Fumonisin B2 Induces Mitochondrial Stress and Mitophagy in Human Embryonic Kidney (Hek293) Cells—A Preliminary Study

 ,
,  and
and
Abstract
:1. Introduction
2. Results
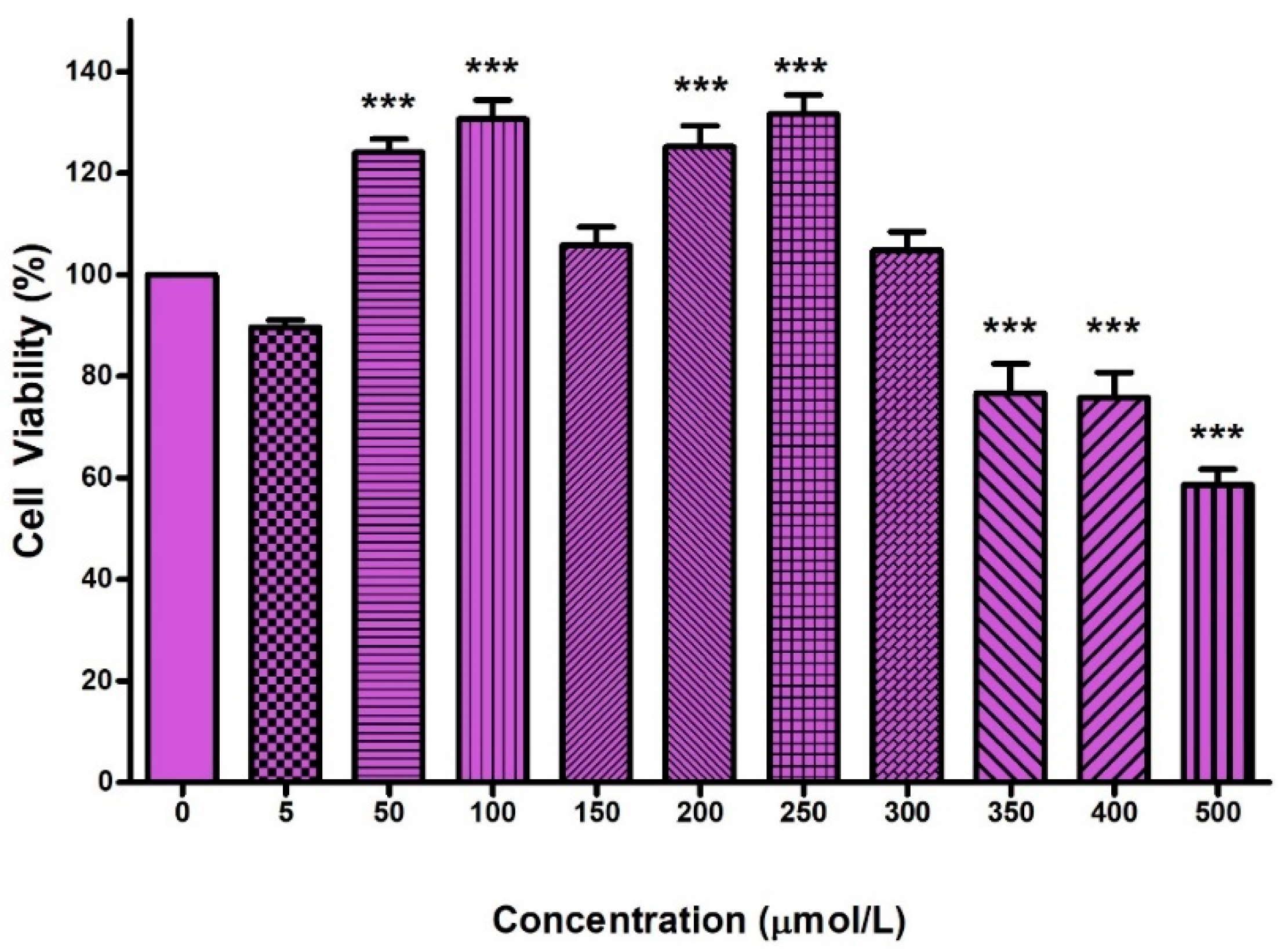
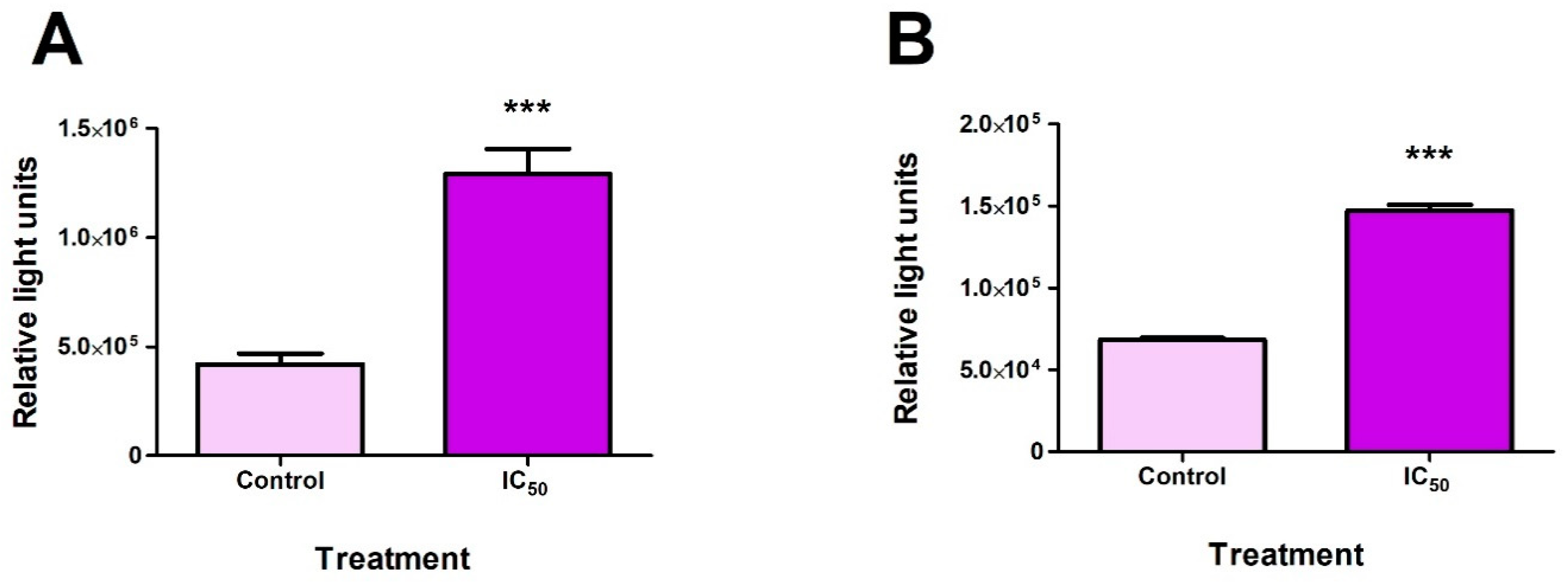
2.1. Cytotoxicity of FB2 in Hek293 Cells
2.2. FB2 Increased ROS Production and Mitochondrial Membrane Depolarisation in Hek293 Cells
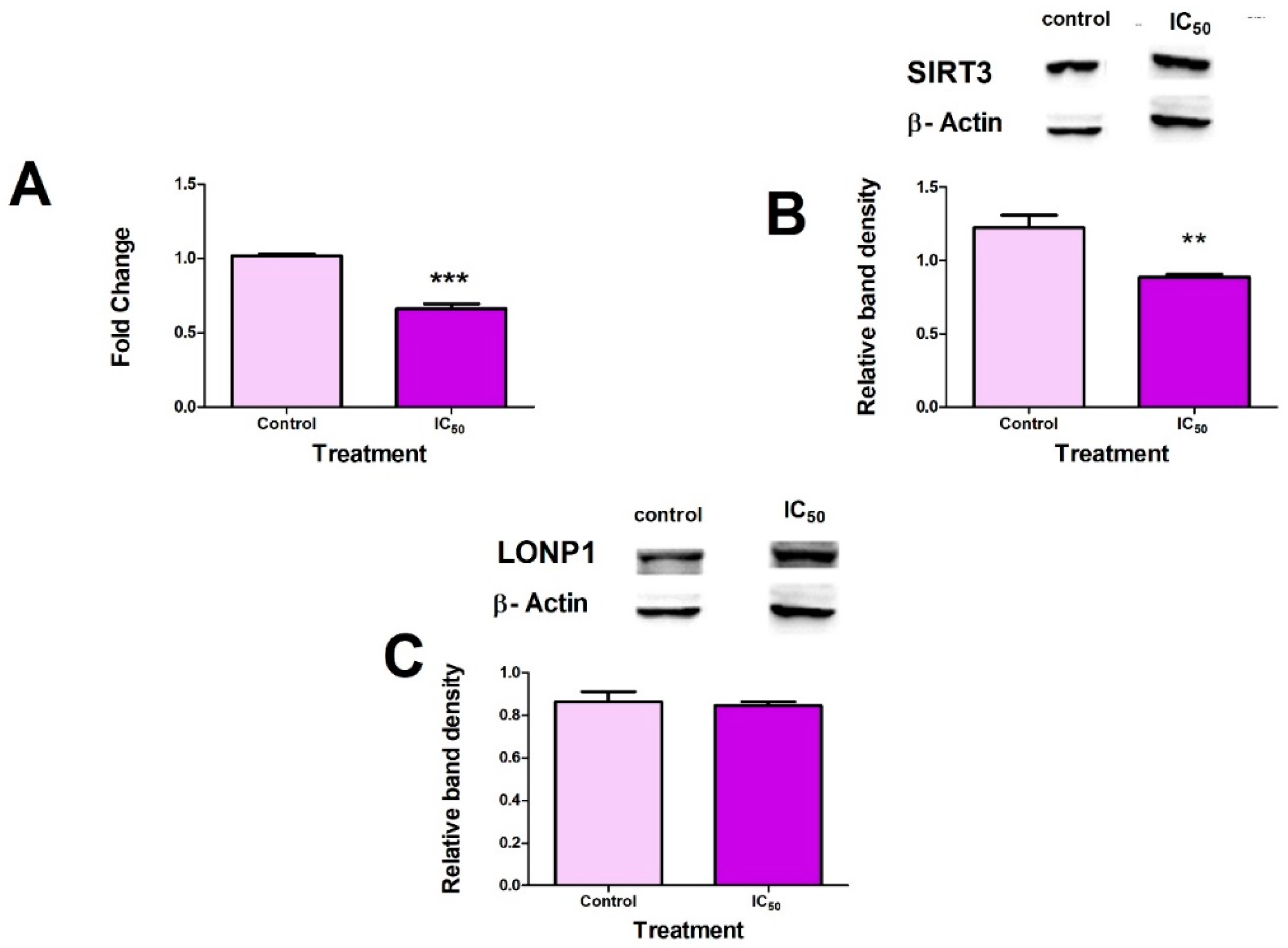
2.3. FB2 Induces Mitochondrial Stress in Hek293 Cells
2.4. FB2 Suppresses Mitochondrial Stress Responses in Hek293 Cells
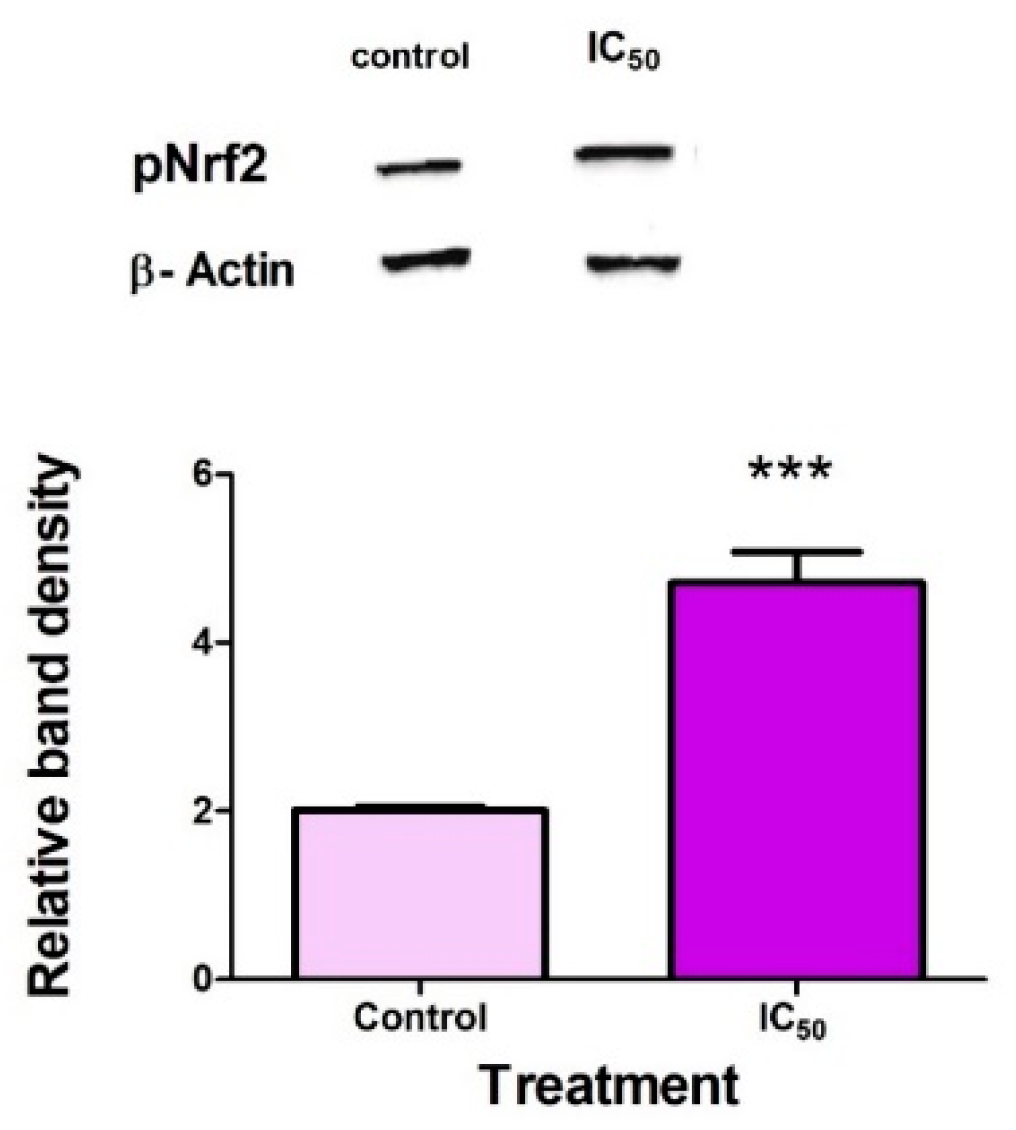
2.5. FB2 Activates Nrf2 in Hek293 Cells
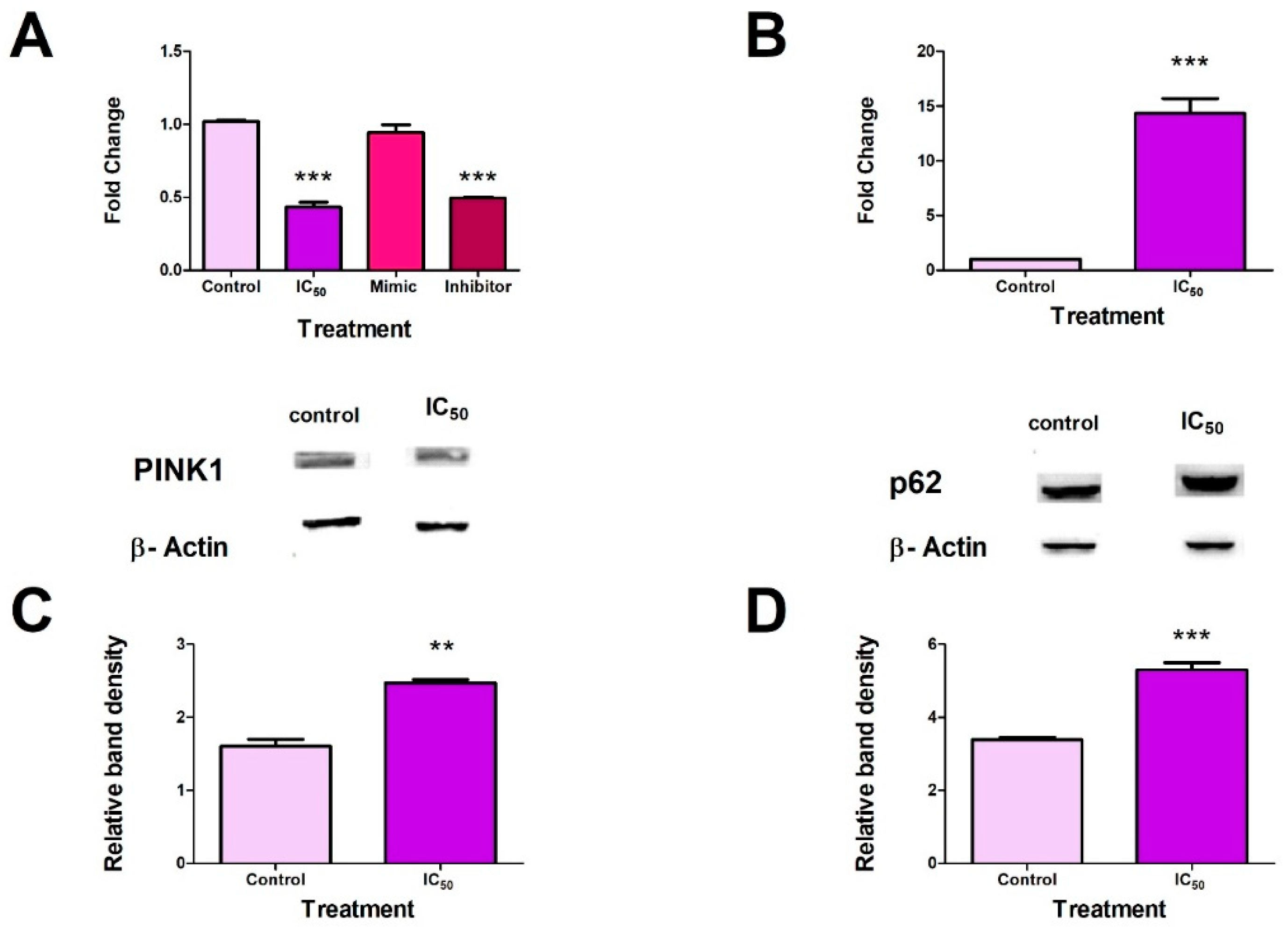
2.6. FB2 induced Mitophagy in Hek293 Cells
3. Discussion
4. Future Recommendations and Limitations
5. Materials and Methods
5.1. Materials
5.2. Cell Culture and Treatment
5.3. Methyl Thiazol Tetrazolium (MTT) Assay
5.4. ATP Assay
5.5. 2′,7′-Dichlorodihydrofluorescein Diacetate (H2DCFDA) Assay
5.6. Mitochondrial Membrane Depolarisation—JC1-Mitoscreen
5.7. Western Blot
5.8. Quantitative PCR
5.8.1. Treatment with the miR-27b Mimic and Inhibitor
5.8.2. RNA Isolation and Quantification
5.8.3. Quantification of mRNA Expression
5.8.4. Quantification of miR-27b Expression
5.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3′-untranslated region | 3′-UTR |
| Dulbecco’s minimum essentials medium | DMEM |
