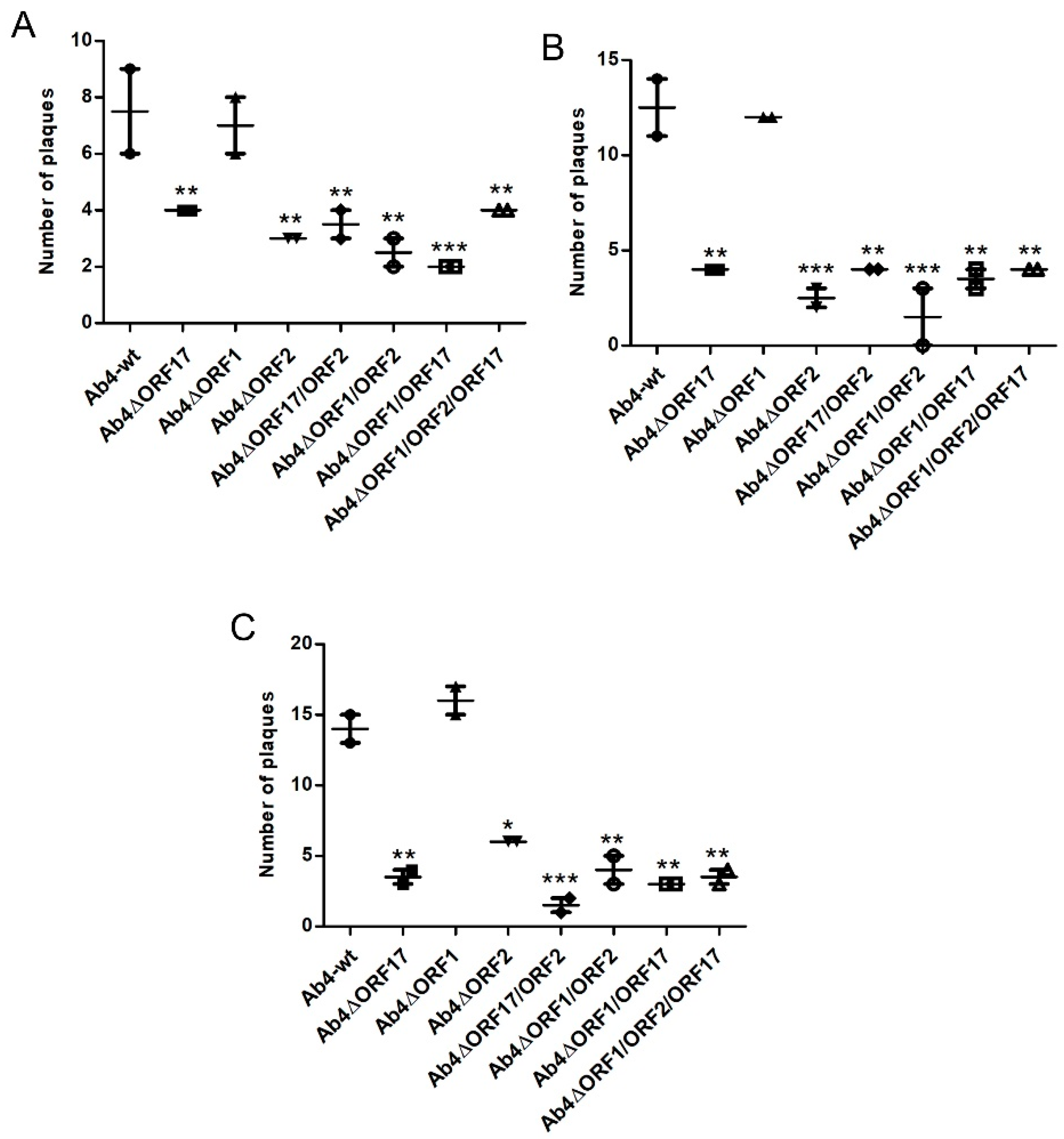
2.4. Co-Cultivation Assay
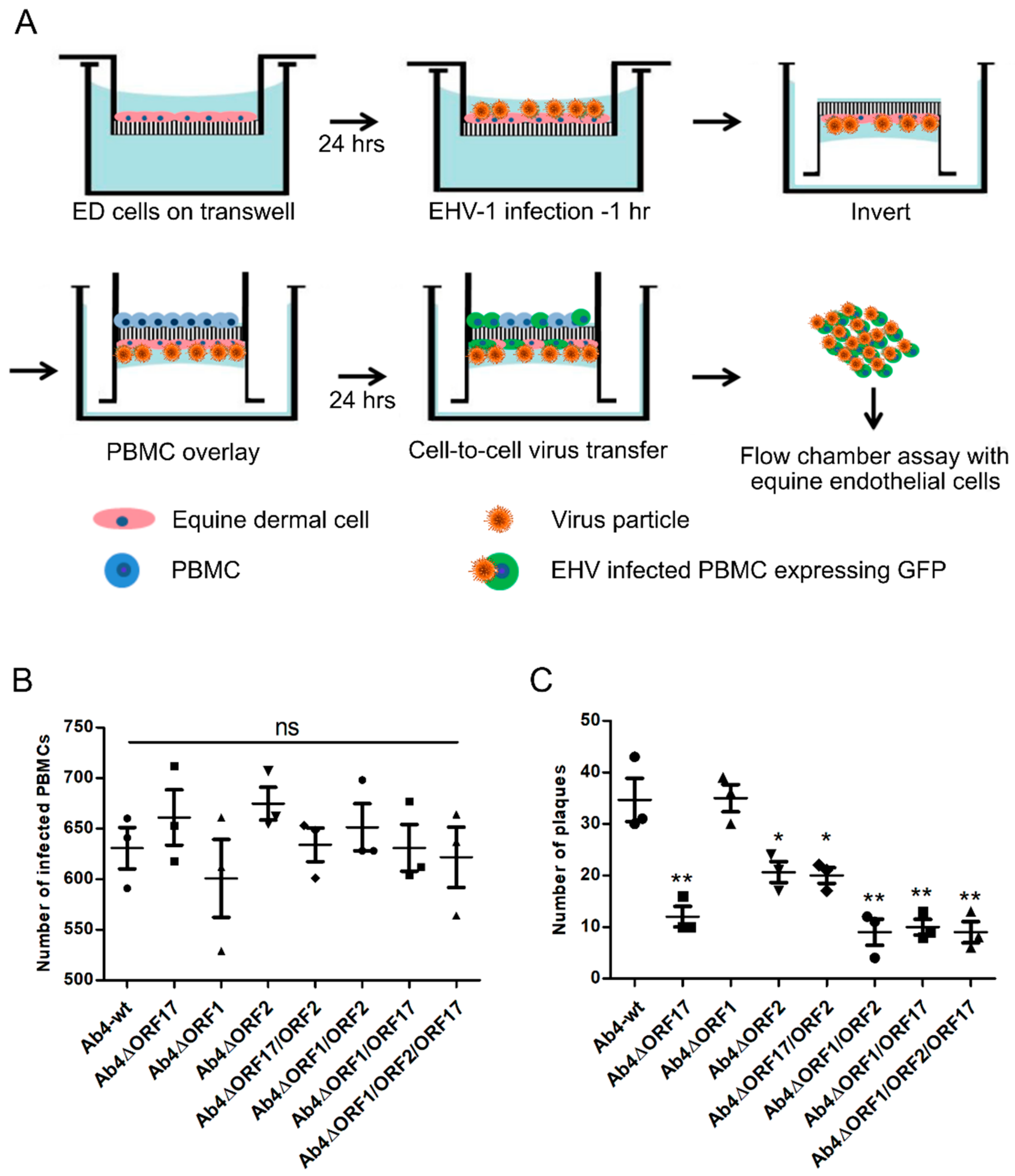
2.5. Flow Chamber Assay
2.6. Infection of Equine PBMC Subpopulations
2.7. Equine Epithelial Cell-PBMC Contact Assay
2.8. Whole-Cell Proteomic Analysis
2.8.1. Sample Preparation
2.8.2. Data Analysis and Interpretation
2.9. Multiplex Equine Cytokine Assay
2.10. Statistical Analysis
3. Results
3.1. The ORF1, ORF2 and ORF17 Genes are Dispensable of EHV-1 Replication
3.2. ORF2 and ORF17 Genes Are Important for Transfer to EC
3.3. Virus Transfer to EC through Different PBMC Subpopulations
3.4. Mimicking the In Vivo Pathway of Virus Spread from Epithelial Cells to PBMC and from PBMC to EC
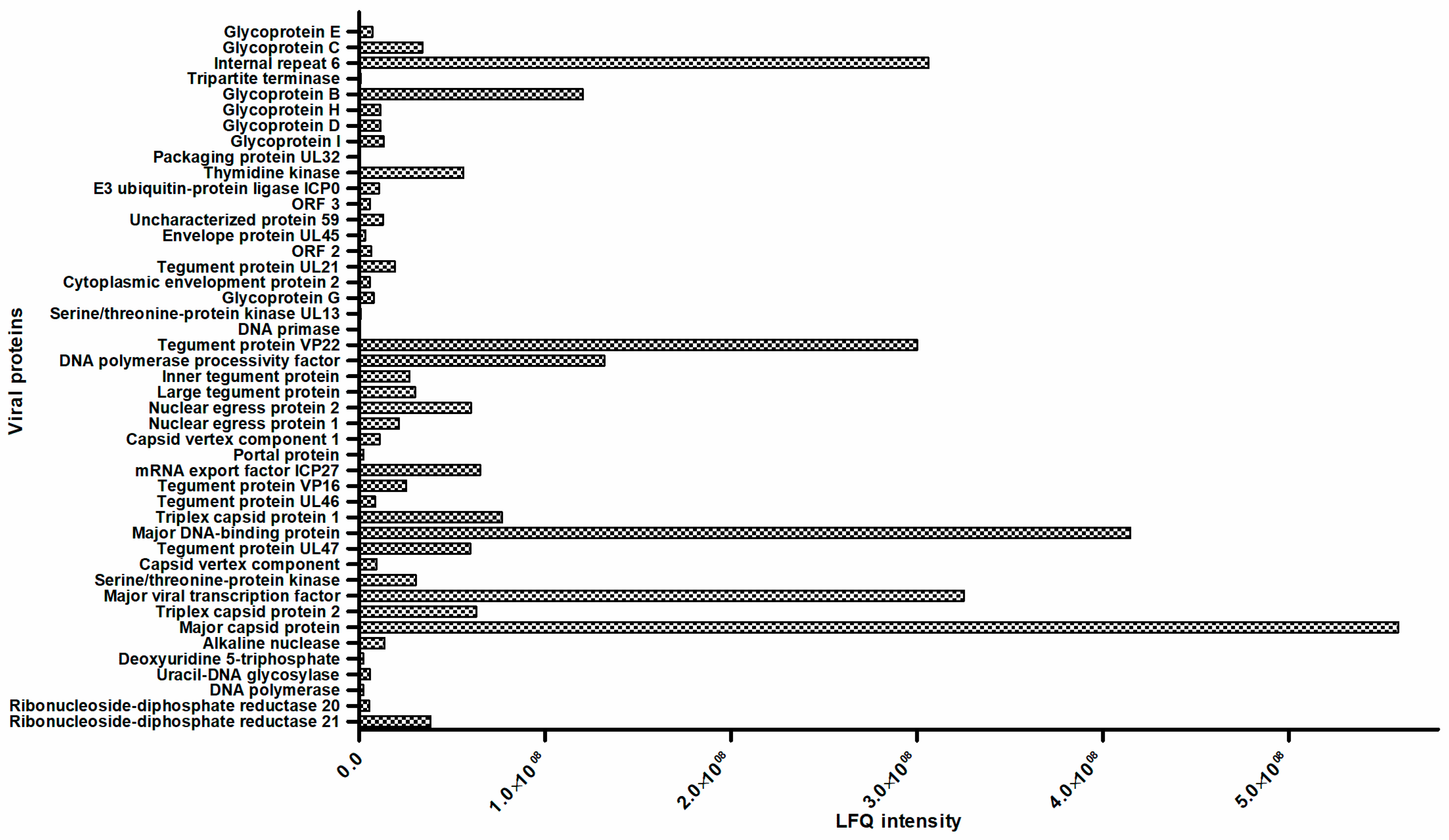
3.5. Comparative Proteomic Analysis
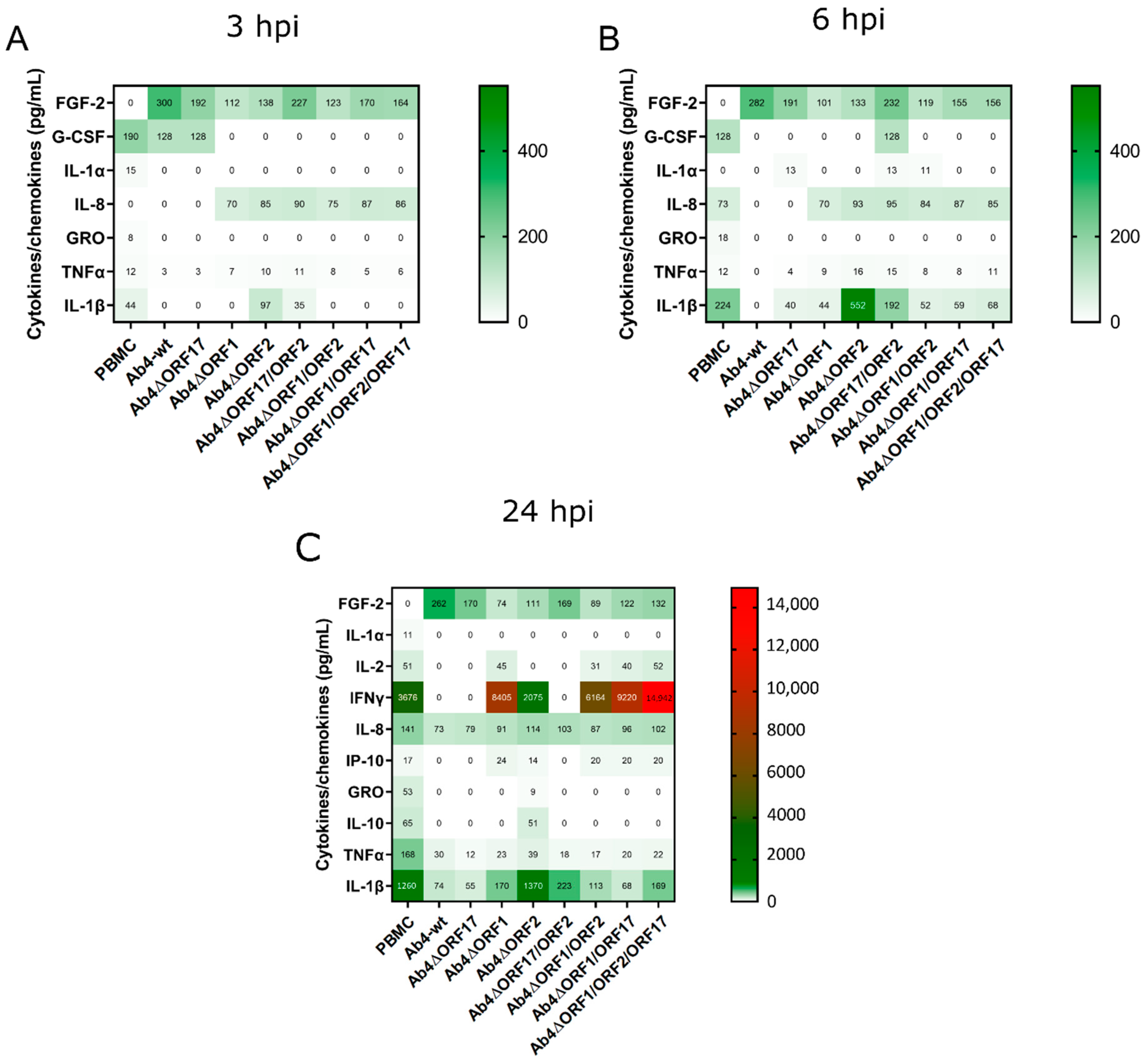
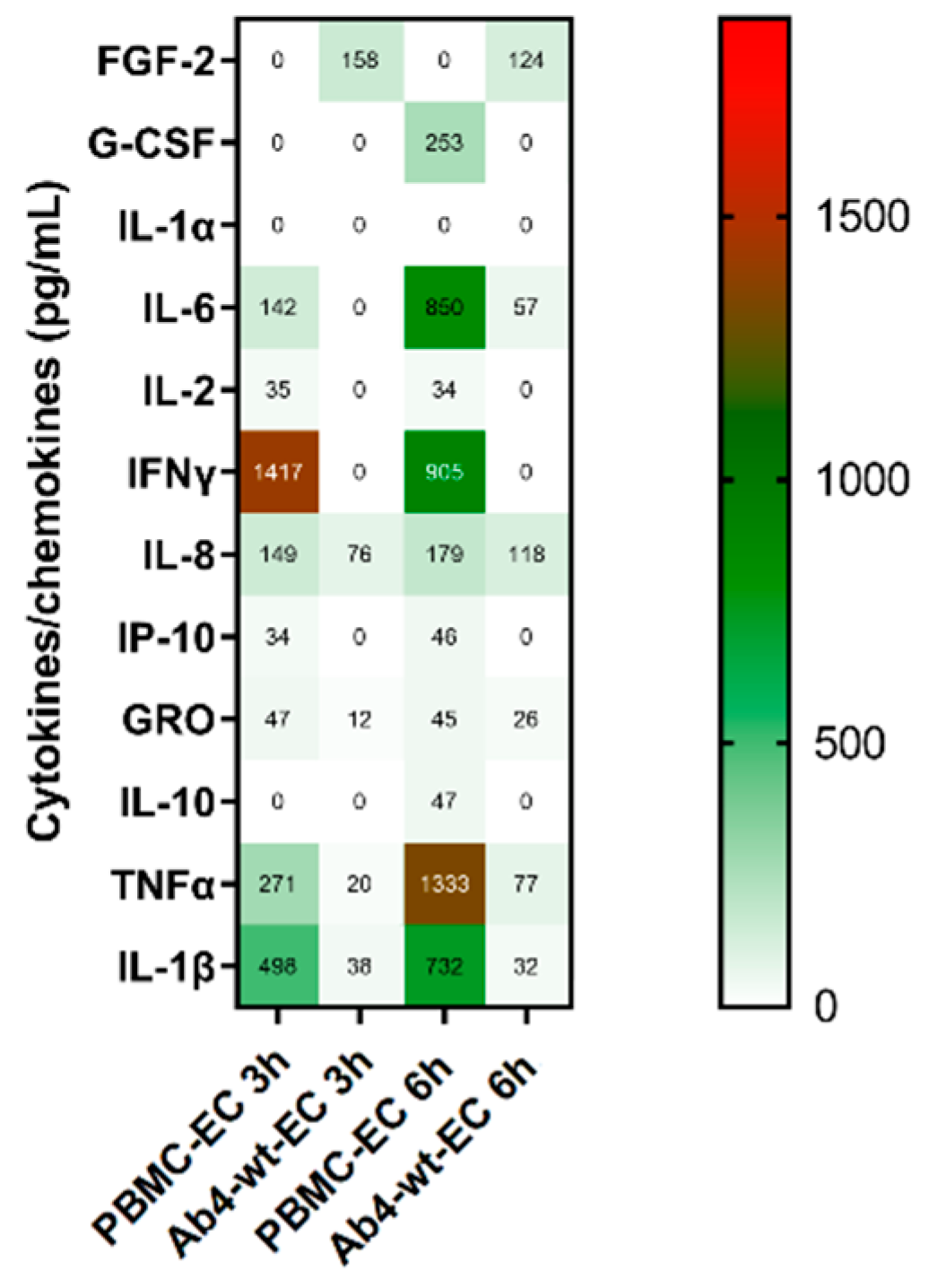
3.6. EHV-1 Infection Modulates Cytokine and Chemokine Profiles of PBMC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wozniakowski, G.; Samorek-Salamonowicz, E. Animal herpesviruses and their zoonotic potential for cross-species infection. Ann. Agric. Environ. Med. 2015, 22, 191–194. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, S.; Kumar, D.; Rouse, B.T. Herpesviruses: Harmonious Pathogens but Relevant Cofactors in Other Diseases? Front. Cell. Infect. Microbiol. 2018, 8, 177. [Google Scholar] [CrossRef] [PubMed]
- Louten, J. Herpesviruses. In Essential Human Virology, 1st ed.; Louten, J., Ed.; Elsevier BV: Amsterdam, The Netherlands, 2016; pp. 235–256. [Google Scholar] [CrossRef]
- Azab, W.; Osterrieder, K. Initial Contact: The First Steps in Herpesvirus Entry. In Cell Biology of Herpes Viruses; Osterrieder, K., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–27. [Google Scholar] [CrossRef]
- Davison, A.J. Evolution of the herpesviruses. Vet. Microbiol. 2002, 86, 69–88. [Google Scholar] [CrossRef]
- Mogensen, T.H.; Paludan, S.R. Virus-cell interactions: Impact on cytokine production, immune evasion and tumor growth. Eur. Cytokine Netw. 2001, 12, 382–390. [Google Scholar] [PubMed]
- Bego, M.G.; St Jeor, S. Human cytomegalovirus infection of cells of hematopoietic origin: HCMV-induced immunosuppression, immune evasion, and latency. Exp. Hematol. 2006, 34, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Berges, B.K.; Tanner, A. Modelling of human herpesvirus infections in humanized mice. J. Gen. Virol. 2014, 95, 2106–2117. [Google Scholar] [CrossRef]
