A Screening Tool for the Direct Analysis of Marine and Freshwater Phycotoxins in Organic SPATT Extracts from the Chesapeake Bay


Abstract
:1. Introduction
2. Results and Discussion
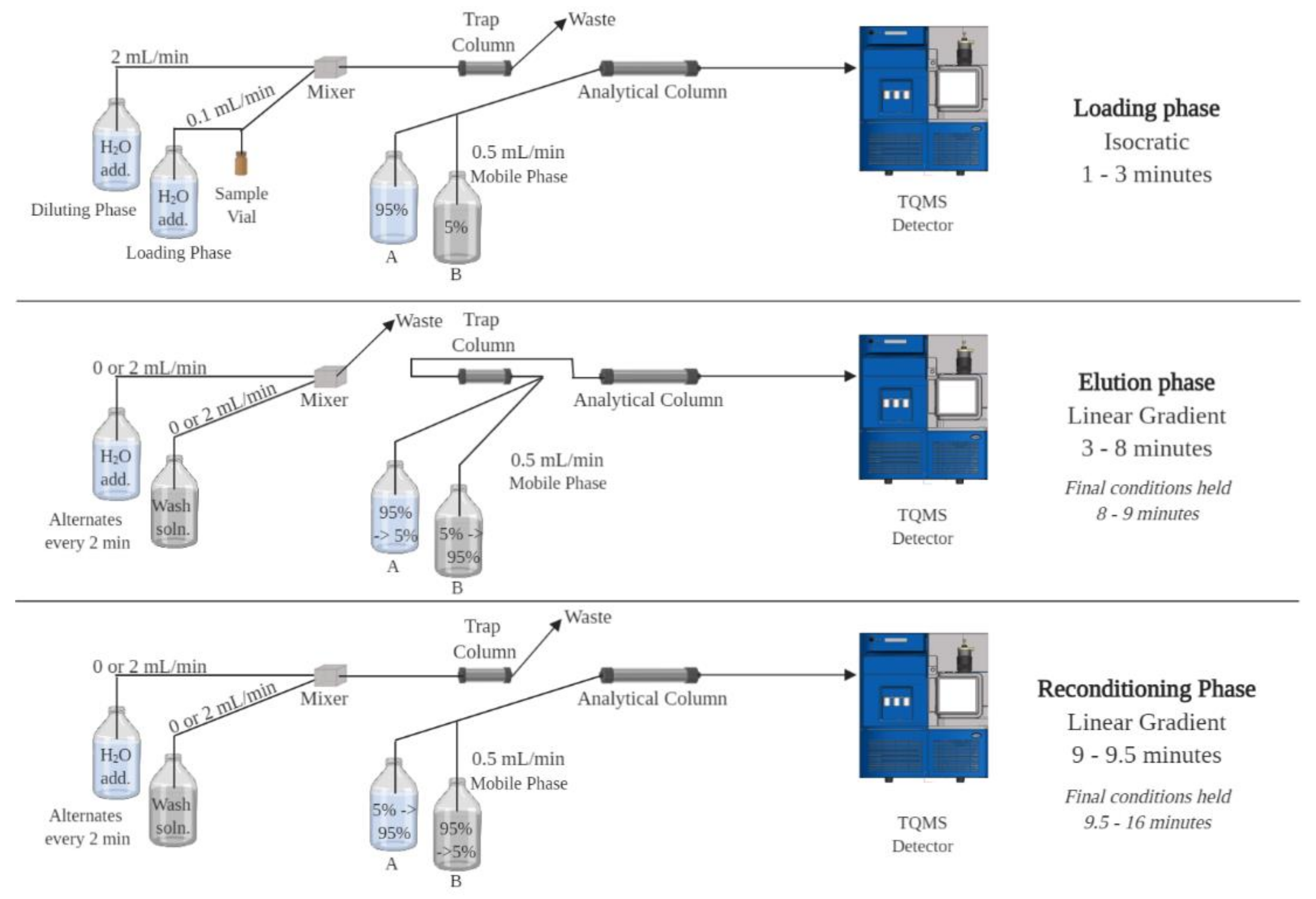
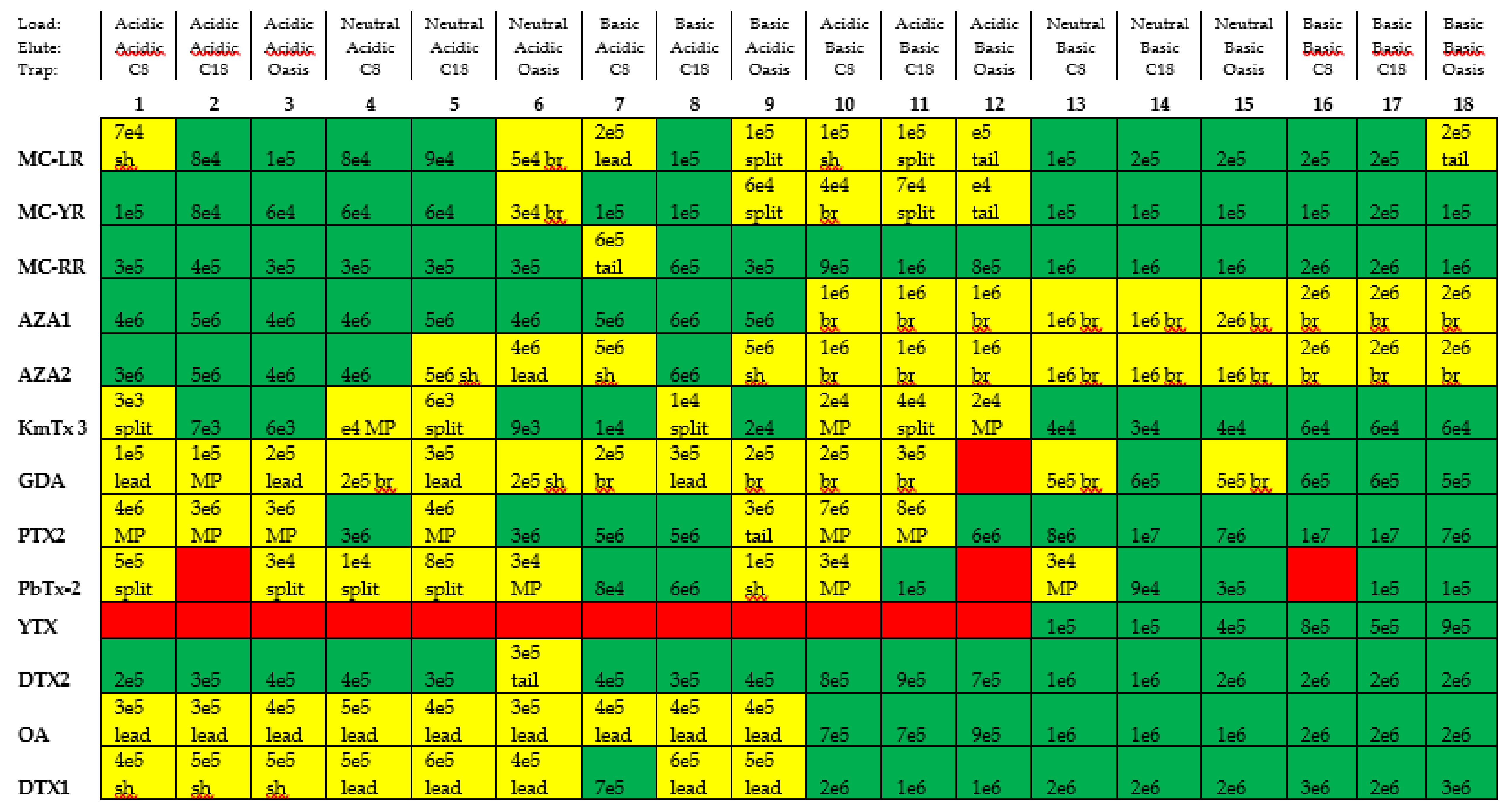
2.1. Chromatographic Method Development
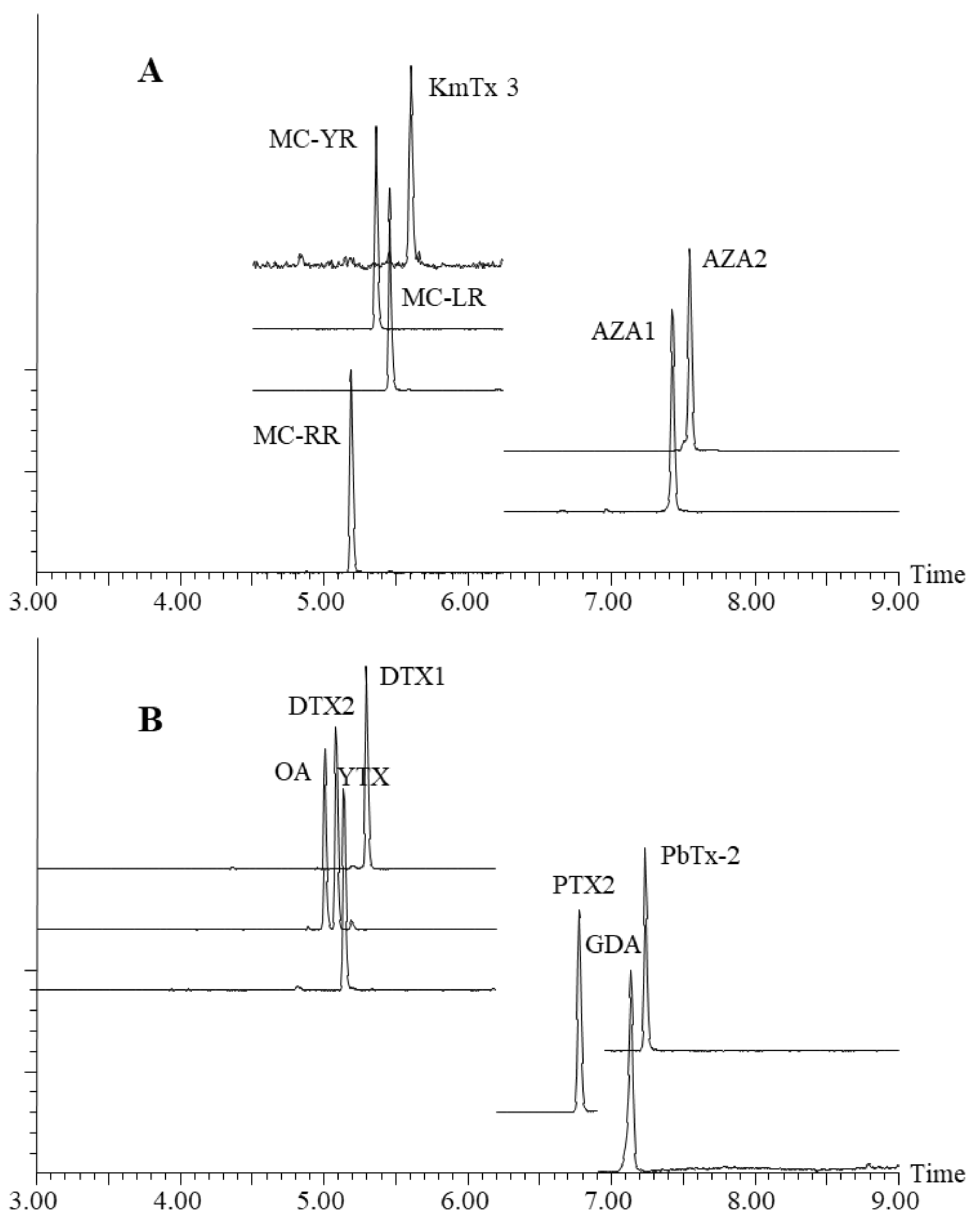
2.2. Final Chromatography Methods
2.3. Reduction of Carryover
2.4. Method Performance Characteristics
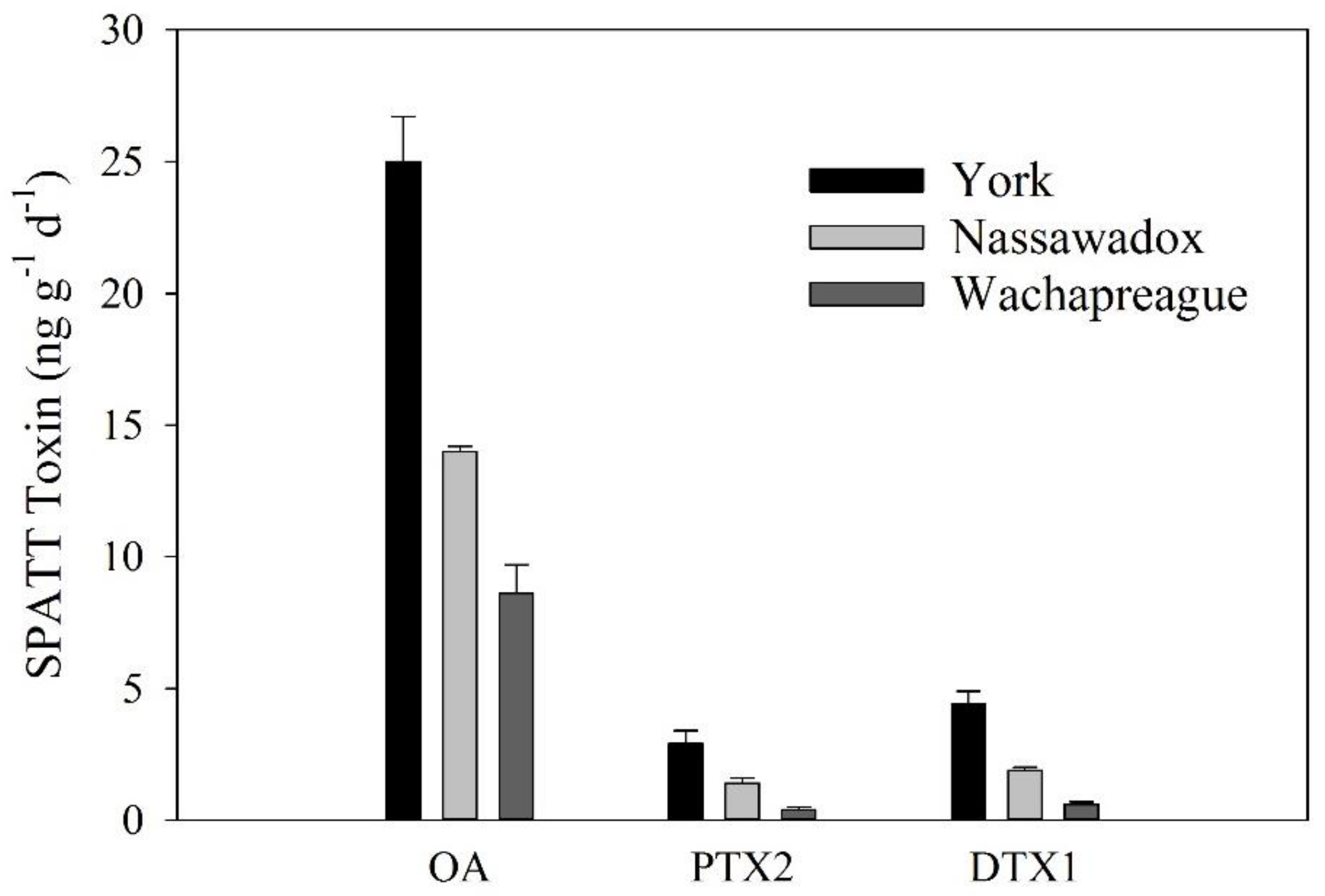
2.5. SPATT Analysis
3. Conclusions
4. Materials and Methods
4.1. Reagents and Analytical Toxin Standards
4.2. Instrumentation
4.2.1. Mass Spectrometry Conditions
4.2.2. Chromatographic Conditions
4.3. Development and Optimization
4.3.1. Carryover Management
4.4. Method Performance Characteristics
4.4.1. Repeatability
4.4.2. Limits of Detection and Quantification
4.4.3. Linearity
4.5. SPATT Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Landsberg, J.H. The Effects of Harmful Algal Blooms on Aquatic Organisms. Rev. Fish. Sci. 2002, 10, 113–390. [Google Scholar] [CrossRef]
- Landsberg, J.; Van Dolah, F.; Doucette, G. Marine and Estuarine Harmful Algal Blooms: Impacts on Human and Animal Health. In Oceans and Health: Pathogens in the Marine Environment; Belkin, S., Colwell, R.R., Eds.; Springer: Boston, MA, USA, 2005; pp. 165–215. [Google Scholar] [CrossRef]
- Visciano, P.; Schirone, M.; Berti, M.; Milandri, A.; Tofalo, R.; Suzzi, G. Marine Biotoxins: Occurrence, Toxicity, Regulatory Limits and Reference Methods. Front. Microbiol. 2016, 7, 390. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Kudela, R.M.; Mekebri, A.; Crane, D.; Oates, S.C.; Tinker, M.T.; Staedler, M.; Miller, W.A.; Toy-Choutka, S.; Dominik, C.; et al. Evidence for a Novel Marine Harmful Algal Bloom: Cyanotoxin (Microcystin) Transfer from Land to Sea Otters. PLoS ONE 2010, 5, e12576. [Google Scholar] [CrossRef] [PubMed]
- Falconer, I.R. An Overview of problems caused by toxic blue–green algae (cyanobacteria) in drinking and recreational water. Environ. Toxicol. 1999, 14, 5–12. [Google Scholar] [CrossRef]