| Electron transport chain | ETC |
| Fumonisin B1 | FB1 |
| Fumonisin B2 | FB2 |
| Heat shock protein 60 | HSP60 |
| Human embryonic kidney cells | Hek293 |
| Lon protease 1 | LONP1 |
| Messenger RNA | mRNA |
| Methylthiazol tetrazolium | MTT |
| Micro-RNA 27b | miR-27b |
| Micro-RNA | miRNA |
| Nuclear factor (erythroid-derived 2)-like 2 | Nrf2 |
| Phosphate buffered saline | PBS |
| Phosphorylated Nrf2 (Ser40) | Nrf2 |
| PTEN-induced putative kinase 1 | PINK1 |
| Quantitative PCR | qPCR |
| Reactive oxygen species | ROS |
| Sirtuin 3 | SIRT3 |
| The half-maximal inhibitory concentration | IC50 |
| Ubiquitin-binding adaptor p62 | p62 |
Appendix A

References
- Ülger, T.G.; Uçar, A.; Çakıroğlu, F.P.; Yilmaz, S. Genotoxic effects of mycotoxins. Toxicon 2020, 185, 104–113. [Google Scholar] [CrossRef]
- Alshannaq, A.; Yu, J.-H. Occurrence, toxicity, and analysis of major mycotoxins in food. Int. J. Environ. Res. Public Health 2017, 14, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockmann-Juvala, H.; Savolainen, K. A review of the toxic effects and mechanisms of action of fumonisin B1. Hum. Exp. Toxicol. 2008, 27, 799–809. [Google Scholar] [CrossRef]
- IARC. Iarc Monographs on the Evaluation of Carcinogenic Risks to cells. World Health Organ. Int. Agency Res. Cancer 2002, 82, 301–366. [Google Scholar]