- Huang, T.; Ma, G.; Osterrieder, N. Equine Herpesvirus 1 Multiply Inserted Transmembrane Protein pUL43 Cooperates with pUL56 in Downregulation of Cell Surface Major Histocompatibility Complex Class, I. J. Virol. 2015, 89, 6251–6263. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Khanna, R. Immune regulation of human herpesviruses and its implications for human transplantation. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2013, 13 (Suppl. 3), 9–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Rao, Y.; Tian, M.; Zhang, S.; Feng, P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses 2019, 11, 572. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H.; Paludan, S.R. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 2001, 65, 131–150. [Google Scholar] [CrossRef] [Green Version]
- Roizman, B. The Family Herpesviridae: General Description, Taxonomy, and Classification. In The Herpesviruses; Roizman, B., Ed.; Springer: Boston, MA, USA, 1982; pp. 1–23. [Google Scholar] [CrossRef]
- Davison, A.J. Herpesvirus systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, S.M.; Toribio, R.E. Equine herpesvirus 1 and 4. Vet. Clin. N. Am Equine Pract. 2004, 20, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Baghi, H.B.; Nauwynck, H.J. Impact of equine herpesvirus type 1 (EHV-1) infection on the migration of monocytic cells through equine nasal mucosa. Comp. Immunol. Microbiol. Infect. Dis. 2014, 37, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Kydd, J.H.; Smith, K.C.; Hannant, D.; Livesay, G.J.; Mumford, J.A. Distribution of equid herpesvirus-1 (EHV-1) in respiratory tract associated lymphoid tissue: Implications for cellular immunity. Equine Vet. J. 1994, 26, 470–473. [Google Scholar] [CrossRef]
- Scott, J.C.; Dutta, S.K.; Myrup, A.C. In vivo harboring of equine herpesvirus-1 in leukocyte populations and subpopulations and their quantitation from experimentally infected ponies. Am. J. Vet. Res. 1983, 44, 1344–1348. [Google Scholar]
- Hussey, S.B.; Clark, R.; Lunn, K.F.; Breathnach, C.; Soboll, G.; Whalley, J.M.; Lunn, D.P. Detection and quantification of equine herpesvirus-1 viremia and nasal shedding by real-time polymerase chain reaction. J. Vet. Diagn. Investig. 2006, 18, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Wilsterman, S.; Soboll-Hussey, G.; Lunn, D.P.; Ashton, L.V.; Callan, R.J.; Hussey, S.B.; Rao, S.; Goehring, L.S. Equine herpesvirus-1 infected peripheral blood mononuclear cell subpopulations during viremia. Vet. Microbiol. 2011, 149, 40–47. [Google Scholar] [CrossRef]
- Goehring, L.S.; Hussey, G.S.; Ashton, L.V.; Schenkel, A.R.; Lunn, D.P. Infection of central nervous system endothelial cells by cell-associated EHV-1. Vet. Microbiol. 2011, 148, 389–395. [Google Scholar] [CrossRef]
- van der Meulen, K.M.; Nauwynck, H.J.; Pensaert, M.B. Increased susceptibility of peripheral blood mononuclear cells to equine herpes virus type 1 infection upon mitogen stimulation: A role of the cell cycle and of cell-to-cell transmission of the virus. Vet. Microbiol. 2002, 86, 157–163. [Google Scholar] [CrossRef]
