Analysis of α- and β-amanitin in Human Plasma at Subnanogram per Milliliter Levels by Reversed Phase Ultra-High Performance Liquid Chromatography Coupled to Orbitrap Mass Spectrometry


Abstract
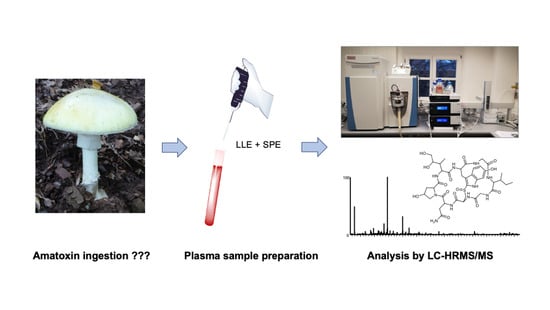
:
1. Introduction
2. Results
2.1. Method Development
2.2. Method Validation
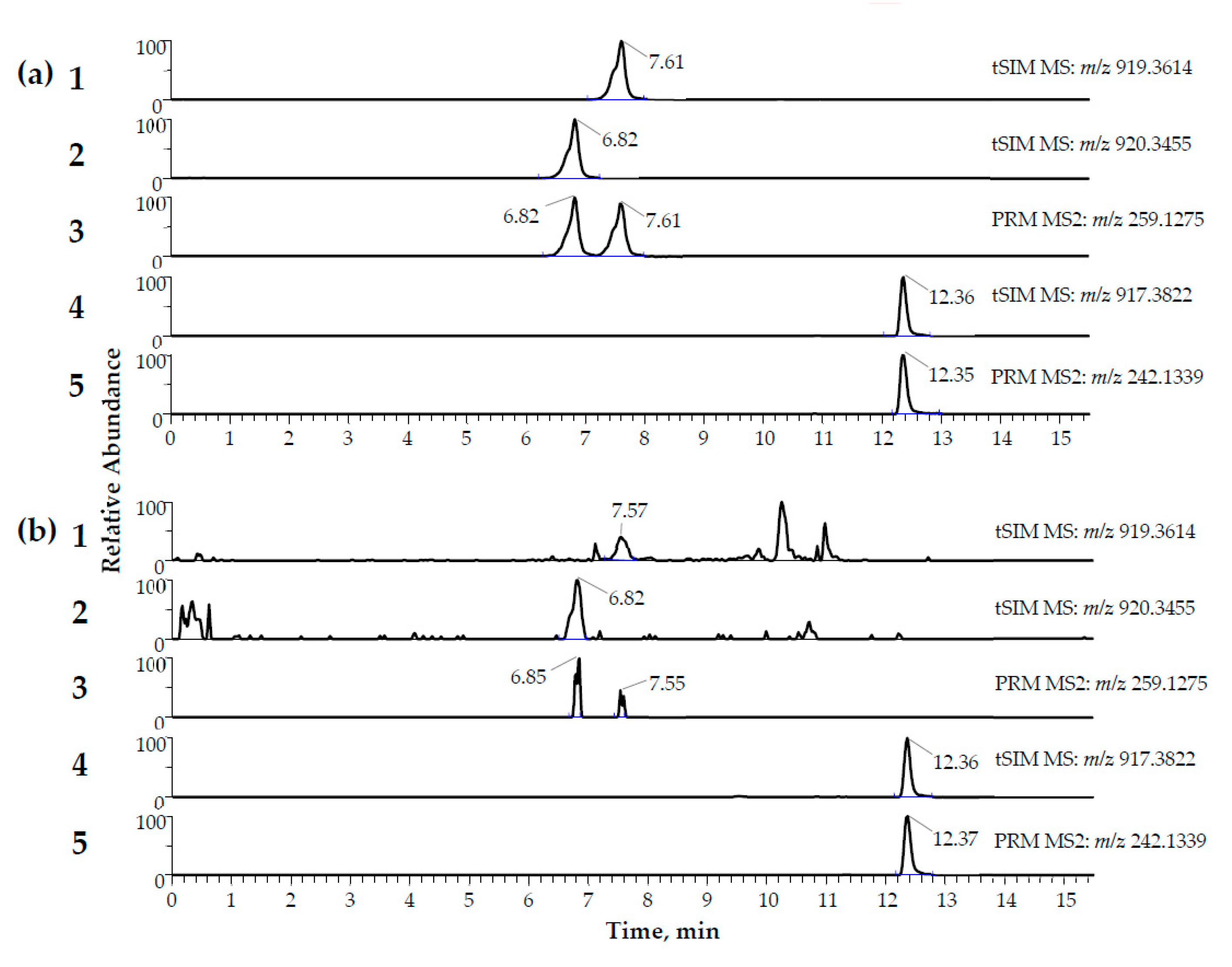
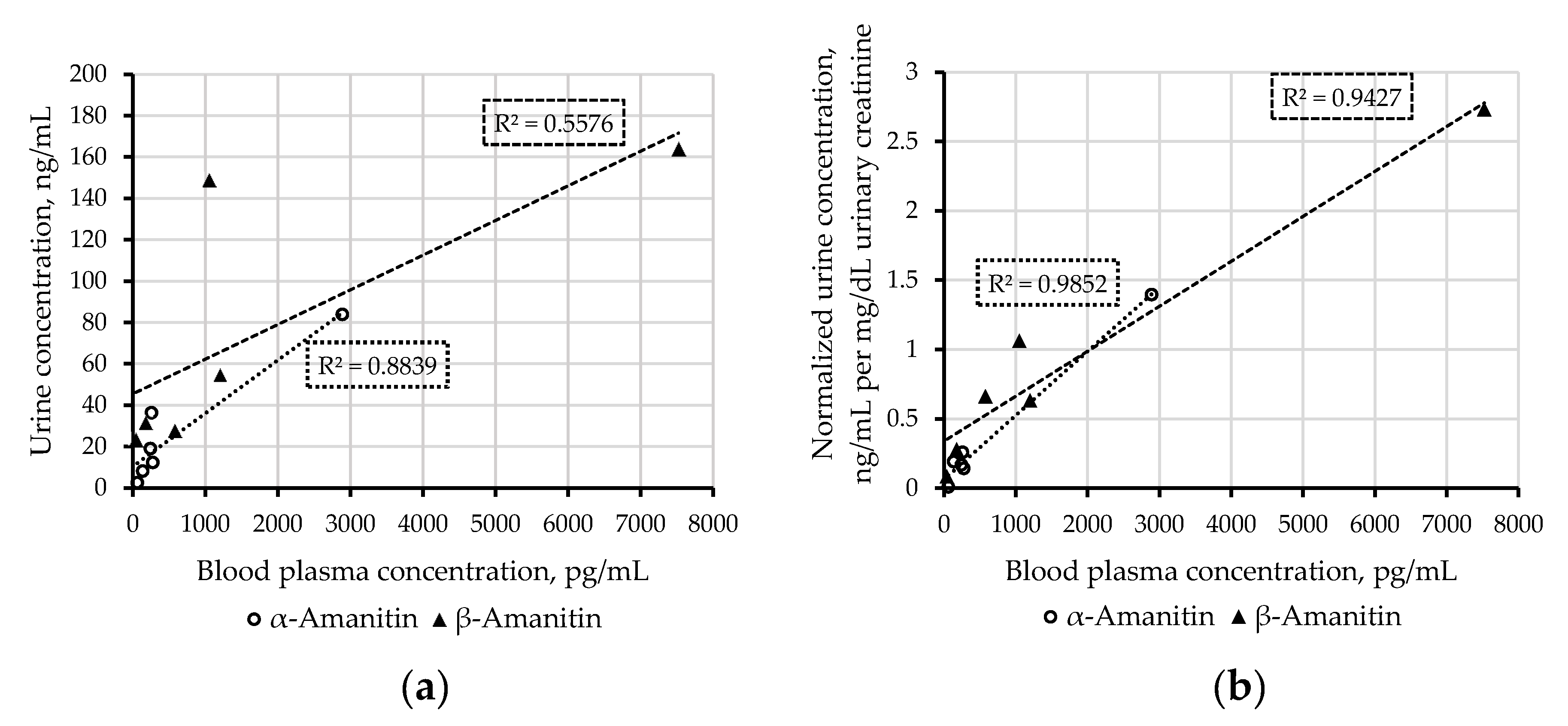
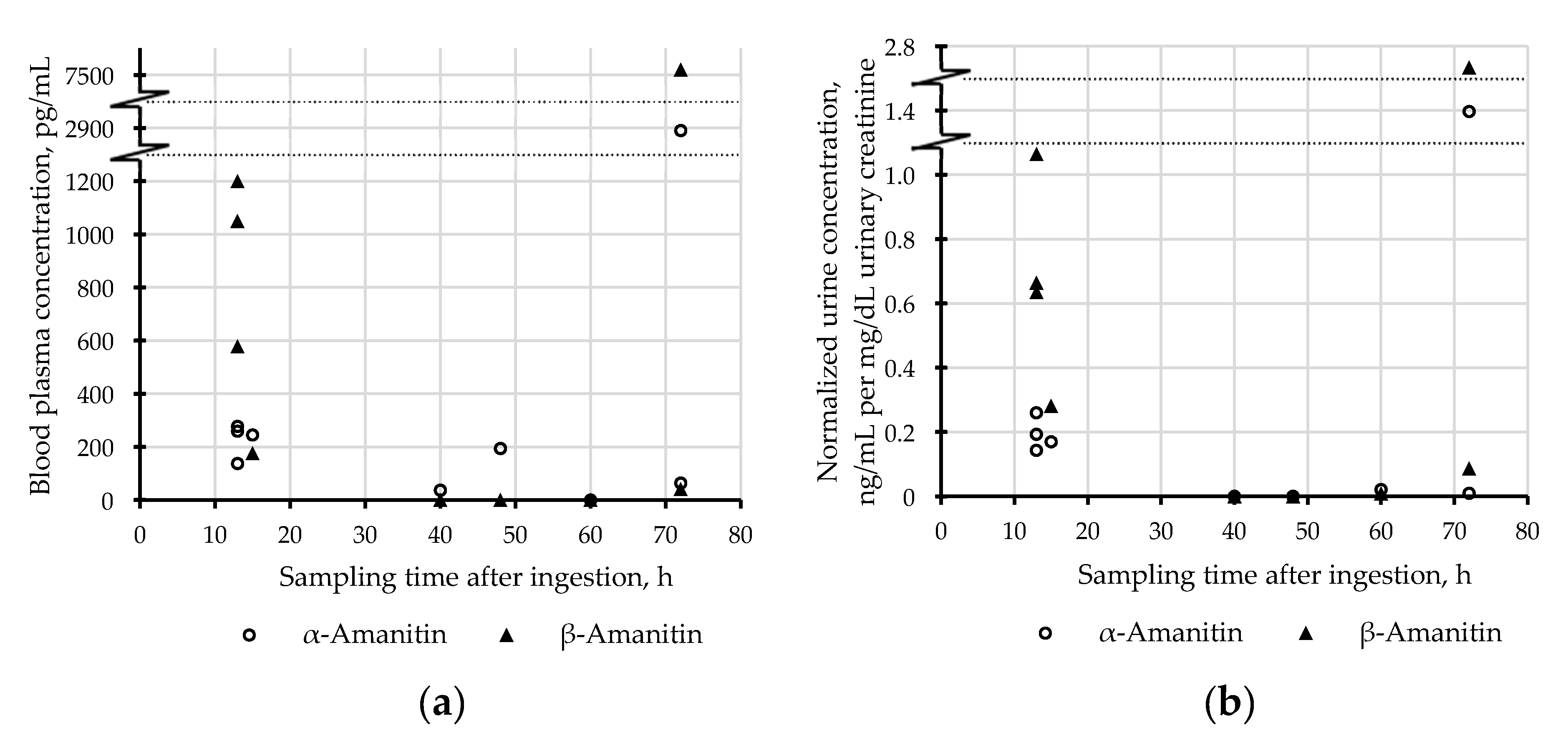
2.3. Applicability
3. Discussion
3.1. Method Development
3.2. Method Validation
3.3. Applicability
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Stock Solutions for Calibration and Control Samples
5.3. LC-HRMS/MS Apparatus
5.4. Chromatography Setup
5.5. Analysis Raw Data Handling and Processing
5.6. Sample Preparation
5.6.1. A-LLE
5.6.2. SPE
5.7. Method Validation
5.8. Applicability
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Persson, H. Mushrooms. Medicine 2016, 44, 116–119. [Google Scholar] [CrossRef]
- Gonmori, K.; Fujita, H.; Yokoyama, K.; Watanabe, K.; Suzuki, O. Mushroom toxins: A forensic toxicological review. Forensic Toxicol. 2011, 29, 85–94. [Google Scholar] [CrossRef]
- Brandenburg, W.E.; Ward, K.J. Mushroom poisoning epidemiology in the United States. Mycologia 2018, 110, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Govorushko, S.; Rezaee, R.; Dumanov, J.; Tsatsakis, A. Poisoning associated with the use of mushrooms: A review of the global pattern and main characteristics. Food Chem. Toxicol. 2019, 128, 267–279. [Google Scholar] [CrossRef] [PubMed]
- White, J.; Weinstein, S.A.; De Haro, L.; Bedry, R.; Schaper, A.; Rumack, B.H.; Zilker, T. Mushroom poisoning: A proposed new clinical classification. Toxicon 2019, 157, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Poucheret, P.; Fons, F.; Dore, J.C.; Michelot, D.; Rapior, S. Amatoxin poisoning treatment decision-making: Pharmaco-therapeutic clinical strategy assessment using multidimensional multivariate statistic analysis. Toxicon 2010, 55, 1338–1345. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Gao, Y.; Walline, J.H.; Lu, X.; Yu, S.; Zhao, L.; Ge, Z.; Li, Y. N-acetylcysteine as a treatment for amatoxin poisoning: A systematic review. Clin. Toxicol. 2020, 58, 1–8. [Google Scholar] [CrossRef]