- Marasas, W.F.O.; Kellerman, T.S.; Gelderblom, W.C.; Thiel, P.; Van der Lugt, J.J.; Coetzer, J.A. Leukoencephalomalacia in a horse induced by fumonisin B₁ isolated from Fusarium moniliforme. Onderstepoort J. Vet. Res. 1988, 55, 197–203. [Google Scholar] [PubMed]
- Colvin, B.M.; Harrison, L.R. Fumonisin-induced pulmonary edema and hydrothorax in swine. Mycopathologia 1992, 117, 79–82. [Google Scholar] [CrossRef]
- Rheeder, J.P.; Marasas, W.F.O.; Thiel, P.G.; Sydenham, E.W.; Shephard, G.S.; Van Schalkwyk, D.J. Fusarium moniliforme and fumonisins in corn in relation to human esophageal cancer in Transkei. Phytopathology 1992, 82, 353–357. [Google Scholar] [CrossRef]
- Missmer, S.A.; Suarez, L.; Felkner, M.; Wang, E.; Merrill, A.H., Jr.; Rothman, K.J.; Hendricks, K.A. Exposure to fumonisins and the occurrence of neural tube defects along the Texas–Mexico border. Environ. Health Perspect. 2005, 114, 237–241. [Google Scholar] [CrossRef]
- Stockmann-Juvala, H.; Mikkola, J.; Naarala, J.; Loikkanen, J.; Elovaara, E.; Savolainen, K. Fumonisin B1-induced toxicity and oxidative damage in U-118MG glioblastoma cells. Toxicology 2004, 202, 173–183. [Google Scholar] [CrossRef]
- Domijan, A.-M.; Abramov, A.Y. Fumonisin B1 inhibits mitochondrial respiration and deregulates calcium homeostasis—Implication to mechanism of cell toxicity. Int. J. Biochem. Cell Biol. 2011, 43, 897–904. [Google Scholar] [CrossRef]