- Pierce, R.H.; Henry, M.S. Harmful algal toxins of the Florida red tide (Karenia brevis): Natural chemical stressors in South Florida coastal ecosystems. Ecotoxicology 2008, 17, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Glibert, P.M.; Burkholder, J.M. Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries 2002, 25, 704–726. [Google Scholar] [CrossRef]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: A formidable predictive challenge. J. Phycol. 2010, 46, 220–235. [Google Scholar] [CrossRef]
- Van Dolah, F.M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Fire, S.E.; Wang, Z.; Byrd, M.; Whitehead, H.R.; Paternoster, J.; Morton, S.L. Co-occurrence of multiple classes of harmful algal toxins in bottlenose dolphins (Tursiops truncatus) stranding during an unusual mortality event in Texas, USA. Harmful Algae 2011, 10, 330–336. [Google Scholar] [CrossRef]
- Fux, E.; Biré, R.; Hess, P. Comparative accumulation and composition of lipophilic marine biotoxins in passive samplers and in mussels (M. edulis) on the West Coast of Ireland. Harmful Algae 2009, 8, 523–537. [Google Scholar] [CrossRef]
- Garcia, C.; Mardones, P.; Sfeir, A.; Lagos, N. Simultaneous presence of Paralytic and Diarrheic Shellfish Poisoning toxins in Mytilus chilensis samples collected in the Chiloe Island, Austral Chilean Fjords. Biol. Res. 2004, 37, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Hattenrath-Lehmann, T.K.; Marcoval, M.A.; Berry, D.L.; Fire, S.E.; Wang, Z.; Morton, S.L.; Gobler, C.J. The emergence of Dinophysis acuminata blooms and DSP toxins in shellfish in New York waters. Harmful Algae 2013, 26, 33–44. [Google Scholar] [CrossRef]
- Jester, R.; Lefebvre, K.; Langlois, G.; Vigilant, V.; Baugh, K.; Silver, M.W. A shift in the dominant toxin-producing algal species in central California alters phycotoxins in food webs. Harmful Algae 2009, 8, 291–298. [Google Scholar] [CrossRef]