- Nauwynck, H.J.; Pensaert, M.B. Virus production and viral antigen expression in porcine blood monocytes inoculated with pseudorabies virus. Arch. Virol. 1994, 137, 69–79. [Google Scholar] [CrossRef]
- Mainka, C.; Fuss, B.; Geiger, H.; Hofelmayr, H.; Wolff, M.H. Characterization of viremia at different stages of varicella-zoster virus infection. J. Med. Virol. 1998, 56, 91–98. [Google Scholar] [CrossRef]
- Vandekerckhove, A.P.; Glorieux, S.; Gryspeerdt, A.C.; Steukers, L.; Van Doorsselaere, J.; Osterrieder, N.; Van de Walle, G.R.; Nauwynck, H.J. Equine alphaherpesviruses (EHV-1 and EHV-4) differ in their efficiency to infect mononuclear cells during early steps of infection in nasal mucosal explants. Vet. Microbiol. 2011, 152, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, T.; Sugiura, T.; Imagawa, H.; Fukunaga, Y.; Kamada, M. Epizootiological aspects of type 1 and type 4 equine herpesvirus infections among horse populations. J. Vet. Med. Sci. 1992, 54, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.R.; Foldi, J.; Bateman, H.; Williams, J.; Didlick, S.; Stark, R. Equid herpesvirus (EHV-1) live vaccine strain C147: Efficacy against respiratory diseases following EHV types 1 and 4 challenges. Vet. Microbiol. 2003, 92, 1–17. [Google Scholar] [CrossRef]
- Laval, K.; Favoreel, H.W.; Poelaert, K.C.; Van Cleemput, J.; Nauwynck, H.J. Equine Herpesvirus Type 1 Enhances Viral Replication in CD172a+ Monocytic Cells upon Adhesion to Endothelial Cells. J. Virol. 2015, 89, 10912–10923. [Google Scholar] [CrossRef] [Green Version]
- Poelaert, K.C.K.; Van Cleemput, J.; Laval, K.; Favoreel, H.W.; Couck, L.; Van den Broeck, W.; Azab, W.; Nauwynck, H.J. Equine Herpesvirus 1 Bridles T Lymphocytes to Reach Its Target Organs. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, N.; Landis, R.C. Adhesion molecules in cell interactions. Curr. Opin. Immunol. 1993, 5, 383–390. [Google Scholar] [CrossRef]
- Smith, D.; Hamblin, A.; Edington, N. Equid herpesvirus 1 infection of endothelial cells requires activation of putative adhesion molecules: An in vitro model. Clin. Exp. Immunol. 2002, 129, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Spiesschaert, B.; Goldenbogen, B.; Taferner, S.; Schade, M.; Mahmoud, M.; Klipp, E.; Osterrieder, N.; Azab, W. Role of gB and pUS3 in Equine Herpesvirus 1 Transfer between Peripheral Blood Mononuclear Cells and Endothelial Cells: A Dynamic In Vitro Model. J. Virol. 2015, 89, 11899–11908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiesschaert, B.; Osterrieder, N.; Azab, W. Comparative analysis of glycoprotein B (gB) of equine herpesvirus type 1 and type 4 (EHV-1 and EHV-4) in cellular tropism and cell-to-cell transmission. Viruses 2015, 7, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, A.; Braun, B.; Brandmuller, C.; Kaaden, O.R.; Osterrieder, N. Analysis of the contributions of the equine herpesvirus 1 glycoprotein gB homolog to virus entry and direct cell-to-cell spread. Virology 1997, 227, 281–294. [Google Scholar] [CrossRef]