- Chuturgoon, A.; Phulukdaree, A.; Moodley, D. Fumonisin B1 induces global DNA hypomethylation in HepG2 cells–An alternative mechanism of action. Toxicology 2014, 315, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Pillay, Y.; Ghazi, T.; Sheik-Abdul, N.; Nagiah, S.; Chuturgoon, A. Fumonisin B1-induced oxidative stress triggers Nrf2-mediated anti-oxidant response in human hepatocellular carcinoma (HepG2) cells. Mycotoxin Res. 2018, 35, 99–109. [Google Scholar] [CrossRef]
- EFSA; Knutsen, H.K.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B. Appropriateness to set a group health-based guidance value for fumonisins and their modified forms. Efsa J. 2018, 16, e05172. [Google Scholar] [CrossRef] [PubMed]
- Shephard, G.; Marasas, W.; Burger, H.-M.; Somdyala, N.; Rheeder, J.; Van der Westhuizen, L.; Gatyeni, P.; Van Schalkwyk, D. Exposure assessment for fumonisins in the former Transkei region of South Africa. Food Addit. Contam. 2007, 24, 621–629. [Google Scholar] [CrossRef]
- Gelderblom, W.; Cawood, M.; Snyman, S.; Vleggaar, R.; Marasas, W. Structure-activity relationships of fumonisins in short-term carcinogenesis and cytotoxicity assays. Food Chem. Toxicol. 1993, 31, 407–414. [Google Scholar] [CrossRef]