- De Olano, J.; Wang, J.J.; Villeneuve, E.; Gosselin, S.; Biary, R.; Su, M.K.; Hoffman, R.S. Current fatality rate of suspected cyclopeptide mushroom poisoning in the United States. Clin. Toxicol. 2020, 1–4. [Google Scholar] [CrossRef]
- Diaz, J.H. Amatoxin-containing mushroom poisonings: Species, toxidromes, treatments, and outcomes. Wilderness Environ. Med. 2018, 29, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, P.; Zhang, Z. Investigation and analysis of 102 mushroom poisoning cases in Southern China from 1994 to 2012. Fungal Divers. 2013, 64, 123–131. [Google Scholar] [CrossRef]
- Tang, S.; Zhou, Q.; He, Z.; Luo, T.; Zhang, P.; Cai, Q.; Yang, Z.; Chen, J.; Chen, Z. Cyclopeptide toxins of lethal amanitas: Compositions, distribution and phylogenetic implication. Toxicon 2016, 120, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Cui, Y.Y.; Yang, Z.L. Lethal Amanita species in China. Mycologia 2016, 108, 993–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilker, T.; Faulstich, H. Cyclopeptide-Containing Mushrooms: The Deadly Amanitas. In Critical Care Toxicology: Diagnosis and Management of the Critically Poisoned Patient, 2nd ed.; Brent, J., Burkhart, K., Dargan, P., Hatten, B., Megarbane, B., Palmer, R., White, J., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 2129–2148. [Google Scholar] [CrossRef]
- Karlson-Stiber, C.; Persson, H. Cytotoxic fungi—An overview. Toxicon 2003, 42, 339–349. [Google Scholar] [CrossRef]
- Enjalbert, F.; Rapior, S.; Nouguier-Soule, J.; Guillon, S.; Amouroux, N.; Cabot, C. Treatment of Amatoxin poisoning: 20-year retrospective analysis. J. Toxicol. Clin. Toxicol. 2002, 40, 715–757. [Google Scholar] [CrossRef]
- Hallen, H.E.; Watling, R.; Adams, G.C. Taxonomy and toxicity of Conocybe lactea and related species. Mycol. Res. 2003, 107, 969–979. [Google Scholar] [CrossRef]
- Jahn, W.; Faulstich, H.; Wieland, T. Pharmacokinetics of [3H]methyl-dehydroxymethyl-amanitin in the Isolated Perfused Rat Liver, and the Influence of Several Drugs. In Amanita Toxins and Poisonings; Faulstich, H., Kommerell, B., Wieland, T., Eds.; Verlag Gerhard Witzstrock: Baden-Baden, Köln, Germany; New York, NY, USA, 1980; pp. 79–85. [Google Scholar]