- Paerl, H.; Joyner, J.; Joyner, A.; Arthur, K.; Paul, V.; O’Neil, J.; Heil, C. Co-occurrence of dinoflagellate and cyanobacterial harmful algal blooms in southwest Florida coastal waters: Dual nutrient (N and P) input controls. Mar. Ecol. Prog. Ser. 2008, 371, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Peacock, M.B.; Gibble, C.M.; Senn, D.B.; Cloern, J.E.; Kudela, R.M. Blurred lines: Multiple freshwater and marine algal toxins at the land-sea interface of San Francisco Bay, California. Harmful Algae 2018, 73, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Gibble, C.M.; Kudela, R.M. Detection of persistent microcystin toxins at the land–sea interface in Monterey Bay, California. Harmful Algae 2014, 39, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gao, L.; Li, Z.; Wang, S.; Li, J.; Cao, W.; Sun, C.; Zheng, L.; Wang, X. Simultaneous screening for lipophilic and hydrophilic toxins in marine harmful algae using a serially coupled reversed-phase and hydrophilic interaction liquid chromatography separation system with high-resolution mass spectrometry. Anal. Chim. Acta 2016, 914, 117–126. [Google Scholar] [CrossRef]
- Dahlmann, J.; Budakowski, W.R.; Luckas, B. Liquid chromatography–electrospray ionisation-mass spectrometry based method for the simultaneous determination of algal and cyanobacterial toxins in phytoplankton from marine waters and lakes followed by tentative structural elucidation of microcystins. J. Chromatogr. A 2003, 994, 45–57. [Google Scholar] [CrossRef]
- Rodríguez, I.; Alfonso, A.; González-Jartín, J.M.; Vieytes, M.R.; Botana, L.M. A single run UPLC-MS/MS method for detection of all EU-regulated marine toxins. Talanta 2018, 189, 622–628. [Google Scholar] [CrossRef]
- Zendong, Z.; McCarron, P.; Herrenknecht, C.; Sibat, M.; Amzil, Z.; Cole, R.B.; Hess, P. High resolution mass spectrometry for quantitative analysis and untargeted screening of algal toxins in mussels and passive samplers. J. Chromatogr. A 2015, 1416, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Mallet, C.; Botch-Jones, S. Illicit Drug Analysis Using Two-Dimension Liquid Chromatography/Tandem Mass Spectrometry. J. Anal. Toxicol. 