- Wellington, J.E.; Love, D.N.; Whalley, J.M. Evidence for involvement of equine herpesvirus 1 glycoprotein B in cell-cell fusion. Arch. Virol. 1996, 141, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Colle, C.F., 3rd; Flowers, C.C.; O’Callaghan, D.J. Open reading frames encoding a protein kinase, homolog of glycoprotein gX of pseudorabies virus, and a novel glycoprotein map within the unique short segment of equine herpesvirus type 1. Virology 1992, 188, 545–557. [Google Scholar] [CrossRef]
- Proft, A.; Spiesschaert, B.; Izume, S.; Taferner, S.; Lehmann, M.J.; Azab, W. The Role of the Equine Herpesvirus Type 1 (EHV-1) US3-Encoded Protein Kinase in Actin Reorganization and Nuclear Egress. Viruses 2016, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Soboll Hussey, G.; Ashton, L.V.; Quintana, A.M.; Van de Walle, G.R.; Osterrieder, N.; Lunn, D.P. Equine herpesvirus type 1 pUL56 modulates innate responses of airway epithelial cells. Virology 2014, 464–465, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, A.; Azab, W.; Damiani, A.; Osterrieder, N. Equine herpesvirus type 4 UL56 and UL49.5 proteins downregulate cell surface major histocompatibility complex class I expression independently of each other. J. Virol. 2012, 86, 8059–8071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, G.; Feineis, S.; Osterrieder, N.; Van de Walle, G.R. Identification and characterization of equine herpesvirus type 1 pUL56 and its role in virus-induced downregulation of major histocompatibility complex class I. J. Virol. 2012, 86, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Lehmann, M.J.; Said, A.; Ma, G.; Osterrieder, N. Major histocompatibility complex class I downregulation induced by equine herpesvirus type 1 pUL56 is through dynamin-dependent endocytosis. J. Virol. 2014, 88, 12802–12815. [Google Scholar] [CrossRef] [Green Version]
- Fruh, K.; Gruhler, A.; Krishna, R.M.; Schoenhals, G.J. A comparison of viral immune escape strategies targeting the MHC class I assembly pathway. Immunol. Rev. 1999, 168, 157–166. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Hill, A.B. Viral interference with antigen presentation. Nat. Immunol. 2002, 3, 1019–1025. [Google Scholar] [CrossRef]
- Ziegler, H.; Thale, R.; Lucin, P.; Muranyi, W.; Flohr, T.; Hengel, H.; Farrell, H.; Rawlinson, W.; Koszinowski, U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 1997, 6, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Wiertz, E.J.; Jones, T.R.; Sun, L.; Bogyo, M.; Geuze, H.J.; Ploegh, H.L. The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 1996, 84, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.R.; Wiertz, E.J.; Sun, L.; Fish, K.N.; Nelson, J.A.; Ploegh, H.L. Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. USA 1996, 93, 11327–11333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telford, E.A.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef]