- Bushnell, D.A.; Cramer, P.; Kornberg, R.D. Structural basis of transcription: Alpha-amanitin-RNA polymerase II cocrystal at 2.8 A resolution. Proc. Natl. Acad. Sci. USA 2002, 99, 1218–1222. [Google Scholar] [CrossRef] [Green Version]
- Letschert, K.; Faulstich, H.; Keller, D.; Keppler, D. Molecular characterization and inhibition of amanitin uptake into human hepatocytes. Toxicol. Sci. 2006, 91, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk-Jaeger, K.M.; Rauber-Luthy, C.; Bodmer, M.; Kupferschmidt, H.; Kullak-Ublick, G.A.; Ceschi, A. Mushroom poisoning: A study on circumstances of exposure and patterns of toxicity. Eur. J. Intern. Med. 2012, 23, e85–e91. [Google Scholar] [CrossRef] [PubMed]
- Gawlikowski, T.; Romek, M.; Satora, L. Edible mushroom-related poisoning: A study on circumstances of mushroom collection, transport, and storage. Hum. Exp. Toxicol. 2015, 34, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Costa, V.M.; Carvalho, A.; Baptista, P.; de Pinho, P.G.; de Lourdes Bastos, M.; Carvalho, F. Amanita phalloides poisoning: Mechanisms of toxicity and treatment. Food Chem. Toxicol. 2015, 86, 41–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganzert, M.; Felgenhauer, N.; Schuster, T.; Eyer, F.; Gourdin, C.; Zilker, T. Amanita poisoning—comparison of silibinin with a combination of silibinin and penicillin. Dtsch. Med. Wochenschr. 2008, 133, 2261–2267. [Google Scholar] [CrossRef] [PubMed]
- Saller, R.; Meier, R.; Brignoli, R. The use of silymarin in the treatment of liver diseases. Drugs 2001, 61, 2035–2063. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Liu, Z. Management of Amanita phalloides poisoning: A literature review and update. J. Crit. Care 2018, 46, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, A.; Jehl, F.; Flesch, F.; Sauder, P.; Kopferschmitt, J. Kinetics of amatoxins in human poisoning: Therapeutic implications. J. Toxicol. Clin. Toxicol. 