- Riley, R.T.; Showker, J.L.; Owens, D.L.; Ross, P.F. Disruption of sphingolipid metabolism and induction of equine leukoencephalomalacia by Fusarium proliferatum culture material containing fumonisin B2 or B3. Environ. Toxicol. Pharmacol. 1997, 3, 221–228. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.-P.; Hussain, S.N.; Barreiro, E.; Gouspillou, G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int. J. Mol. Sci. 2021, 22, 8179. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9. [Google Scholar] [CrossRef]
- Ngo, J.K.; Davies, K.J. Importance of the lon protease in mitochondrial maintenance and the significance of declining lon in aging. Ann. N. Y. Acad. Sci. 2007, 1119, 78–87. [Google Scholar] [CrossRef]
- Bause, A.S.; Haigis, M.C. SIRT3 regulation of mitochondrial oxidative stress. Exp. Gerontol. 2013, 48, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.-Z.; Song, C.-Q.; Xu, Y.-M.; Wang, R.; Liu, W.; Liu, Z.; Dong, X.-S. Involvement of PINK1/Parkin-mediated mitophagy in paraquat-induced apoptosis in human lung epithelial-like A549 cells. Toxicol. In Vitro 2018, 53, 148–159. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119. [Google Scholar] [CrossRef]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.-H.; Sakaguchi, M. NRF2 regulates PINK1 expression under oxidative stress conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.-X.; Kim, C.; Nelson, P.T.; Springer, W.; Kim, J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Chollate, S.; Jung, M.-Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and inhibitors of NRF2: A review of their potential for clinical development. Oxidative Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef]