2016, 40, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fux, E.; Marcaillou, C.; Mondeguer, F.; Biré, R.; Hess, P. Field and mesocosm trials on passive sampling for the study of adsorption and desorption behaviour of lipophilic toxins with a focus on OA and DTX1. Harmful Algae 2008, 7, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Kudela, R.M. Characterization and deployment of Solid Phase Adsorption Toxin Tracking (SPATT) resin for monitoring of microcystins in fresh and saltwater. Harmful Algae 2011, 11, 117–125. [Google Scholar] [CrossRef]
- Hattenrath-Lehmann, T.K.; Lusty, M.W.; Wallace, R.B.; Haynes, B.; Wang, Z.; Broadwater, M.; Deeds, J.R.; Morton, S.L.; Hastback, W.; Porter, L.; et al. Evaluation of Rapid, Early Warning Approaches to Track Shellfish Toxins Associated with Dinophysis and Alexandrium Blooms. Mar. Drugs 2018, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.Q.; Roddam, C.M.; Langlois, G.W.; Kudela, R.M. Application of Solid Phase Adsorption Toxin Tracking (SPATT) for field detection of the hydrophilic phycotoxins domoic acid and saxitoxin in coastal California. Limnol. Oceanogr. Methods 2010, 8, 645–660. [Google Scholar] [CrossRef]
- MacKenzie, L.A. In situ passive solid-phase adsorption of micro-algal biotoxins as a monitoring tool. Curr. Opin. Biotechnol. 2010, 21, 326–331. [Google Scholar] [CrossRef]
- Marine Biotoxins. FAO Food and Nutrition Paper 80; Food and Agriculture Organization of the United Nations: Rome, Italy, 2004. [Google Scholar]
- Pease, S.K.D. Alexandrium Monilatum in the Lower Chesapeake Bay: Sediment Cyst Distribution and Potential Health Impacts on Crassostrea Virginica. Master’s Thesis, Virginia Institute of Marine Science, Gloucester Point, VA, USA, 2016. [Google Scholar] [CrossRef]
- Suzuki, T.; Beuzenberg, V.; MacKenzie, L.; Quilliam, M. Liquid chromatography-mass spectrometry of spiroketal stereoisomers of pectenotoxins and the analysis of novel pectenotoxin isomers in the toxic dinoflagellate Dinophysis acuta from New Zealand. J. Chromatogr. A 2003, 992, 141–150. [Google Scholar] [CrossRef]
- Gerssen, A.; Mulder, P.; McElhinney, M.A.; De Boer, J. Liquid chromatography–tandem mass spectrometry method for the detection of marine lipophilic toxins under alkaline conditions. J. Chromatogr. A 2009, 1216, 1421–1430. [Google Scholar] [CrossRef]
