Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
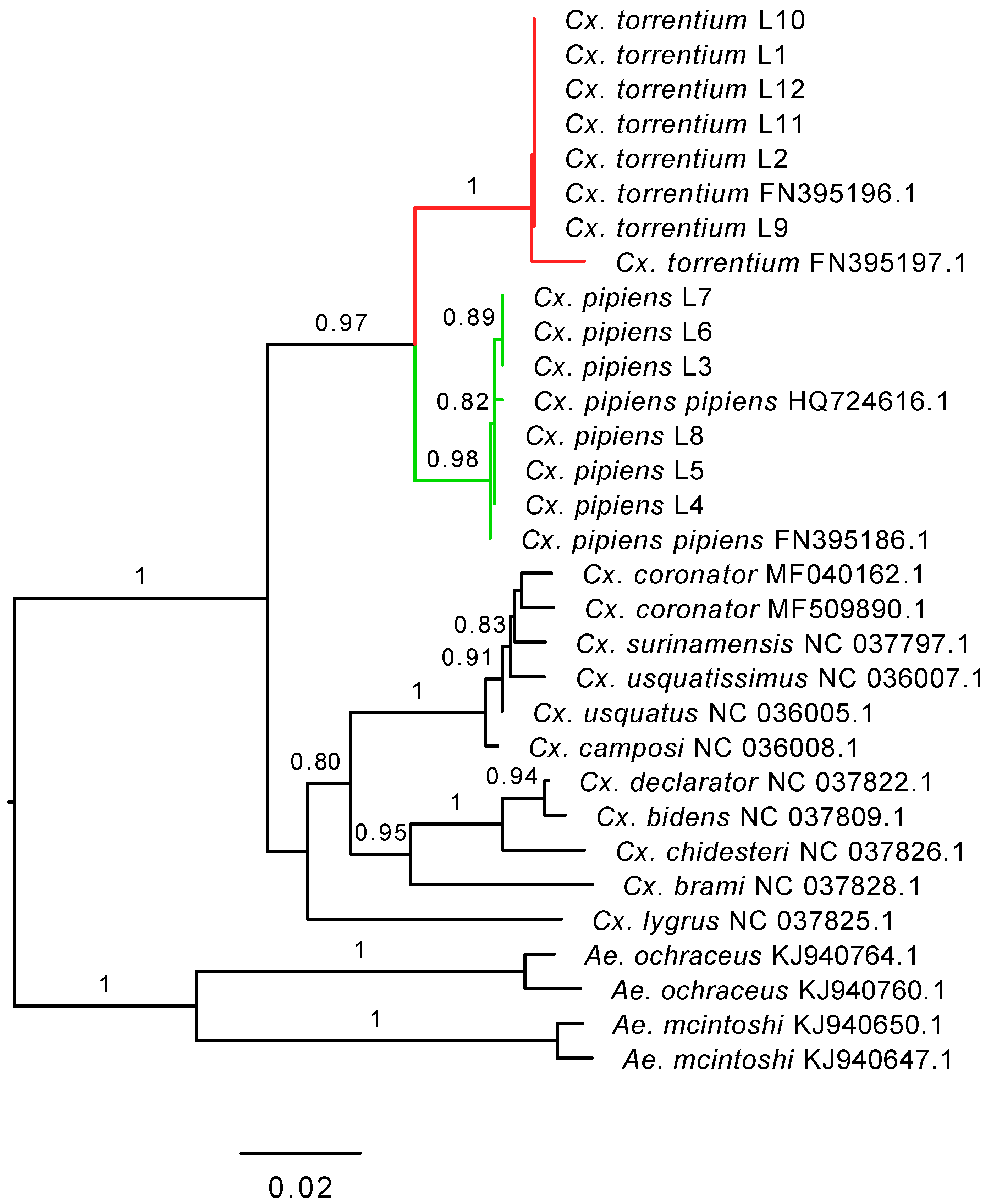
2.1. Mosquito Collection
2.2. Sample Processing and Sequencing
2.3. Identification of Viruses and Wolbachia Bacteria
2.4. Inference of Virus Evolutionary History and Host Associations
3. Results
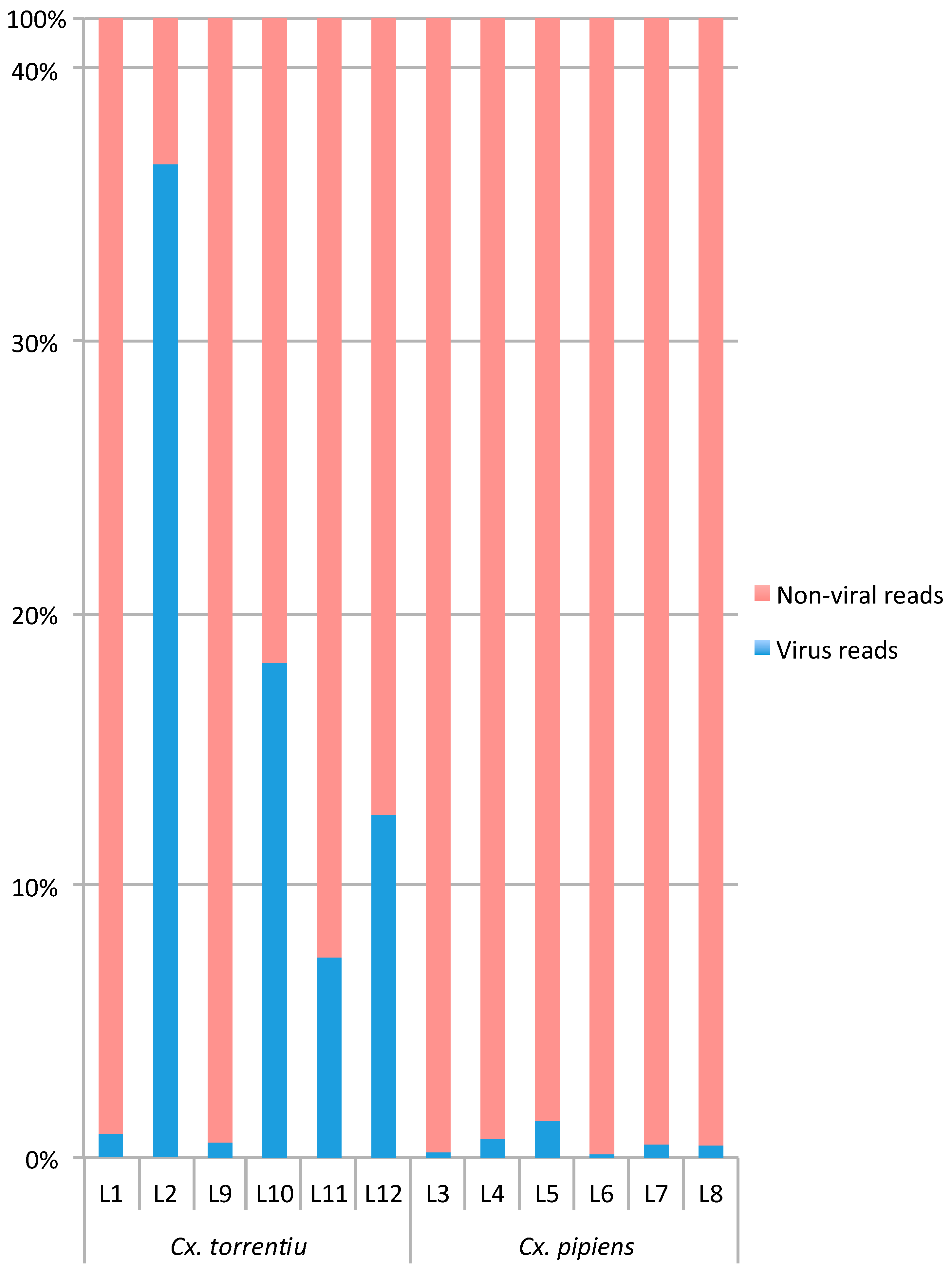
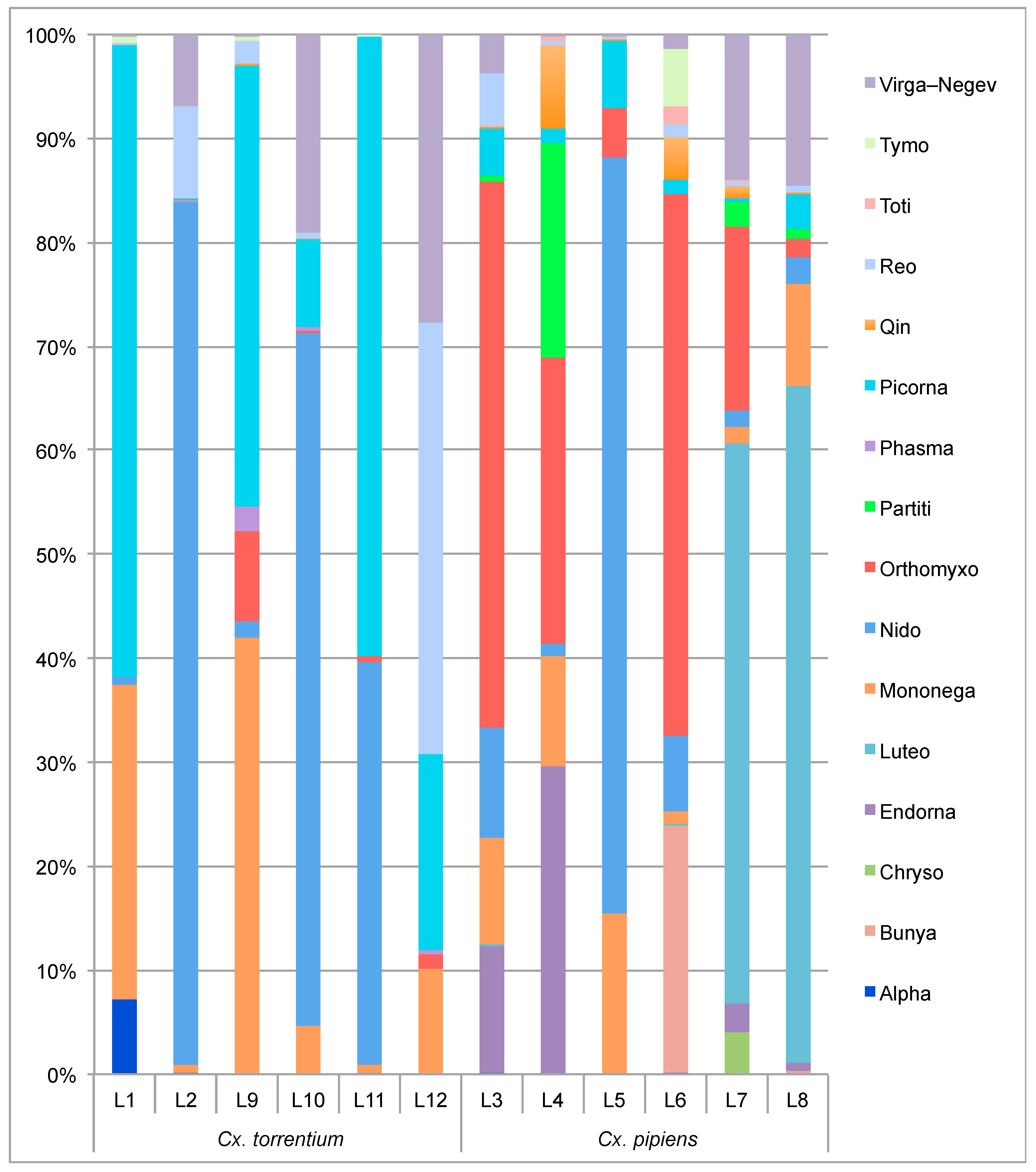
3.1. RNA Virome Characterization
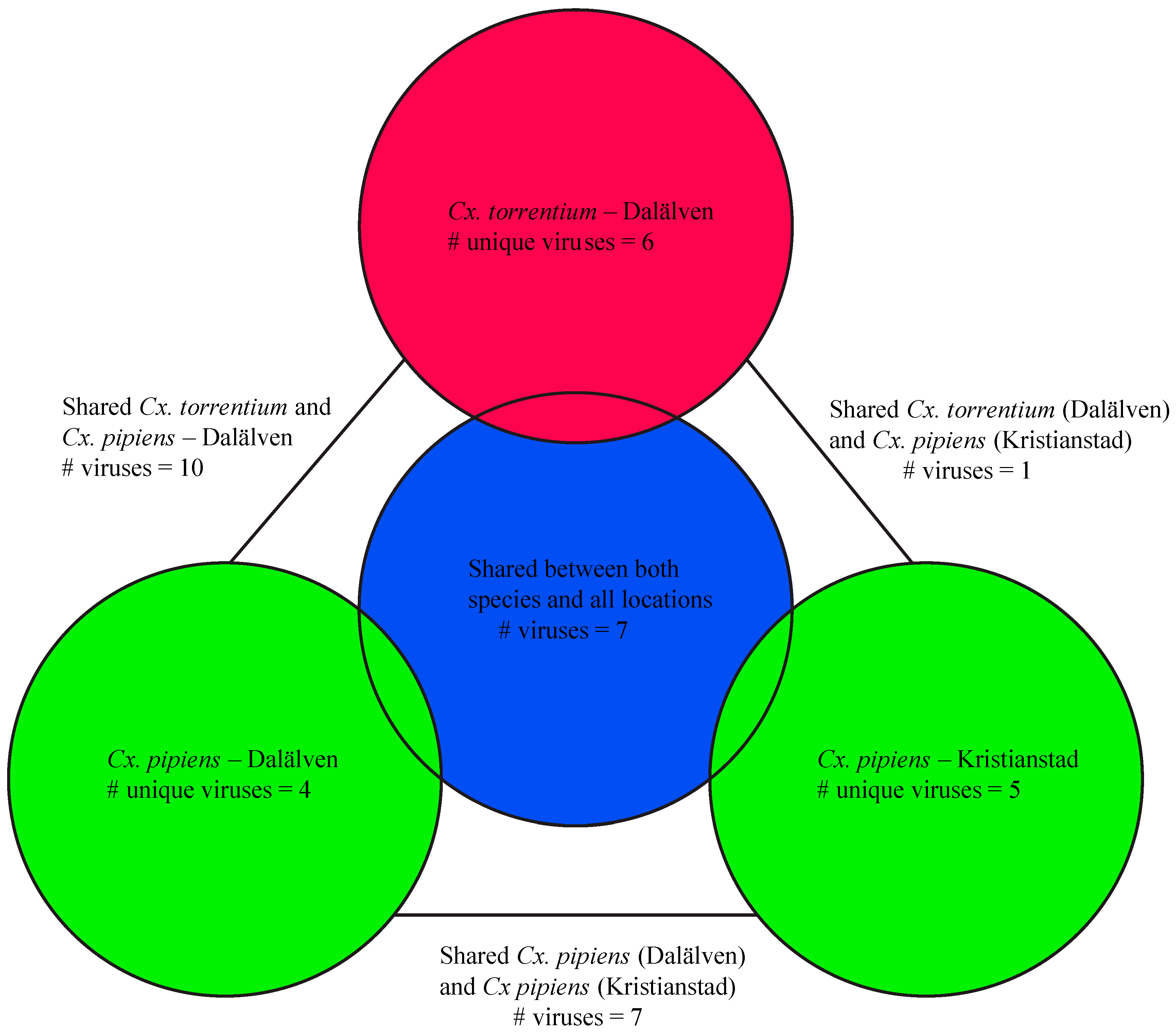
3.2. Virome Comparison between Mosquito Species and Geographical Regions
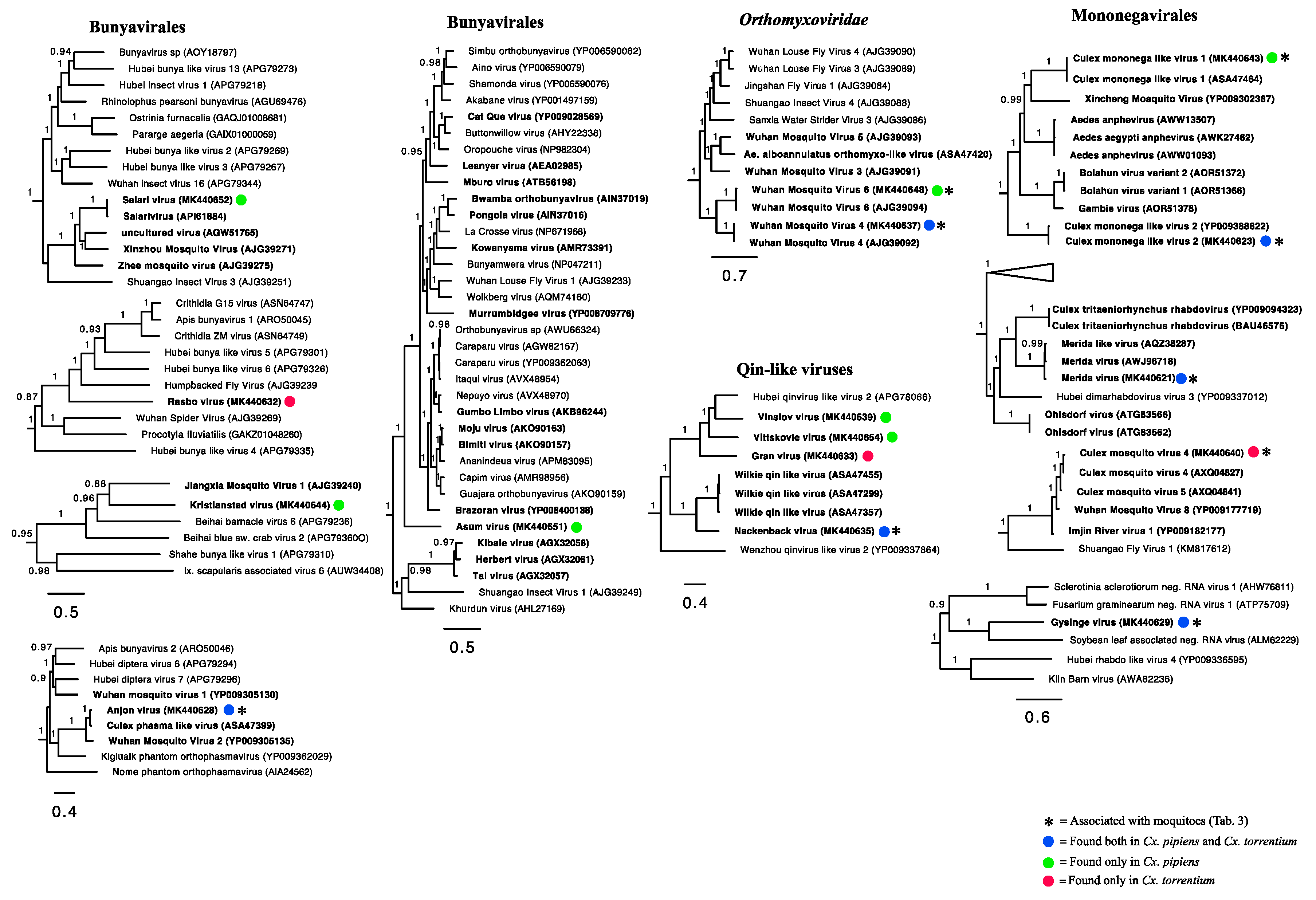
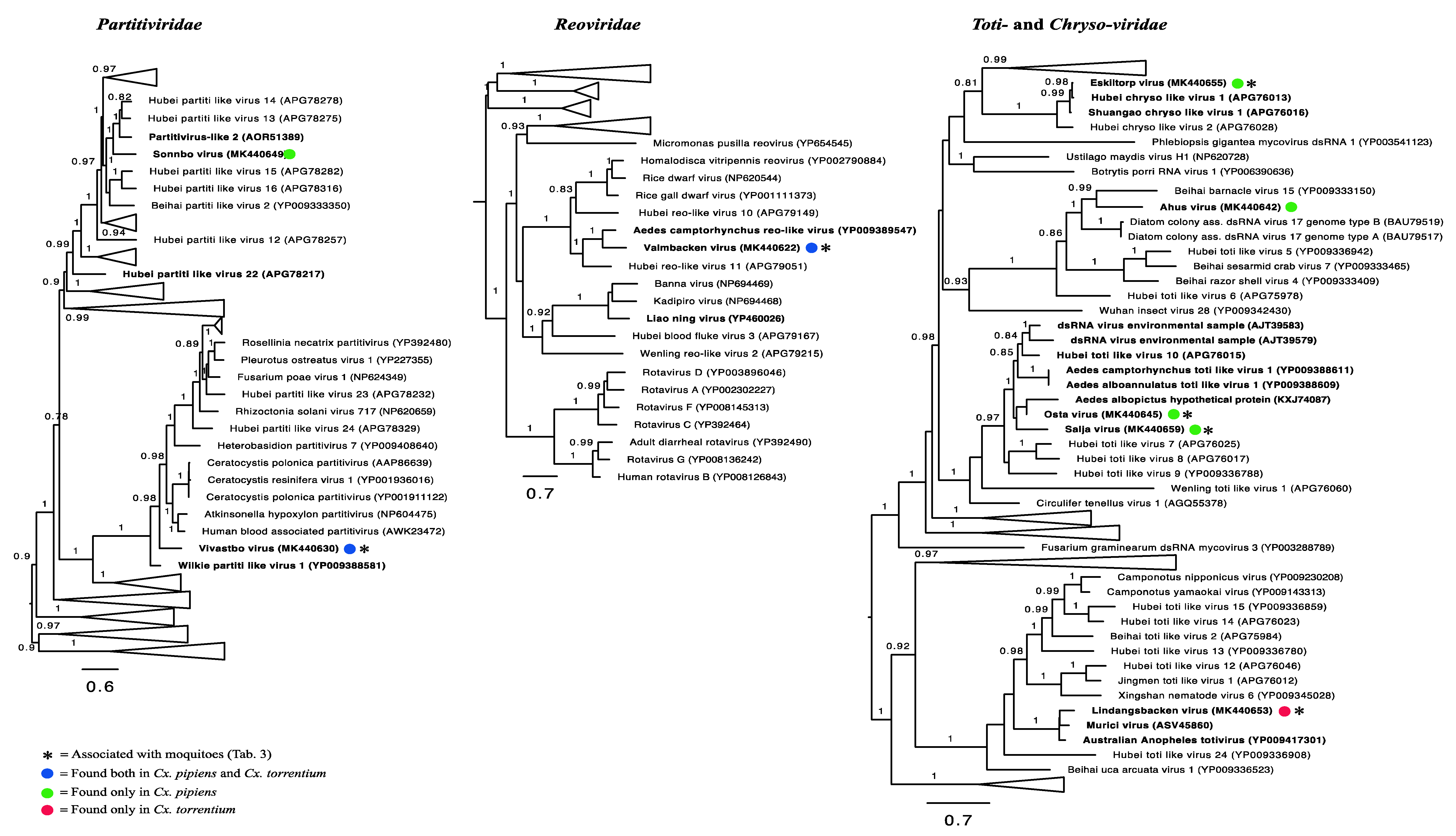
3.3. Evolutionary History and Host Associations of the Discovered RNA Viruses
3.4. Positive-Sense RNA Viruses
3.5. Negative-Sense RNA Viruses
3.6. Double-Stranded RNA Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mullen, G.R.; Durden, L. Medical and Veterinary Entomology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 261–325. ISBN 978-0-12-372500-4. [Google Scholar]