1993, 31, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Butera, R.; Locatelli, C.; Coccini, T.; Manzo, L. Diagnostic accuracy of urinary amanitin in suspected mushroom poisoning: A pilot study. J. Toxicol. Clin. Toxicol. 2004, 42, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Freitas, A.; Azul, A.M.; Barbosa, J.; Costa, S.; Ramos, F. Development, optimization and application of an analytical methodology by ultra performance liquid chromatography-tandem mass spectrometry for determination of amanitins in urine and liver samples. Anal. Chim. Acta 2013, 799, 77–87. [Google Scholar] [CrossRef]
- Abbott, N.L.; Hill, K.L.; Garrett, A.; Carter, M.D.; Hamelin, E.I.; Johnson, R.C. Detection of α-, β-, and γ-amanitin in urine by LC-MS/MS using 15N10-α-amanitin as the internal standard. Toxicon 2018, 152, 71–77. [Google Scholar] [CrossRef]
- Bambauer, T.P.; Wagmann, L.; Maurer, H.H.; Weber, A.A.; Meyer, M.R. Development and application of a strategy for analyzing eight biomarkers in human urine to verify toxic mushroom or ricinus communis ingestions by means of hydrophilic interaction LC coupled to HRMS/MS. Talanta 2020, 213, 120847. [Google Scholar] [CrossRef]
- Robinson-Fuentes, V.A.; Jaime-Sanchez, J.L.; Garcia-Aguilar, L.; Gomez-Peralta, M.; Vazquez-Garciduenas, M.S.; Vazquez-Marrufo, G. Determination of α-and β-amanitin in clinical urine samples by Capillary Zone Electrophoresis. J. Pharm. Biomed. Anal. 2008, 47, 913–917. [Google Scholar] [CrossRef]
- Garcia, J.; Costa, V.M.; Baptista, P.; de Bastos, M.L.; Carvalho, F. Quantification of alpha-amanitin in biological samples by HPLC using simultaneous UV- diode array and electrochemical detection. J. Chromatogr. B 2015, 997, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhao, Y.; Li, H.; Zhou, S.; Chen, D.; Zhang, Y.; Yao, Q.; Sun, C. A simple and high-throughput analysis of amatoxins and phallotoxins in human plasma, serum and urine using UPLC-MS/MS combined with PRiME HLB μElution platform. Toxins 2016, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonderschmitt, D.J.; Gautschi, K.; Frei, W.; Andres, R.Y. Radioimmunoassay for amatoxins by use of a rapid, 125I-tracer-based system. Clin. Chem. 1986, 32, 1751–1755. [Google Scholar] [CrossRef]