- Wimer, C.L.; Schnabel, C.L.; Perkins, G.; Babasyan, S.; Freer, H.; Stout, A.E.; Rollins, A.; Osterrieder, N.; Goodman, L.B.; Glaser, A.; et al. The deletion of the ORF1 and ORF71 genes reduces virulence of the neuropathogenic EHV-1 strain Ab4 without compromising host immunity in horses. PLoS ONE 2018, 13, e0206679. [Google Scholar] [CrossRef]
- Schnabel, C.L.; Wimer, C.L.; Perkins, G.; Babasyan, S.; Freer, H.; Watts, C.; Rollins, A.; Osterrieder, N.; Wagner, B. Deletion of the ORF2 gene of the neuropathogenic equine herpesvirus type 1 strain Ab4 reduces virulence while maintaining strong immunogenicity. BMC Vet. Res. 2018, 14, 245. [Google Scholar] [CrossRef] [Green Version]
- Soboll Hussey, G.; Hussey, S.B.; Wagner, B.; Horohov, D.W.; Van de Walle, G.R.; Osterrieder, N.; Goehring, L.S.; Rao, S.; Lunn, D.P. Evaluation of immune responses following infection of ponies with an EHV-1 ORF1/2 deletion mutant. Vet. Res. 2011, 42, 23. [Google Scholar] [CrossRef] [Green Version]
- Dietze, K.; Slosarek, I.; Fuhrmann-Selter, T.; Hopperdietzel, C.; Plendl, J.; Kaessmeyer, S. Isolation of equine endothelial cells and life cell angiogenesis assay. Clin. Hemorheol. Microcirc. 2014, 58, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Edington, N.; Bridges, C.G.; Patel, J.R. Endothelial cell infection and thrombosis in paralysis caused by equid herpesvirus-1: Equine stroke. Arch. Virol. 1986, 90, 111–124. [Google Scholar] [CrossRef]
- Goodman, L.B.; Loregian, A.; Perkins, G.A.; Nugent, J.; Buckles, E.L.; Mercorelli, B.; Kydd, J.H.; Palu, G.; Smith, K.C.; Osterrieder, N.; et al. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog 2007, 3, e160. [Google Scholar] [CrossRef]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar] [PubMed]
- Tischer, B.K.; Smith, G.A.; Osterrieder, N. En passant mutagenesis: A two step markerless red recombination system. Methods Mol. Biol. 2010, 634, 421–430. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. (Eds.) Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Birnboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, M.S.K. Development and Molecular Studies on Equine Herpesvirus Type 1 (EHV-1) Vaccine against Foot and Mouth Disease Virus; Freien Universität: Berlin, Germany, 2018. [Google Scholar]
- Azab, W.; Osterrieder, N. Glycoproteins D of equine herpesvirus type 1 (EHV-1) and EHV-4 determine cellular tropism independently of integrins. J. Virol. 2012, 86, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Image Processing and Analysis in Java. Available online: http://rsb.info.nih.gov/ij/ (accessed on 9 August 2020).