- Gould, E.; Pettersson, J.; Higgs, S.; Charrel, R.; de Lamballerie, X. Emerging arboviruses: Why today? One Health 2017, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Lecuit, M. Chikungunya virus and the global spread of a mosquito-borne disease. N. Engl. J. Med. 2015, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Hesson, J.C.; Rettich, F.; Merdić, E.; Vignjević, G.; Ostman, O.; Schäfer, M.; Schaffner, F.; Foussadier, R.; Besnard, G.; Medlock, J.; et al. The arbovirus vector Culex torrentium is more prevalent than Culex pipiens in northern and central Europe. Med. Vet. Entomol. 2014, 28, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Kurkela, S.; Helve, T.; Vaheri, A.; Vapalahti, O. Arthritis and arthralgia three years after Sindbis virus infection: Clinical follow-up of a cohort of 49 patients. Scand. J. Infect. Dis. 2008, 40, 167–173. [Google Scholar] [CrossRef]
- Hesson, J.C.; Verner-Carlsson, J.; Larsson, A.; Ahmed, R.; Lundkvist, Å.; Lundström, J.O. Culex torrentium Mosquito Role as Major Enzootic Vector Defined by Rate of Sindbis Virus Infection, Sweden, 2009. Emerg. Infect. Dis. 2015, 21, 875–878. [Google Scholar] [CrossRef]
- Hesson, J.C.; Lundström, J.O.; Tok, A.; Östman, Ö.; Lundkvist, Å. Temporal variation in Sindbis virus antibody prevalence in bird hosts in an endemic area in Sweden. PLoS ONE 2016, 11, e0162005. [Google Scholar] [CrossRef]
- Komar, N.; Langevin, S.; Hinten, S.; Nemeth, N.; Edwards, E.; Hettler, D.; Davis, B.; Bowen, R.; Bunning, M. Experimental infection of North American birds with the New York 1999 strain of West Nile virus. Emerg. Infect. Dis. 2003, 9, 311–322. [Google Scholar] [CrossRef]
- Leggewie, M.; Krumkamp, R.; Badusche, M.; Heitmann, A.; Jansen, S.; Schmidt-Chanasit, J.; Tannich, E.; Becker, S.C. Culex torrentium mosquitoes from Germany are negative for Wolbachia. Med. Vet. Entomol. 2018, 32, 115–120. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic virome analysis of Culex mosquitoes from Kenya and China. Viruses 2018, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of >12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, F.; Liu, A.; Lin, X.; Fu, S.; Song, J.; Liu, G.; Shao, N.; Tao, Z.; Wang, Q.; et al. Identification and genetic analysis of Kadipiro virus isolated in Shandong province, China. Virol. J. 2018, 15, 64. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.H. Why are there so many species in the tropics? J. Biogeogr. 2014, 41, 8–22. [Google Scholar] [CrossRef]
- Foley, D.H.; Rueda, L.M.; Wilkerson, R.C. Insight into global mosquito biogeography from country species records. J. Med. Entomol. 2007, 44, 554–567. [Google Scholar] [CrossRef]
- Becker, N.; Petrić, D.; Boase, C.; Lane, J.; Zgomba, M.; Dahl, C.; Kaiser, A. Mosquitoes and Their Control; Springer US: Boston, MA, USA, 2003; pp. 113–286. ISBN 978-1-4757-5897-9. [Google Scholar]
- Hesson, J.C.; Lundström, J.O.; Halvarsson, P.; Erixon, P.; Collado, A. A sensitive and reliable restriction enzyme assay to distinguish between the mosquitoes Culex torrentium and Culex pipiens. Med. Vet. Entomol. 2010, 24, 142–149. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Prot. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersson, J.H.-O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Marzano, S.-Y.L.; Domier, L.L. Novel mycoviruses discovered from metatranscriptomics survey of soybean phyllosphere phytobiomes. Virus Res. 2016, 213, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Charles, J.; Firth, A.E.; Loroño-Pino, M.A.; Garcia-Rejon, J.E.; Farfan-Ale, J.A.; Lipkin, W.I.; Blitvich, B.J.; Briese, T. Merida virus, a putative novel rhabdovirus discovered in Culex and Ochlerotatus spp. mosquitoes in the Yucatan Peninsula of Mexico. J. Gen. Virol. 2016, 97, 977–987. [Google Scholar] [CrossRef]
- Hesson, J.C. Bloodmeal analyses of Sindbis virus vectors. Unpublished.