- Xu, W.; Chen, Q.; Zhang, T.; Cai, Z.; Jia, X.; Xie, Q.; Ren, Y. Development and application of ultra performance liquid chromatography–electrospray ionization tandem triple quadrupole mass spectrometry for determination of seven microcystins in water samples. Anal. Chim. Acta 2008, 626, 28–36. [Google Scholar] [CrossRef]
- Fux, E.; McMillan, D.; Biré, R.; Hess, P. Development of an ultra-performance liquid chromatography–mass spectrometry method for the detection of lipophilic marine toxins. J. Chromatogr. A 2007, 1157, 273–280. [Google Scholar] [CrossRef]
- Orellana, G.; Van Meulebroek, L.; Van Vooren, S.; De Rijcke, M.; Vandegehuchte, M.; Janssen, C.R.; Vanhaecke, L. Quantification and profiling of lipophilic marine toxins in microalgae by UHPLC coupled to high-resolution orbitrap mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 6345–6356. [Google Scholar] [CrossRef] [PubMed]
- Krock, B.; Tillmann, U.; Wen, Y.; Hansen, P.J.; Larsen, T.O.; Andersen, A.J. Development of a LC-MS/MS method for the quantification of goniodomins A and B and its application to Alexandrium pseudogonyaulax strains and plankton field samples of Danish coastal waters. Toxicon 2018, 155, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachvaroff, T.R.; Adolf, J.E.; Squier, A.H.; Harvey, H.R.; Place, A.R. Characterization and quantification of karlotoxins by liquid chromatography–mass spectrometry. Harmful Algae 2008, 7, 473–484. [Google Scholar] [CrossRef]
- Harris, C.M.; Reece, K.S.; Stec, D.F.; Scott, G.P.; Jones, W.M.; Hobbs, P.L.; Harris, T.M. The toxin goniodomin, produced by Alexandrium spp., is identical to goniodomin A. Harmful Algae 2020, 92, 101707. [Google Scholar] [CrossRef]
- Gerssen, A.; Van Olst, E.H.W.; Mulder, P.P.J.; De Boer, J. In-house validation of a liquid chromatography tandem mass spectrometry method for the analysis of lipophilic marine toxins in shellfish using matrix-matched calibration. Anal. Bioanal. Chem. 2010, 397, 3079–3088. [Google Scholar] [CrossRef] [Green Version]