- Yin, S.; Guo, X.; Li, J.; Fan, L.; Hu, H. Fumonisin B1 induces autophagic cell death via activation of ERN1-MAPK8/9/10 pathway in monkey kidney MARC-145 cells. Arch. Toxicol. 2016, 90, 985–996. [Google Scholar] [CrossRef]
- Voss, K.A.; Riley, R.T.; Gardner, N.M.; Gelineau-van Waes, J. Fumonisins. Reprod. Dev. Toxicol. 2017, 2, 925–943. [Google Scholar] [CrossRef]
- Ying, W. NAD+ and NADH in cellular functions and cell death. Front. Biosci. 2006, 11, 3129–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological effects of exogenous NAD on mitochondrial bioenergetics, DNA repair, and apoptosis. Mol. Pharmacol. 2011, 80, 1136–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, L.R.; Imai, S.-I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef]
- Mahoney, D.E.; Hiebert, J.B.; Thimmesch, A.; Pierce, J.T.; Vacek, J.L.; Clancy, R.L.; Sauer, A.J.; Pierce, J.D. Understanding d-ribose and mitochondrial function. Adv. Biosci. Clin. Med. 2018, 6, 1–5. [Google Scholar] [CrossRef]
- Lin, S.; Xing, H.; Zang, T.; Ruan, X.; Wo, L.; He, M. Sirtuins in mitochondrial stress: Indispensable helpers behind the scenes. Ageing Res. Rev. 2018, 44, 22–32. [Google Scholar] [CrossRef]