- Ling, J.; Smura, T.; Lundström, J.O.; Pettersson, J.H.-O.; Sironen, T.; Vapalahti, O.; Lundkvist, Å.; Hesson, J.C. The introduction and dispersal of Sindbis virus from central Africa to Europe. J. Virol. 2019, 93, e00620-19. [Google Scholar] [CrossRef]
- Blitvich, B.J.; Firth, A.E. Insect-specific flaviviruses: A systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses 2015, 7, 1927–1959. [Google Scholar] [CrossRef]
- Huhtamo, E.; Putkuri, N.; Kurkela, S.; Manni, T.; Vaheri, A.; Vapalahti, O.; Uzcategui, N.Y. Characterization of a novel flavivirus from mosquitoes in northern Europe that is related to mosquito-borne flaviviruses of the tropics. J. Virol. 2009, 83, 9532–9540. [Google Scholar] [CrossRef]
- Huhtamo, E.; Moureau, G.; Cook, S.; Julkunen, O.; Putkuri, N.; Kurkela, S.; Uzcátegui, N.Y.; Harbach, R.E.; Gould, E.A.; Vapalahti, O.; et al. Novel insect-specific flavivirus isolated from northern Europe. Virology 2012, 433, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, K.E.; Bonizzoni, M. Nonretroviral integrated RNA viruses in arthropod vectors: An occasional event or something more? Curr. Opin. Insect Sci. 2017, 22, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Holmes, E.C. Endogenous RNA viruses of plants in insect genomes. Virology 2012, 427, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.N. The Impact of Wolbachia on Virus Infection in Mosquitoes. Viruses 2015, 7, 5705–5717. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cx. torrentium | Cx. pipiens | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Library | L1 | L2 | L9 | L10 | L11 | L12 | L3 | L4 | L5 | L6 | L7 | L8 |
| Total virus reads | 311,893 | 22,961,076 | 258,016 | 9,604,141 | 4,654,540 | 7,452,859 | 45,019 | 279,568 | 565,968 | 57,867 | 227,988 | 186,792 |
| Host COX1 reads | 586 | 265 | 322 | 2427 | 2254 | 3117 | 126 | 2529 | 317 | 2850 | 860 | 417 |
| Total reads | 34,150,856 | 62,820,620 | 43,914,132 | 52,916,282 | 62,936,342 | 59,016,596 | 39,231,440 | 41,210,662 | 41,328,330 | 40,762,624 | 44,703,752 | 46,526,884 |
| Virus % | 0.9133 | 36.5502 | 0.5875 | 18.1497 | 7.3956 | 12.6284 | 0.1148 | 0.6784 | 1.3694 | 0.1420 | 0.5100 | 0.4015 |
| Host COX1 % | 0.0017 | 0.0004 | 0.0007 | 0.0046 | 0.0036 | 0.0053 | 0.0003 | 0.0061 | 0.0008 | 0.0070 | 0.0019 | 0.0009 |
| Other % | 99.0850 | 63.4494 | 99.4117 | 81.8457 | 92.6008 | 87.3663 | 99.8849 | 99.3155 | 98.6298 | 99.8510 | 99.4881 | 99.5976 |
| Cx. torrentium | Cx. pipiens | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SINV+ | SINV Unscreened | SINV+ | SINV Unscreened | ||||||||||||
| Location | Dalälven | Dalälven | Dalälven | Dalälven | Dalälven | Dalälven | Dalälven | Dalälven | Dalälven | Kristianstad | Kristianstad | Dalälven | |||
| Virus | Threshold | # Mosq | 10 | 10 | 50 | 50 | 15 | 15 | 10 | 15 | 15 | 15 | 15 | 50 | |
| * | ** | Length | L1 | L2 | L9 | L10 | L11 | L12 | L3 | L4 | L5 | L6 | L7 | L8 | |
| Sindbis virus | 0672 | 1 | 11,688 | 671,784 | 417,443 | 0387 | 0151 | 0540 | 0474 | 2523 | 0995 | 0339 | 2012 | 0380 | 0107 |
| Nam Dinh virus | 303,146 | 1 | 20,240 | 73,673 | 303,145,830 | 85,644 | 120,821,017 | 28,690,339 | 95,244 | 137,084 | 83,376 | 10,006,671 | 116,626 | 87,174 | 106,154 |
| Biggie virus | 35,064 | 1 | 9207 | 16,720 | 15,525,826 | 15,439 | 34,543,772 | 23,405 | 35,063,781 | 46,391 | 18,709 | 20,978 | 18,301 | 21,363 | 28,930 |
| Negev virus | 9715 | 1 | 9493 | 1171 | 9,715,234 | 1070 | 2192 | 1748 | 1915 | 2651 | 1432 | 1670 | 2723 | 1946 | 582,029 |
| Rinkaby virus | 0706 | 1 | 14,498 | 0000 | 0159 | 0068 | 0151 | 0127 | 0034 | 0280 | 0000 | 0387 | 0196 | 706,361 | 0172 |
| Kerstinbo virus | 1825 | 1 | 11,280 | 038 | 0207 | 0250 | 10,734 | 117,913 | 0136 | 156,456 | 1,825,401 | 0266 | 0098 | 146,475 | 20,633 |
| Forneby virus | 0138 | 1 | 8255 | 0000 | 0064 | 0000 | 3288 | 0000 | 0000 | 0051 | 138,241 | 0000 | 0049 | 0000 | 6362 |
| Osterfarnebo virus | 0087 | 1 | 5357 | 0000 | 0000 | 0046 | 1077 | 0000 | 0000 | 0000 | 87,016 | 0000 | 0000 | 0000 | 4900 |
| Hallsjon virus | 0014 | 1 | 2847 | 0000 | 0000 | 0000 | 0491 | 13,712 | 0000 | 0000 | 0340 | 0048 | 0442 | 0000 | 0000 |