- Maurer, H.H.; Schmitt, C.J.; Weber, A.A.; Kraemer, T. Validated electrospray liquid chromatographic-mass spectrometric assay for the determination of the mushroom toxins alpha- and beta-amanitin in urine after immunoaffinity extraction. J. Chromatogr. B Biomed. Sci. Appl. 2000, 748, 125–135. [Google Scholar] [CrossRef]
- Abuknesha, R.A.; Maragkou, A. A highly sensitive and specific enzyme immunoassay for detection of β-amanitin in biological fluids. Anal. Bioanal. Chem. 2004, 379, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Bever, C.S.; Swanson, K.D.; Hamelin, E.I.; Filigenzi, M.; Poppenga, R.H.; Kaae, J.; Cheng, L.W.; Stanker, L.H. Rapid, sensitive, and accurate point-of-care detection of lethal amatoxins in urine. Toxins 2020, 12, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieck, W.; Platt, D. High-performance liquid chromatographic method for the determination of α-amanitin and phalloidin in human plasma using the column-switching technique and its application in suspected cases of poisoning by the green species of amanita mushroom (Amanita phalloides). J. Chromatogr. B Biomed. Sci. Appl. 1988, 425, 121–134. [Google Scholar] [CrossRef]
- Nomura, M.; Suzuki, Y.; Kaneko, R.; Ogawa, T.; Hattori, H.; Seno, H.; Ishii, A. Simple and rapid analysis of amatoxins using UPLC–MS–MS. Forensic Toxicol. 2012, 30, 185–192. [Google Scholar] [CrossRef]
- Tanahashi, M.; Kaneko, R.; Hirata, Y.; Hamajima, M.; Arinobu, T.; Ogawa, T.; Ishii, A. Simple analysis of α-amanitin and β-amanitin in human plasma by liquid chromatography-mass spectrometry. Forensic Toxicol. 2010, 28, 110–114. [Google Scholar] [CrossRef]
- Filigenzi, M.S.; Poppenga, R.H.; Tiwary, A.K.; Puschner, B. Determination of α-amanitin in serum and liver by multistage linear ion trap mass spectrometry. J. Agric. Food Chem. 2007, 55, 2784–2790. [Google Scholar] [CrossRef]
- Helfer, A.G.; Meyer, M.R.; Michely, J.A.; Maurer, H.H. Direct analysis of the mushroom poisons alpha- and beta-amanitin in human urine using a novel on-line turbulent flow chromatography mode coupled to liquid chromatography-high resolution-mass spectrometry/mass spectrometry. J. Chromatogr. A 2014, 1325, 92–98. [Google Scholar] [CrossRef]