- Blay, P.; Hui, J.P.M.; Chang, J.; Melanson, J. Screening for multiple classes of marine biotoxins by liquid chromatography–high-resolution mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 577–585. [Google Scholar] [CrossRef]
- Bosch-Orea, C.; Sanchís, J.; Farré, M.; Barceló, J. Analysis of lipophilic marine biotoxins by liquid chromatography coupled with high-resolution mass spectrometry in seawater from the Catalan Coast. Anal. Bioanal. Chem. 2017, 409, 5451–5462. [Google Scholar] [CrossRef]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phycotoxin Congener Abbreviation (Group) | Producer | Human Syndrome or Toxicity |
|---|---|---|
| GDA (goniodomins) | Alexandrium monilatum | Ichthyotoxic |
| OA; DTX1; DTX2 (okadaic acid and dinophysistoxins) | Dinophysis spp., Prorocentrum lima | Diarrhetic shellfish poisoning (DSP) |
| PTX2 (pectenotoxins *) | Dinophysis spp. | Acute toxicity in vertebrate model with i.p. injection |
| YTX (yessotoxin *) | Protoceratium reticulatum, Gymnodinium catenatum, Pyrodinium bahamense, Gonyaulax spp. Lingulodinium polyedrum | Acute toxicity in vertebrate model with i.p. injection |
| AZA-1; AZA-2 (azaspiracids) | Amphidoma languida, Azadinium spp. | Azaspiracid shellfish poisoning (AZP) |
| KmTx 3 (karlotoxins) | Karlodinium spp. | Ichthyotoxic |
| MC-LR; MC-YR; MC-RR (microcystins) | Microcystis spp., Anabaena spp., Oscillatoria spp. Planktothrix spp. | Hepatotoxic |
| PbTx-2 (brevetoxins) | Karenia spp. | Neurotoxic shellfish poisoning (NSP), ichthyotoxic |
| Analyte | Trapping Column | Loading Conditions | Separation Column | Elution Conditions |
|---|---|---|---|---|
| MC-LR MC-YR MC-RR AZA1 AZA2 KmTx 3 | XBridge BEH C18 130 Å 10 µm, 2.1 × 30 mm | H2O + 50 mM formic acid + 2 mM ammonium formate Isocratic for 3 min | Acquity BEH C18 130 Å 1.7 µm, 2.1 × 50 mm | Acidic A: H2O + 50 mM formic acid + 2 mM ammonium formate B: ACN + 50 mM formic acid + 2 mM ammonium formate Linear gradient 5% to 95% B over 5 min Isocratic 95% B for 1 min |