- Hudetz, A.G.; Feher, G.; Kampine, J.P. Heterogeneous autoregulation of cerebrocortical capillary flow: Evidence for functional thoroughfare channels? Microvasc. Res. 1996, 51, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Soboll Hussey, G.; Ashton, L.V.; Quintana, A.M.; Lunn, D.P.; Goehring, L.S.; Annis, K.; Landolt, G. Innate immune responses of airway epithelial cells to infection with equine herpesvirus-1. Vet. Microbiol. 2014, 170, 28–38. [Google Scholar] [CrossRef]
- Hughes, C.S.; Moggridge, S.; Muller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Murugaiyan, J.; Eravci, M.; Weise, C.; Roesler, U. Mass spectrometry data from label-free quantitative proteomic analysis of harmless and pathogenic strains of infectious microalgae, Prototheca spp. Data Brief. 2017, 12, 320–326. [Google Scholar] [CrossRef]
- Dunn, J.; Ferluga, S.; Sharma, V.; Futschik, M.; Hilton, D.A.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Proteomic analysis discovers the differential expression of novel proteins and phosphoproteins in meningioma including NEK9, HK2 and SET and deregulation of RNA metabolism. EBioMedicine 2019, 40, 77–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, R.; Lu, C.; Han, X.; Guo, S.; Yan, S.; Liu, L.; Fu, X.; Chen, N.; Guo, X.; Chi, H.; et al. Comparative proteomic analysis of maize (Zea mays L.) seedlings under rice black-streaked dwarf virus infection. BMC Plant Biol. 2018, 18, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.M.; Messenger, K.M.; Sheats, M.K.; Jones, S.L. Misoprostol Inhibits Lipopolysaccharide-Induced Pro-inflammatory Cytokine Production by Equine Leukocytes. Front. Vet. Sci. 2017, 4, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, J. Chapter 14—Equine Herpesviruses. In Equine Infectious Diseases, 2nd ed.; Sellon, D.C., Long, M.T., Eds.; W.B. Saunders: St. Louis, MI, USA, 2014; pp. 151–168.e158. [Google Scholar] [CrossRef]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhong, P.; Agosto, L.M.; Munro, J.B.; Mothes, W. Cell-to-cell transmission of viruses. Curr. Opin. Virol. 2013, 3, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Rappocciolo, G.; Birch, J.; Ellis, S.A. Down-regulation of MHC class I expression by equine herpesvirus-1. J. Gen. Virol. 2003, 84, 293–300. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Verweij, M.C.; Lipinska, A.D.; Wang, Y.; Quinten, E.; Reits, E.A.; Koch, J.; Loch, S.; Marcondes Rezende, M.; Daus, F.; et al. Varicellovirus UL 49.5 proteins differentially affect the function of the transporter associated with antigen processing, TAP. PLoS Pathog 2008, 4, e1000080. [Google Scholar] [CrossRef]
- Schnabel, C.L.; Babasyan, S.; Rollins, A.; Freer, H.; Wimer, C.L.; Perkins, G.A.; Raza, F.; Osterrieder, N.; Wagner, B. An equine herpesvirus type 1 (EHV-1) Ab4 open reading frame (ORF)2 deletion mutant provides immunity and protection from EHV-1 infection and disease. J. Virol. 2019. [Google Scholar] [CrossRef]
- Klupp, B.G.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Identification and characterization of the pseudorabies virus UL43 protein. Virology 2005, 334, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.H.; Ding, Q.; Yang, Y.; Sun, H. Analysis of contributions of herpes simplex virus type 1 UL43 protein to induction of cell-cell fusion. Trop. J. Pharm. Res. 2016, 15, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.F. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 2009, 6, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalan, P.K.; Jones, D.A.; McIntire, L.V.; Smith, C.W. Cell adhesion under hydrodynamic flow conditions. Curr. Protoc. Immunol. 2001, 15, 7.29.1–7.29.23. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, A.P.; Glorieux, S.; Gryspeerdt, A.C.; Steukers, L.; Duchateau, L.; Osterrieder, N.; Van de Walle, G.R.; Nauwynck, H.J. Replication kinetics of neurovirulent versus non-neurovirulent equine herpesvirus type 1 strains in equine nasal mucosal explants. J. Gen. Virol. 2010, 91, 2019–2028. [Google Scholar] [CrossRef]
- Zhao, J.; Poelaert, K.C.K.; Van Cleemput, J.; Nauwynck, H.J. CCL2 and CCL5 driven attraction of CD172a(+) monocytic cells during an equine herpesvirus type 1 (EHV-1) infection in equine nasal mucosa and the impact of two migration inhibitors, rosiglitazone (RSG) and quinacrine (QC). Vet. Res. 2017, 48, 14. [Google Scholar] [CrossRef] [Green Version]
- Gryspeerdt, A.C.; Vandekerckhove, A.P.; Garre, B.; Barbe, F.; Van de Walle, G.R.; Nauwynck, H.J. Differences in replication kinetics and cell tropism between neurovirulent and non-neurovirulent EHV1 strains during the acute phase of infection in horses. Vet. Microbiol. 2010, 142, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Leick, M.; Azcutia, V.; Newton, G.; Luscinskas, F.W. Leukocyte recruitment in inflammation: Basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell. Tissue Res. 2014, 355, 647–656. [Google Scholar] [CrossRef]
- van Der Meulen, K.M.; Nauwynck, H.J.; Buddaert, W.; Pensaert, M.B. Replication of equine herpesvirus type 1 in freshly isolated equine peripheral blood mononuclear cells and changes in susceptibility following mitogen stimulation. J. Gen. Virol. 2000, 81, 21–25. [Google Scholar] [CrossRef]
- Paillot, R.; Daly, J.M.; Juillard, V.; Minke, J.M.; Hannant, D.; Kydd, J.H. Equine interferon gamma synthesis in lymphocytes after in vivo infection and in vitro stimulation with EHV-1. Vaccine 2005, 23, 4541–4551. [Google Scholar] [CrossRef]