- Tomkova, J.; Ondra, P.; Valka, I. Simultaneous determination of mushroom toxins α-amanitin, β-amanitin and muscarine in human urine by solid-phase extraction and ultra-high-performance liquid chromatography coupled with ultra-high-resolution TOF mass spectrometry. Forensic Sci. Int. 2015, 251, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Sgambelluri, R.M.; Epis, S.; Sassera, D.; Luo, H.; Angelos, E.R.; Walton, J.D. Profiling of amatoxins and phallotoxins in the genus Lepiota by liquid chromatography combined with UV absorbance and mass spectrometry. Toxins 2014, 6, 2336–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köppel, C. Clinical symptomatology and management of mushroom poisoning. Toxicon 1993, 31, 1513–1540. [Google Scholar] [CrossRef]
- Busi, C.; Flume, L.; Constantino, D.; Borroni, M.; Ambrosino, G.; Olivotto, A.; Bernardini, D. Détermination des amanitines dans le sérum de patients intoxiqués par l’amanite phalloïde [The determination of amanitines in the serum of patients poisoned by Amanita phalloides (author’s transl)]. Nouv. Presse Med. 1977, 6, 2855–2857. [Google Scholar] [PubMed]
- Luo, H.; DuBois, B.; Sgambelluri, R.M.; Angelos, E.R.; Li, X.; Holmes, D.; Walton, J.D. Production of 15N-labeled α-amanitin in Galerina marginata. Toxicon 2015, 103, 60–64. [Google Scholar] [CrossRef] [Green Version]
- Maurer, H.H.; Kraemer, T.; Ledvinka, O.; Schmitt, C.J.; Weber, A.A. Gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) in toxicological analysis. Studies on the detection of clobenzorex and its metabolites within a systematic toxicological analysis procedure by GC-MS and by immunoassay and studies on the detection of α- and β-amanitin in urine by atmospheric pressure ionization electrospray LC-MS. J. Chromatogr. B Biomed. Sci. Appl. 1997, 689, 81–89. [Google Scholar] [CrossRef]
- ICH. M10 Guideline on Bioanalytical Method Validation Step 2b. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf (accessed on 5 August 2020).