| Tarnsjo virus (variant 1) | 0063 | 1 | 7890 | 49,047 | 0000 | 11,955 | 27,629 | 63,064 | 2864 | 0051 | 0000 | 0024 | 14,253 | 0045 | 0086 |
| Tarnsjo virus (variant 2) | 0076 | 1 | 7838 | 7701 | 0000 | 2095 | 3553 | 10,471 | 0491 | 0000 | 0000 | 0048 | 76,075 | 0000 | 0021 |
| Culex associated luteo like virus | 1101 | 1 | 2790 | 0322 | 0127 | 0182 | 0113 | 0127 | 0322 | 0510 | 0170 | 0387 | 0368 | 1,101,496 | 109,421 |
| Berrek virus | 0014 | 1 | 2807 | 0000 | 0000 | 0046 | 0000 | 0000 | 0034 | 0000 | 0000 | 0000 | 0000 | 0000 | 13,906 |
| Fagle virus | 0002 | 1 | 1453 | 0000 | 1544 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 |
| Marma virus | 2602 | 1 | 3151 | 0703 | 0493 | 0934 | 1077 | 0604 | 0729 | 1963 | 0849 | 0919 | 1251 | 1,737,371 | 2,601,593 |
| Merida virus | 12,423 | 1 | 11,785 | 2,327,789 | 2,408,604 | 2,230,876 | 7,176,373 | 7420 | 12,423,082 | 16,084 | 9803 | 2,037,222 | 6526 | 6219 | 301,911 |
| Culex mononega like virus 2 | 1216 | 1 | 13,316 | 467,748 | 157,082 | 285,831 | 1,216,374 | 553,242 | 311,692 | 1096 | 467,088 | 87,857 | 13,296 | 0761 | 23,513 |
| Gysinge virus | 0260 | 1 | 9532 | 0088 | 0032 | 0091 | 0170 | 7245 | 0102 | 115,749 | 259,544 | 0073 | 0147 | 68,093 | 0086 |
| Culex mosquito virus 4 | 0453 | 1 | 11,954 | 0059 | 452,511 | 0068 | 1928 | 0079 | 1881 | 0102 | 0000 | 0097 | 0270 | 0045 | 0129 |
| Culex mononega like virus 1 | 0083 | 1 | 6604 | 0000 | 0000 | 0046 | 0038 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 83,113 |
| Valmbacken virus | 52,265 | 1 | 4315 | 19,092 | 32,360,489 | 139,363 | 1,158,301 | 29,172 | 52,264,756 | 66,401 | 24,678 | 21,462 | 19,331 | 20,983 | 30,993 |
| Jotan virus | 20,309 | 1 | 9112 | 16,808 | 18,433 | 15,712 | 7,758,198 | 41,738,015 | 20,308,796 | 50,878 | 91,457 | 912,788 | 18,718 | 18,119 | 22,224 |
| Ista virus | 5605 | 1 | 9551 | 5,605,247 | 576,435 | 2,529,573 | 7,361,250 | 2,402,094 | 3,661,343 | 8131 | 8129 | 4065 | 3950 | 4094 | 116,771 |
| Wuhan Mosquito Virus 4 | 1900 | 1 | 2445 | 1318 | 229,558 | 520,561 | 642,373 | 332,654 | 1,679,358 | 677,569 | 1,900,406 | 1742 | 325,740 | 930,772 | 70,003 |
| Wuhan Mosquito Virus 6 | 0636 | 1 | 2440 | 0117 | 0127 | 0774 | 0189 | 0127 | 0102 | 0102 | 0097 | 636,319 | 510,664 | 2349 | 1891 |
| Vivastbo virus | 1338 | 1 | 2157 | 0000 | 0096 | 0091 | 34,791 | 3972 | 0169 | 7341 | 1,338,440 | 0266 | 0393 | 119,475 | 26,974 |
| Sonnbo virus | 0088 | 1 | 1737 | 0000 | 0032 | 0000 | 0038 | 0064 | 0034 | 0000 | 87,720 | 0048 | 0000 | 0000 | 15,561 |
| Rasbo virus | 0017 | 1 | 5974 | 0000 | 0000 | 4964 | 7257 | 4798 | 17,368 | 0051 | 0049 | 0000 | 0000 | 0000 | 0043 |
| Kristianstad virus | 0022 | 1 | 5406 | 0000 | 0000 | 0000 | 0038 | 0000 | 0000 | 0000 | 0000 | 0000 | 22,104 | 0000 | 0000 |
| Asum virus | 0362 | 1 | 7184 | 0000 | 0032 | 0410 | 0397 | 0016 | 0051 | 0051 | 0218 | 0097 | 361,949 | 0045 | 0043 |
| Salari virus | 0014 | 1 | 6630 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0051 | 0000 | 0000 | 0000 | 0045 | 13,906 |
| Anjon virus | 0407 | 1 | 6495 | 3485 | 406,952 | 140,775 | 725,051 | 6864 | 537,290 | 0918 | 0582 | 1089 | 0589 | 0224 | 0645 |
| Gran virus | 0012 | 1 | 5622 | 0000 | 0032 | 9473 | 5858 | 3559 | 11,912 | 0000 | 0000 | 0048 | 0000 | 0000 | 0021 |
| Nackenback virus | 0559 | 1 | 6128 | 0059 | 0064 | 0091 | 0113 | 8374 | 0102 | 0102 | 559,443 | 0048 | 0123 | 65,498 | 0107 |
| Vinslov virus | 0047 | 1 | 5590 | 0000 | 0000 | 0046 | 0000 | 0000 | 0000 | 0000 | 0340 | 8082 | 47,445 | 0000 | 0000 |
| Vittskovle virus | 0019 | 1 | 5671 | 0000 | 0684 | 0046 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 19,380 | 0000 | 0000 |
| Ahus virus | 0027 | 1 | 7732 | 0000 | 0032 | 0000 | 0038 | 0000 | 0000 | 0000 | 0243 | 4404 | 26,691 | 0000 | 0000 |
| Osta virus | 0019 | 1 | 5398 | 0000 | 0032 | 0000 | 0000 | 0000 | 0000 | 0102 | 19,364 | 0000 | 0049 | 10,111 | 0043 |
| Lindangsbacken virus | 0105 | 1 | 6171 | 0000 | 104,711 | 0046 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0045 | 0000 |
| Salja virus | 0002 | 1 | 1286 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 0000 | 1626 | 0000 | 0000 | 1611 | 0000 |