- Gao, J.; Feng, Z.; Wang, X.; Zeng, M.; Liu, J.; Han, S.; Xu, J.; Chen, L.; Cao, K.; Long, J. SIRT3/SOD2 maintains osteoblast differentiation and bone formation by regulating mitochondrial stress. Cell Death Differ. 2018, 25, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Weir, H.J.; Lane, J.D.; Balthasar, N. SIRT3: A central regulator of mitochondrial adaptation in health and disease. Genes Cancer 2013, 4, 118–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Lin, P.; Knoll, E.; Chakrabarti, R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: Computational and experimental studies. PLoS ONE 2014, 9, e107729. [Google Scholar] [CrossRef] [Green Version]
- Pollecker, K.; Sylvester, M.; Voos, W. Proteomic analysis demonstrates the role of the quality control protease LONP1 in mitochondrial protein aggregation. J. Biol. Chem. 2021, 297, 101134. [Google Scholar] [CrossRef]
- Bota, D.A.; Ngo, J.K.; Davies, K.J. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Gibellini, L.; Pinti, M.; Beretti, F.; Pierri, C.L.; Onofrio, A.; Riccio, M.; Carnevale, G.; De Biasi, S.; Nasi, M.; Torelli, F. Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 2014, 18, 76–81. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.-C.; Tian, H.-X.; Yi, T.; Chen, H.