- Li, C.; Wei, F.; Muhammad, S.; Yang, G.; Wang, S.; Liu, X. A cost-effective LC-MS/MS method for identification and quantification of alpha-amanitin in rat plasma: Application to toxicokinetic study. J. Chromatogr. B 2017, 1064, 36–39. [Google Scholar] [CrossRef]
- Jehl, F.; Gallion, C.; Birckel, P.; Jaeger, A.; Flesch, F.; Minck, R. Determination of α-amanitin and β-amanitin in human biological fluids by high-performance liquid chromatography. Anal. Biochem. 1985, 149, 35–42. [Google Scholar] [CrossRef]
- Helfer, A.G.; Michely, J.A.; Weber, A.A.; Meyer, M.R.; Maurer, H.H. Orbitrap technology for comprehensive metabolite-based liquid chromatographic-high resolution-tandem mass spectrometric urine drug screening - exemplified for cardiovascular drugs. Anal. Chim. Acta 2015, 891, 221–233. [Google Scholar] [CrossRef]
- Helfer, A.G.; Michely, J.A.; Weber, A.A.; Meyer, M.R.; Maurer, H.H. Liquid chromatography-high resolution-tandem mass spectrometry using Orbitrap technology for comprehensive screening to detect drugs and their metabolites in blood plasma. Anal. Chim. Acta 2017, 965, 83–95. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Time, min | Duration, sec | Flow Rate, mL/min | Eluent Ratio | |
|---|---|---|---|---|---|
| A a | B b | ||||
| 1 | 0.00 | 60 | 0.30 | 99.0 | 1.0 |
| 2 | 1.00 | 120 | 0.30 | 99.0 | 1.0 |
| 3 | 3.00 | 420 | 0.20 | 85.0 | 15.0 |
| 4 | 10.00 | 150 | 0.20 | 70.0 | 30.0 |
| 5 | 12.50 | 90 | 0.20 | 1.0 | 99.0 |
| 6 | 14.00 | 0 | 0.30 | 1.0 | 99.0 |
| 7 | 14.00 | 89 | 0.30 | 99.0 | 1.0 |
| 8 | 15.48 | 1 | 0.30 | 99.0 | 1.0 |
| 9 | 15.50 | 0 | 0.05 | 99.0 | 1.0 |
| Intra-Day (n = 5) | Nominal Conc., pg/mL | Calculated Conc., pg/mL | Accuracy, % | Precision, % | ||||
|---|---|---|---|---|---|---|---|---|
| α | β | α | β | α | β | α | β | |
| QC1 (LLOQ) | 20 | 20 | 19.5 ± 2.17 | 19.1 ± 2.22 | −2.7 | −4.5 | 11 | 12 |
| QC2 (low) | 60 | 60 | 62.0 ± 5,76 | 53.0 ± 3.89 | 3.4 | −12 | 9.3 | 7.3 |
| QC3 (mid) | 400 | 400 | 421 ± 28.4 | 411 ± 27.4 | 5.3 | 2.8 | 6.7 | 6.7 |
| QC4 (high) | 1600 | 1600 | 1510 ± 23.4 | 1570 ± 15.5 | −5.4 | −1.6 | 1.6 | 1.0 |
| QC Dil F2 a | 1000*2 | 1000*2 | 2140 ± 81.3 | 2065 ± 103 | 6.8 | 3.2 | 3.8 | 5.0 |
| QC Dil F50 b | 40*50 | 40*50 | 1940 ± 130 | 1757 ± 157 | −3.1 | −12 | 6.7 | 8.9 |