| GDA PTX2PbTx-2YTXOA DTX1 DTX2 | XBridge BEH C18 130 Å 10 µm, 2.1 × 30 mm | H2O + 6.7 mM NH4OH Isocratic for 3 min | Acquity BEH C18 130 Å 1.7 µm, 2.1 × 50 mm | Basic A: H2O + 6.7 mM NH4OH B: ACN + 6.7 mM NH4OH Linear gradient 5% to 95% B over 5 min Isocratic 95% B for 1 min |
| Literature | This Study | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | LOD LC-MS Methods (µg/L) | Reference | LOQ (S/N ≥ 10) 50 µL (µg/L) | LOD (S/N ≥ 3) 50 µL (µg/L) | %RSD 50 µL | LOD Calc. 50 µL (µg/L) | LOD Calc. 50 µL (pg on- column) | %RSD 1000 µL | LOD Calc. 1000 µL (µg/L) | LOD Calc. 1000 µL (pg on- column) |
| MC-RR | 0.017 ^ | [32] | 0.31 | 0.13 | 3.81 | 0.07 | 3.5 | 3.66 | 0.007 | 7.5 |
| MC-YR | 0.043 ^ | [32] | 0.15 | 0.13 | 3.23 | 0.24 | 12 | 7.32 | 0.01 | 15 |
| MC-LR | 0.029 ^ | [32] | 0.15 | 0.13 | 3.10 | 0.25 | 13 | 5.80 | 0.01 | 12 |
| AZA1 | 0.033 | [33] | 004 | 0.03 | 3.02 | 0.02 | 1.0 | 2.12 | 0.001 | 1.0 |
| AZA2 | 0.070 | [34] | 0.04 | 0.03 | 2.56 | 0.01 | 0.5 | 3.11 | 0.001 | 1.5 |
| GDA | 2.34 | [35] | 1.98 | 0.60 | 2.43 | 0.39 | 19.5 | 2.45 | 0.019 | 19.2 |
| KmTx3 | 4.0 * | [36] | 1.39 | 0.97 | 5.64 | 0.64 | 32 | 5.64 | 0.05 | 54 |
| OA | 0.483 | [33] | 0.15 | 0.13 | 1.21 | 0.10 | 5.0 | 4.05 | 0.008 | 8.3 |
| DTX1 | 0.030 | [34] | 0.15 | 0.13 | 2.30 | 0.11 | 5.5 | 5.87 | 0.01 | 12 |
| DTX2 | 0.930 | [20] | 1.24 | 0.13 | 1.43 | 0.12 | 6.0 | 3.05 | 0.006 | 6.2 |
| PTX2 | 0.048 | [33] | 0.04 | 0.03 | 1.89 | 0.04 | 2.0 | 3.91 | 0.004 | 3.7 |
| YTX | 0.336 | [33] | 1.24 | 0.50 | 1.19 | 0.14 | 7.0 | 4.27 | 0.03 | 34 |
| PbTx-2 | n.r. | 0.15 | 0.13 | 7.18 | 0.16 | 8.0 | 7.39 | 0.03 | 15 | |
| York | Nassawadox | Wachapreague | ||||
|---|---|---|---|---|---|---|
| P > P (µg/L) | P > D (µg/L) | P > P (µg/L) | P > D (µg/L) | P > P (µg/L) | P > D (µg/L) | |
| OA | 34 ± 2.4 | 27 ± 2.3 | 21 ± 0.4 | 17 ± 0.3 | 13 ± 1.7 | 11 ± 1.2 |
| PTX2 | 3.9 ± 0.7 | 3.5 ± 0.6 | 2.1 ± 0.04 | 1.9 ± 0.6 | 0.6 ± 0.1 | 0.6 ± 0.1 |
| DTX1 | 6.1 ± 0.7 | 6.1 ± 0.7 | 2.8 ± 0.1 | 2.9 ± 0.2 | 1.0 ± 0.2 | 1.0 ± 0.2 |
| Suppression (<100%) or Enhancement (>100%) | |||
|---|---|---|---|
| York (%) | Nassawadox (%) | Wachapreague (%) | |
| MC-RR | 26 ± 1 | 39 ± 3 | 38 ± 7 |