-B. The critical roles of mitophagy in cerebral ischemia. Protein Cell 2016, 7, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Zhang, Y.; Xu, W.; Luo, Y.; Hao, J.; Shen, X.L.; Yang, X.; Li, X.; Huang, K. Zinc protects HepG2 cells against the oxidative damage and DNA damage induced by ochratoxin A. Toxicol. Appl. Pharmacol. 2013, 268, 123–131. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′-3′) | Annealing Temperature (°C) | |
|---|---|---|---|
| SIRT3 | Sense | GAGCGGCCTCTACAGCAAC | 60 |
| Anti-sense | GAGTAGTGAGTGACATTGGG | ||
| PINK1 | Sense | AAGCGAGGCTTTCCCCTAC | 56 |
| Anti-sense | GCACTACATTGACCACCGATTT | ||
| GAPDH | Sense | TCCACCACCCTGTTGCTGTA | - |
| Anti-sense | ACCACAGTCCATGCCATCAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, J.; Sheik Abdul, N.; Nagiah, S.; Ghazi, T.; Chuturgoon, A.A. Fumonisin B2 Induces Mitochondrial Stress and Mitophagy in Human Embryonic Kidney (Hek293) Cells—A Preliminary Study. Toxins 2022, 14, 171. https://doi.org/10.3390/toxins14030171
Mohan J, Sheik Abdul N, Nagiah S, Ghazi T, Chuturgoon AA. Fumonisin B2 Induces Mitochondrial Stress and Mitophagy in Human Embryonic Kidney (Hek293) Cells—A Preliminary Study. Toxins. 2022; 14(3):171. https://doi.org/10.3390/toxins14030171
Chicago/Turabian StyleMohan, Jivanka, Naeem Sheik Abdul, Savania Nagiah, Terisha Ghazi, and Anil A. Chuturgoon. 2022. "Fumonisin B2 Induces Mitochondrial Stress and Mitophagy in Human Embryonic Kidney (Hek293) Cells—A Preliminary Study" Toxins 14, no. 3: 171. https://doi.org/10.3390/toxins14030171