| Eskilstorp virus | 0210 | 1 | 2933 | 0000 | 0000 | 0000 | 0076 | 0032 | 0068 | 0076 | 0000 | 0000 | 0049 | 210,363 | 0129 |
| Wolbachia COX1 | 0001 | 1 | 1573 | 000 | 0.99 | 000 | 000 | 000 | 000 | 000 | 0.05 | 0.05 | 000 | 000 | 000 |
| Wolbachia WSP | 0000 | 1 | 614 | 000 | 0,00 | 000 | 000 | 0.03 | 0.20 | 0.10 | 0.34 | 0.22 | 000 | 000 | 0.09 |
| Host COX1 | 0067 | 1 | 1506 | 17.16 | 4.22 | 7.33 | 4.86 | 35.81 | 52.82 | 14.94 | 6.43 | 7.79 | 59.54 | 50.42 | 66.99 |
| Number of virus species | – | – | – | 6 | 13 | 9 | 17 | 14 | 12 | 10 | 13 | 8 | 10 | 12 | 17 |
| Virus | Virus Family | Cx. torrentium | Cx. pipiens | Abundant? | More Abundant than Host RNA? | Present in >2 Libraries? | Clusters with Mosquito Viruses? | Mosquito Associated? |
|---|---|---|---|---|---|---|---|---|
| Sindbis virus | Alpha | P | P | Yes | Yes | No | Yes | Yes |
| Nam Dinh virus | Nido | P | P | Yes | Yes | Yes | Yes | Yes |
| Biggie virus | Virga–Negev | P | P | Yes | Yes | Yes | Yes | Yes |
| Negev virus | Virga–Negev | P | P | Yes | Yes | No | Yes | Yes |
| Rinkaby virus | Virga–Negev | NP | P | Yes | Yes | No | No | Yes |
| Kerstinbo virus | Endorna | P | P | Yes | Yes | No | No | Yes |
| Forneby virus | Endorna | P | P | No | Yes | No | No | No |
| Osterfarnebo virus | Endorna | P | P | No | Yes | No | No | No |
| Hallsjon virus | Endorna | P | NP | No | No | No | No | No |
| Tarnsjo virus (variant 1) | Tymo | P | P | No | Yes | No | Yes | Yes |
| Tarnsjo virus (variant 2) | Tymo | P | P | No | Yes | No | Yes | Yes |
| Culex associated luteo like virus | Luteo | NP | P | Yes | Yes | No | Yes | Yes |
| Berrek virus | Luteo | NP | P | No | No | No | Yes | No |
| Fagle virus | Luteo | P | NP | No | No | No | No | No |
| Marma virus | Luteo | NP | P | Yes | Yes | No | Yes | Yes |
| Merida virus | Mononega | P | P | Yes | Yes | Yes | Yes | Yes |
| Culex mononega like virus 2 | Mononega | P | P | Yes | Yes | Yes | Yes | Yes |
| Gysinge virus | Mononega | P | P | Yes | Yes | Yes | No | Yes |
| Culex mosquito virus 4 | Mononega | P | NP | Yes | Yes | No | Yes | Yes |
| Culex mononega like virus 1 | Mononega | NP | P | No | Yes | No | Yes | Yes |
| Valmbacken virus | Reo | P | P | Yes | Yes | Yes | Yes | Yes |
| Jotan virus | Picorna | P | P | Yes | Yes | Yes | Yes | Yes |
| Ista virus | Picorna | P | P | Yes | Yes | Yes | No | Yes |
| Wuhan Mosquito Virus 6 | Orthomyxo | P | P | Yes | Yes | Yes | Yes | Yes |
| Wuhan Mosquito Virus 4 | Orthomyxo | NP | P | Yes | No | No | Yes | Yes |
| Vivastbo virus | Partiti | P | P | Yes | Yes | No | No | Yes |
| Sonnbo virus | Partiti | NP | P | No | Yes | No | No | No |
| Rasbo virus | Bunya | P | NP | No | No | No | No | No |
| Kristianstad virus | Bunya | NP | P | No | No | No | Yes | No |
| Asum virus | Bunya | NP | P | Yes | No | No | No | No |
| Salari virus | Bunya | NP | P | No | No | No | Yes | No |
| Anjon virus | Phasma | P | P | Yes | Yes | Yes | Yes | Yes |
| Gran virus | Qin | P | NP | No | No | No | No | No |
| Nackenback virus | Qin | P | P | Yes | Yes | Yes | No | Yes |
| Vinslov virus | Qin | NP | P | No | No | No | No | No |
| Vittskovle virus | Qin | NP | P | No | No | No | No | No |
| Ahus virus | Toti | NP | P | No | No | No | No | No |
| Osta virus | Toti | NP | P | No | No | No | Yes | No |
| Lindangsbacken virus | Toti | P | NP | No | Yes | No | Yes | Yes |
| Salja virus | Toti | NP | P | No | No | No | Yes | No |
| Eskilstorp virus | Chryso | NP | P | No | Yes | No | Yes | Yes |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pettersson, J.H.-O.; Shi, M.; Eden, J.-S.; Holmes, E.C.; Hesson, J.C. Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe. Viruses 2019, 11, 1033. https://doi.org/10.3390/v11111033
Pettersson JH-O, Shi M, Eden J-S, Holmes EC, Hesson JC. Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe. Viruses. 2019; 11(11):1033. https://doi.org/10.3390/v11111033
Chicago/Turabian StylePettersson, John H.-O., Mang Shi, John-Sebastian Eden, Edward C. Holmes, and Jenny C. Hesson. 2019. "Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex pipiens and Culex torrentium in Northern Europe" Viruses 11, no. 11: 1033. https://doi.org/10.3390/v11111033