| MC-LR | 36 ± 0.4 | 50 ± 2 | 47 ± 4 |
| MC-YR | 32 ± 1 | 46 ± 2 | 44 ± 3 |
| AZA1 | 33 ± 1 | 33 ± 2 | 43 ± 12 |
| AZA2 | 49 ± 3 | 44 ± 3 | 52 ± 8 |
| PbTx-2 | 146 ± 7 | 109 ± 13 | 123 ± 5 |
| YTX | 103 ± 2 | 132 ± 10 | 121 ± 18 |
| DTX2 | 69 ± 0.8 | 81 ± 1 | 78 ± 3 |
| OA * | 101 ± 1 | 103 ± 0.7 | 101 ± 1 |
| PTX2 * | 57 ± 2 | 45 ± 0.8 | 36 ± 7 |
| DTX1 * | 98 ± 2 | 96 ± 3 | 91 ± 1 |
| Analyte | Ionization Mode | Adduct | Molecular Ion m/z | Cone Voltage (V) | Dominant Transition Detected | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| MC-RR | ESI+ | 2H+ | 520.0 | 30 | 520.0 > 135.1 | 30 |
| MC-YR | ESI+ | H+ | 1045.5 | 30 | 1045.5 > 135.1 | 85 |
| MC-LR | ESI+ | H+ | 995.5 | 30 | 995.5 > 135.1 | 85 |
| AZA1 | ESI+ | H+ | 842.4 | 30 | 842.4 > 824.6 | 30 |
| AZA2 | ESI+ | H+ | 856.4 | 30 | 856.4 > 838.6 | 30 |
| KmTx 3 | ESI+ | Na+ | 1347.7 | 70 | 1347.7 > 937.7 | 80 |
| GDA | ESI+ | NH4+ | 786.5 | 30 | 786.5 > 139.0 | 40 |
| OA | ESI− | −H | 803.5 | 30 | 803.5 > 255.5 | 60 |
| DTX1 | ESI− | −H | 817.5 | 30 | 817.5 > 113.0 | 70 |
| DTX2 | ESI− | −H | 803.5 | 30 | 803.5 > 255.5 | 60 |
| PTX2 | ESI+ | NH4+ | 876.6 | 30 | 876.6 > 841.5 | 30 |
| YTX | ESI− | −2H | 571.1 | 30 | 571.1 > 467.7 | 30 |
| PbTx-2 | ESI+ | H+ | 895.4 | 40 | 895.4 > 877.3 | 20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onofrio, M.D.; Mallet, C.R.; Place, A.R.; Smith, J.L. A Screening Tool for the Direct Analysis of Marine and Freshwater Phycotoxins in Organic SPATT Extracts from the Chesapeake Bay. Toxins 2020, 12, 322. https://doi.org/10.3390/toxins12050322
Onofrio MD, Mallet CR, Place AR, Smith JL. A Screening Tool for the Direct Analysis of Marine and Freshwater Phycotoxins in Organic SPATT Extracts from the Chesapeake Bay. Toxins. 2020; 12(5):322. https://doi.org/10.3390/toxins12050322
Chicago/Turabian StyleOnofrio, Michelle D., Claude R. Mallet, Allen R. Place, and Juliette L. Smith. 2020. "A Screening Tool for the Direct Analysis of Marine and Freshwater Phycotoxins in Organic SPATT Extracts from the Chesapeake Bay" Toxins 12, no. 5: 322. https://doi.org/10.3390/toxins12050322