| Intra-Day (n = 5), 3 runs | Nominal conc., pg/mL | Calculated conc., pg/mL | Accuracy, % | Precision, % | ||||
|---|---|---|---|---|---|---|---|---|
| α | β | α | β | α | β | α | β | |
| QC1 (LLOQ) | 20 | 20 | 21.0 ± 2.10 | 19.9 ± 2.72 | 4.9 | −0.4 | 10 | 14 |
| QC2 (low) | 60 | 60 | 64.2 ± 4.92 | 55.8 ± 5.60 | 6.9 | −7.0 | 7.7 | 10 |
| QC3 (mid) | 400 | 400 | 409 ± 24.2 | 401 ± 25.0 | 2.2 | 0.3 | 5.9 | 6.2 |
| QC4 (high) | 1600 | 1600 | 1514 ± 111 | 1535 ± 107 | −5.4 | −4.1 | 7.4 | 7.1 |
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Amounts of mushrooms ingested | 3–4 | 2–3 | 2–3 | unk. | 3 | 1 cap | unk. | unk. | unk. | |
| Sampling time after intake a, h | 13 | 13 | 13 | 40 | 48 | 15 | 48-72 | 72 | 72 | |
| Blood plasma concentration, pg/mL | α-Amanitin | 277 | 259 | 137 | 37 | 194 b | 245 | <40 b | 63 | 2890 |
| β-Amanitin | 1200 | 1050 | 578 | <20 | n.i. b | 176 | n.i. b | 41 | 7520 | |
| Urinary concentration, ng/mL | α-Amanitin | 12 | 36 | 8.1 | n.i. | <1 | 19 | 4.6 | 2.5 | 24 |
| β-Amanitin | 55 | 149 | 28 | n.i. | <1 | 32 | 2.0 | 23 | 164 | |
| Urinary creatinine, mg/dL | 86 | 140 | 42 | 169 | 232 | 112 | 220 | 268 | 60 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bambauer, T.P.; Wagmann, L.; Weber, A.A.; Meyer, M.R. Analysis of α- and β-amanitin in Human Plasma at Subnanogram per Milliliter Levels by Reversed Phase Ultra-High Performance Liquid Chromatography Coupled to Orbitrap Mass Spectrometry. Toxins 2020, 12, 671. https://doi.org/10.3390/toxins12110671
Bambauer TP, Wagmann L, Weber AA, Meyer MR. Analysis of α- and β-amanitin in Human Plasma at Subnanogram per Milliliter Levels by Reversed Phase Ultra-High Performance Liquid Chromatography Coupled to Orbitrap Mass Spectrometry. Toxins. 2020; 12(11):671. https://doi.org/10.3390/toxins12110671
Chicago/Turabian StyleBambauer, Thomas P., Lea Wagmann, Armin A. Weber, and Markus R. Meyer. 2020. "Analysis of α- and β-amanitin in Human Plasma at Subnanogram per Milliliter Levels by Reversed Phase Ultra-High Performance Liquid Chromatography Coupled to Orbitrap Mass Spectrometry" Toxins 12, no. 11: 671. https://doi.org/10.3390/toxins12110671