- van der Meulen, K.M.; Nauwynck, H.J.; Pensaert, M.B. Absence of viral antigens on the surface of equine herpesvirus-1-infected peripheral blood mononuclear cells: A strategy to avoid complement-mediated lysis. J. Gen. Virol. 2003, 84, 93–97. [Google Scholar] [CrossRef]
- Engel, E.A.; Song, R.; Koyuncu, O.O.; Enquist, L.W. Investigating the biology of alpha herpesviruses with MS-based proteomics. Proteomics 2015, 15, 1943–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, B.; Gillet, L.; Vanderplasschen, A.; Wattiez, R. Structural Proteomics of Herpesviruses. Viruses 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.Y.; Huang, D.Y.; Ho, F.M.; Huang, K.C.; Lin, W.W. PKC-dependent human monocyte adhesion requires AMPK and Syk activation. PLoS ONE 2012, 7, e40999. [Google Scholar] [CrossRef] [Green Version]
- Filippakis, H.; Spandidos, D.A.; Sourvinos, G. Herpesviruses: Hijacking the Ras signaling pathway. Biochim. Biophys. Acta 2010, 1803, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien. Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Rickinson, A.B. Epstein-Barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Qiu, Y.-Q. KEGG Pathway Database. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 1068–1069. [Google Scholar] [CrossRef]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cohen, J.I. The role of PI3K/Akt in human herpesvirus infection: From the bench to the bedside. Virology 2015, 479–480, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kachi, K.; Yoshiyama, H.; Ohba, Y. Epstein-Barr virus exploits host endocytic machinery for cell-to-cell viral transmission rather than a virological synapse. J. Gen. Virol. 2016, 97, 2989–3006. [Google Scholar] [CrossRef]
- Zhu, Y.; Ramos da Silva, S.; He, M.; Liang, Q.; Lu, C.; Feng, P.; Jung, J.U.; Gao, S.J. An Oncogenic Virus Promotes Cell Survival and Cellular Transformation by Suppressing Glycolysis. PLoS Pathog. 2016, 12, e1005648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavignac, Y.; Esclatine, A. Herpesviruses and autophagy: Catch me if you can! Viruses 2010, 2, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, R.; Sasaki, M.; Sawa, H.; Wada, R.; Umemura, T.; Kimura, T. Infectious entry of equine herpesvirus-1 into host cells through different endocytic pathways. Virology 2009, 393, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Van de Walle, G.R.; Peters, S.T.; VanderVen, B.C.; O’Callaghan, D.J.; Osterrieder, N. Equine herpesvirus 1 entry via endocytosis is facilitated by alphaV integrins and an RSD motif in glycoprotein D. J. Virol. 2008, 82, 11859–11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, A.R., Jr.; Uchida, H.; von Einem, J.; Goins, W.F.; Grandi, P.; Cohen, J.B.; Osterrieder, N.; Glorioso, J.C. Equine herpesvirus type 1 (EHV-1) utilizes microtubules, dynein, and ROCK1 to productively infect cells. Vet. Microbiol. 2010, 141, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Pleschka, S. RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol. Chem. 2008, 389, 1273–1282. [Google Scholar] [CrossRef]
- DuShane, J.K.; Maginnis, M.S. Human DNA Virus Exploitation of the MAPK-ERK Cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar] [CrossRef] [Green Version]
- Ichise, T.; Yoshida, N.; Ichise, H. FGF2-induced Ras-MAPK signalling maintains lymphatic endothelial cell identity by upregulating endothelial-cell-specific gene expression and suppressing TGFbeta signalling through Smad2. J. Cell. Sci. 2014, 127, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Wimer, C.L.; Damiani, A.; Osterrieder, N.; Wagner, B. Equine herpesvirus type-1 modulates CCL2, CCL3, CCL5, CXCL9, and CXCL10 chemokine expression. Vet. Immunol. Immunopathol. 2011, 140, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.; Wimer, C.; Freer, H.; Osterrieder, N.; Erb, H.N. Infection of peripheral blood mononuclear cells with neuropathogenic equine herpesvirus type-1 strain Ab4 reveals intact interferon-alpha induction and induces suppression of anti-inflammatory interleukin-10 responses in comparison to other viral strains. Vet. Immunol. Immunopathol. 2011, 143, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, J.J.; Matikainen, S.; Nyman, T.A. Global secretome characterization of herpes simplex virus 1-infected human primary macrophages. J. Virol. 2012, 86, 12770–12778. [Google Scholar] [CrossRef] [Green Version]
- Milora, K.A.; Miller, S.L.; Sanmiguel, J.C.; Jensen, L.E. Interleukin-1alpha released from HSV-1-infected keratinocytes acts as a functional alarmin in the skin. Nat. Commun. 2014, 5, 5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Primer Name | Nucleotide Sequence |
|---|---|---|
| P1 | ORF17 STOP Fwd | caaaggttggcttgctacatcaaggttatcaatcatgatgtaacagccagatagagagcccggtagggataacagggtaatcgat |
| P2 | ORF17 STOP Rev | gcaccagacacgagtcttcaccgggctctctatctggctgttacatcatgattgataaccttgccagtgttacaaccaattaacc |
| P3 | ORF17 pre For seq | ctttatgtgaattcaccgac |
| P4 | ORF17 post Rev seq | gttttatgactaatacctgg |
| P5 | ORF17 Revertant Fwd | caaaggttggcttgctacatcaaggttatcaatcatgatgtaccagccagatagagagcccggtagggataacagggtaatcgat |
| P6 | ORF17 Revertant Rev | gcaccagacacgagtcttcaccgggctctctatctggctggtacatcatgattgataaccttgccagtgttacaaccaattaacc |
| P7 | ORF1 deletion Fwd | tccacctgcaccttttccatctcctctccaactcgccgccaacgactgtagtaccgcaaaaggatgacgacgataagtaggg |
| P8 | ORF1 deletion Rev | aaaaataaatgcgattaacctttgcggtactacagtcgttggcggcgagttggagaggagcaaccaattaaccaattctgattag |
| P9 | ORF1 pre Fwd seq | ggctcctcccttttggctctgg |
| P10 | ORF1 post Rev seq | tctggtgctgatcggaatagtgta |
| P11 | ORF1 BamH Fwd | attggatccatgagacccgagggagtttc |
| P12 | ORF1 EcoRI Rev | cacgaattcttatttctccttcttgccgt |
| P13 | ORF1 kana Fwd | atttagccttccgctcctgtctgcttacactttacacttttctgctcgtcatgagacccgagggagtttc |
| P14 | ORF1 kana Rev | aggggtgtttgtgaaaataaacataatacaactgtgttgaaccacttgttttatttctccttcttgccgt |
| P15 | ORF1 Revertant Fwd | ttccactttctccacctgcaccttttccatctcctctccaactcgccgccatgagacccgagggagtttc |
| P16 | ORF1 Revertant Rev | gagtgcatgtaaaaataaatgcgattaacctttgcggtactacagtcgttttatttctccttcttgccgt |
| P17 | ORF2 deletion For | aaaacgactgtagtaccgcaaaggttaatcgcatttatttgcttaaacactttggagcgaaggatgacgacgataagtaggg |
| P18 | ORF2 deletion Rev | cgcccccataccccgccccctcgctccaaagtgtttaagcaaataaatgcgattaaccttcaaccaattaaccaattctgattag |
| P19 | ORF2 pre Fwd seq | taacaaacggcaagaaggag |
| P20 | ORF2 post Rev seq | taacgctgtagattgagttt |
| P21 | ORF2 EcoRI Fwd | aattagaattcttacatgcactcctttccaa |
| P22 | ORF2 Xba Rev | atatatctagaatggatccagcgtggaggag |
| P23 | ORF2 Kan Fwd | cgcggggcggccgcactaccatcggaagtttaccaggatgacgacgataagtaggg |
| P24 | ORF2 Kan Rev | ggtagtgcggccgccccgcggtgatttctagtaacaaccaattaaccaattctgattag |
| P25 | ORF2 Revertant Fwd | aaggagaaataaaacgactgtagtaccgcaaaggttaatcgcatttatttttacatgcactcctttccaa |
| P26 | ORF2 Revertant Rev | ttcaggcatacgcccccataccccgccccctcgctccaaagtgtttaagcatggatccagcgtggaggag |
| Cell Marker | Cell | % in PBMC | Rate of Infection in % | % in Infected Population |
|---|---|---|---|---|
| CD14 | Monocyte | 27.1 ± 1.7 | 41.3 ± 2.3 | 66.2 ± 1.1 |
| IgM | B lymphocyte | 9.8 ± 1.1 | 22 ± 3.2 | 12.9 ± 0.8 |
| CD3 | T lymphocyte | 63.2 ± 2.3 | 5.5 ± 0.5 | 20.8 ± 1.5 |
| Type | Name of Viral Proteins | |
|---|---|---|
| Nonstructural protein | Ribonucleoside-diphosphate reductase R1 Ribonucleoside-diphosphate reductase R2 DNA polymerase Uracil-DNA glycosylase Alkaline nuclease Major viral transcription factor Serine/theonine-protein kinase Major DNA-binding protein mRNA export factor ICP27 | Nuclear egress protein 1 Nuclear egress protein 2 DNA polymerase processivity factor DNA primase Thymidine kinase Packaging protein UL32 Tripartite terminase Deoxyuridine 5-triphosphate Internal repeat 6 |
| Structural protein | Tegument proteins Tegument protein UL47 Tegument protein UL46 Tegument protein VP16 Large tegument protein Inner tegument protein Tegument protein VP22 Tegument protein UL21 | Serine/theonine-protein kinase UL13 Cytoplasmic envelopment protein 2 Envelope protein UL45 E3 ubiquitin-protein ligase ICP0 |
| Capsid proteins Major capsid protein Triplex capsid protein 1 Triplex capsid protein 2 | Capsid vertex component Capsid vertex component 1 Portal protein | |
| Envelope proteins Glycoprotein G Glycoprotein I Glycoprotein D Glycoprotein H | Glycoprotein B Glycoprotein C Glycoprotein E | |
| Uncharacterized proteins | ORF protein 2 ORF protein 59 ORF protein 3 | |
| Infected Population | Pathways Upregulated | Pathways Downregulated |
|---|---|---|
| PBMC vs. Ab4-wt | Lysosome cAMP signaling Ras signaling pathway Endocytosis Platelet activation Leukocyte transendothelial migration Oxydative phosphorylation Fatty acid elongation | Herpesvirus infection Spliceosome Chemokine signaling pathway RNA degradation Apoptosis |
| PBMC vs. Ab4∆ORF17 | Lysosome Herpesvirus infection Oxydative phosphorylation Protein processing in endoplasmic reticulum Fc epsilon RI signaling Metabolic pathways | Chemokine signaling pathway MAPK signaling pathway Spliceosome RNA transport |
| PBMC vs. Ab4∆ORF1 | mTOR signaling pathway Endocytosis Lysosome Focal adhesion Ras signaling pathway Herpesvirus infection Chemokine signaling pathway Leukocyte transendothelial migration Platelet activation | Spliceosome RNA degradation Metabolic pathways Aminoacyl-tRNA biosynthesis |
| PBMC vs. Ab4∆ORF 2 | Herpesvirus infection mTOR signaling pathway Regulation of actin cytoskeleton Chemokine signaling pathway | Spliceosome RNA degradation MAPK signaling pathway |
| PBMC vs. Ab4∆ORF 1/ORF2/ORF17 | Endocytosis Lysosome Herpesvirus infection T cell signaling Chemokine signaling pathway | Spliceosome RNA degradation MAPK signaling pathway |
| PBMC Samples | Number of Detected Cytokines | Co-Culture Samples | Number of Detected Cytokines | |||
|---|---|---|---|---|---|---|
| 3 hpi | 6 hpi | 12 hpi | 3 hpi | 6 hpi | ||
| PBMC | 5 | 5 | 9 | PBMC-EC | 8 | 10 |
| Ab4-wt | 3 | 1 | 4 | Ab4-wt-EC | 5 | 6 |
| Ab4∆ORF17 | 3 | 4 | 4 | |||
| Ab4∆ORF1 | 3 | 4 | 7 | |||
| Ab4∆ORF2 | 4 | 4 | 8 | |||
| Ab4∆ORF17/ORF2 | 4 | 6 | 4 | |||
| Ab4∆ORF1/ORF2 | 3 | 5 | 7 | |||
| Ab4∆ORF1/ORF17 | 3 | 4 | 7 | |||
| Ab4∆ORF1/ORF2/ORF17 | 3 | 4 | 7 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavulraj, S.; Kamel, M.; Stephanowitz, H.; Liu, F.; Plendl, J.; Osterrieder, N.; Azab, W. Equine Herpesvirus Type 1 Modulates Cytokine and Chemokine Profiles of Mononuclear Cells for Efficient Dissemination to Target Organs. Viruses 2020, 12, 999. https://doi.org/10.3390/v12090999
Pavulraj S, Kamel M, Stephanowitz H, Liu F, Plendl J, Osterrieder N, Azab W. Equine Herpesvirus Type 1 Modulates Cytokine and Chemokine Profiles of Mononuclear Cells for Efficient Dissemination to Target Organs. Viruses. 2020; 12(9):999. https://doi.org/10.3390/v12090999
Chicago/Turabian StylePavulraj, Selvaraj, Mohamed Kamel, Heike Stephanowitz, Fan Liu, Johanna Plendl, Nikolaus Osterrieder, and Walid Azab. 2020. "Equine Herpesvirus Type 1 Modulates Cytokine and Chemokine Profiles of Mononuclear Cells for Efficient Dissemination to Target Organs" Viruses 12, no. 9: 999. https://doi.org/10.3390/v12090999