Mechanism of Vascular Toxicity in Rats Subjected to Treatment with a Tyrosine Kinase Inhibitor


 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Blood Pressure and Tissue Preparations
2.3. Measurement of the Levels of Superoxide Anion and Nitric Oxide Concentration
2.4. Measurement of NADPH Oxidase Activity
2.5. RNA Extraction and Real-Time PCR
2.6. Western Blotting
2.7. Vascular Reactivity
2.8. Histomorphometric Studies
2.9. Immunofluorescence Studies
2.10. Statistics
3. Results
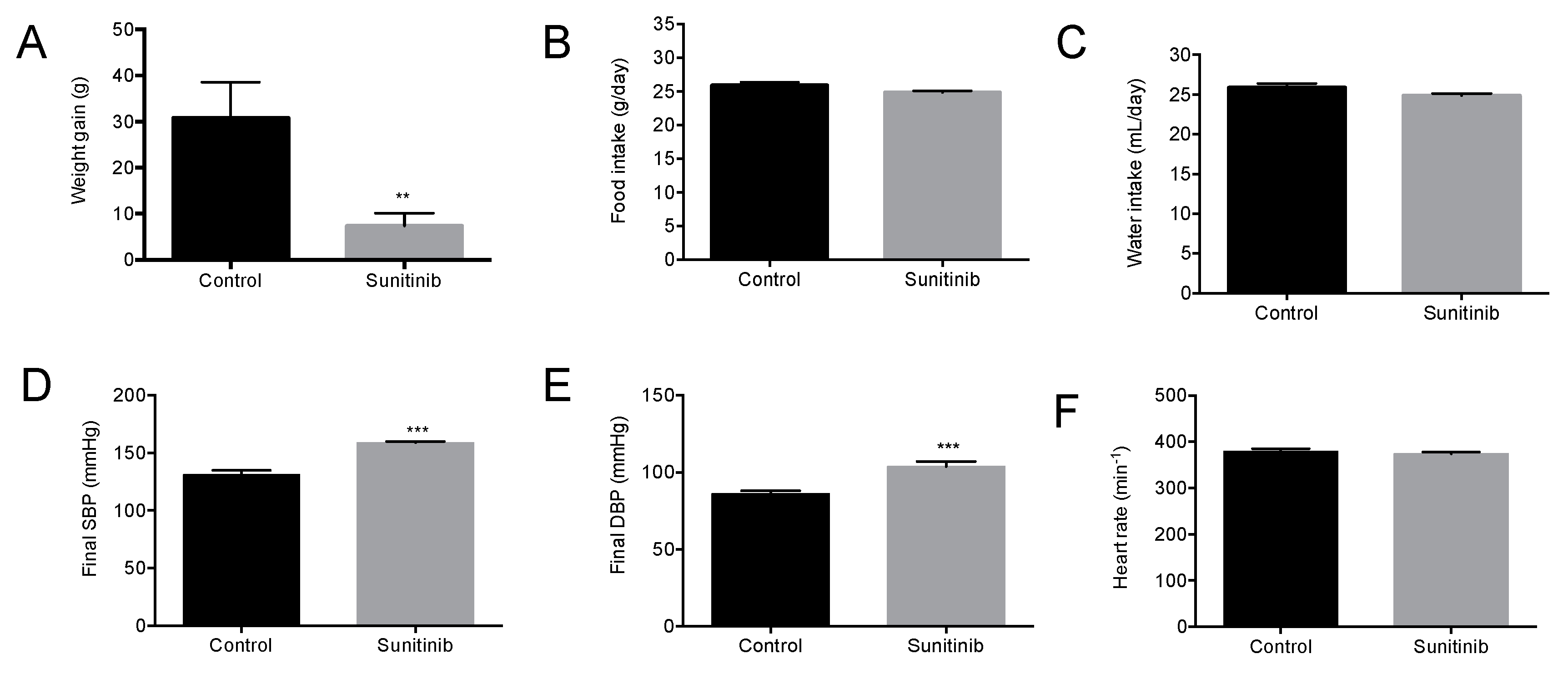
3.1. General Characteristics of Animals
3.2. Endothelial Dysfunction in Su-treated Rats
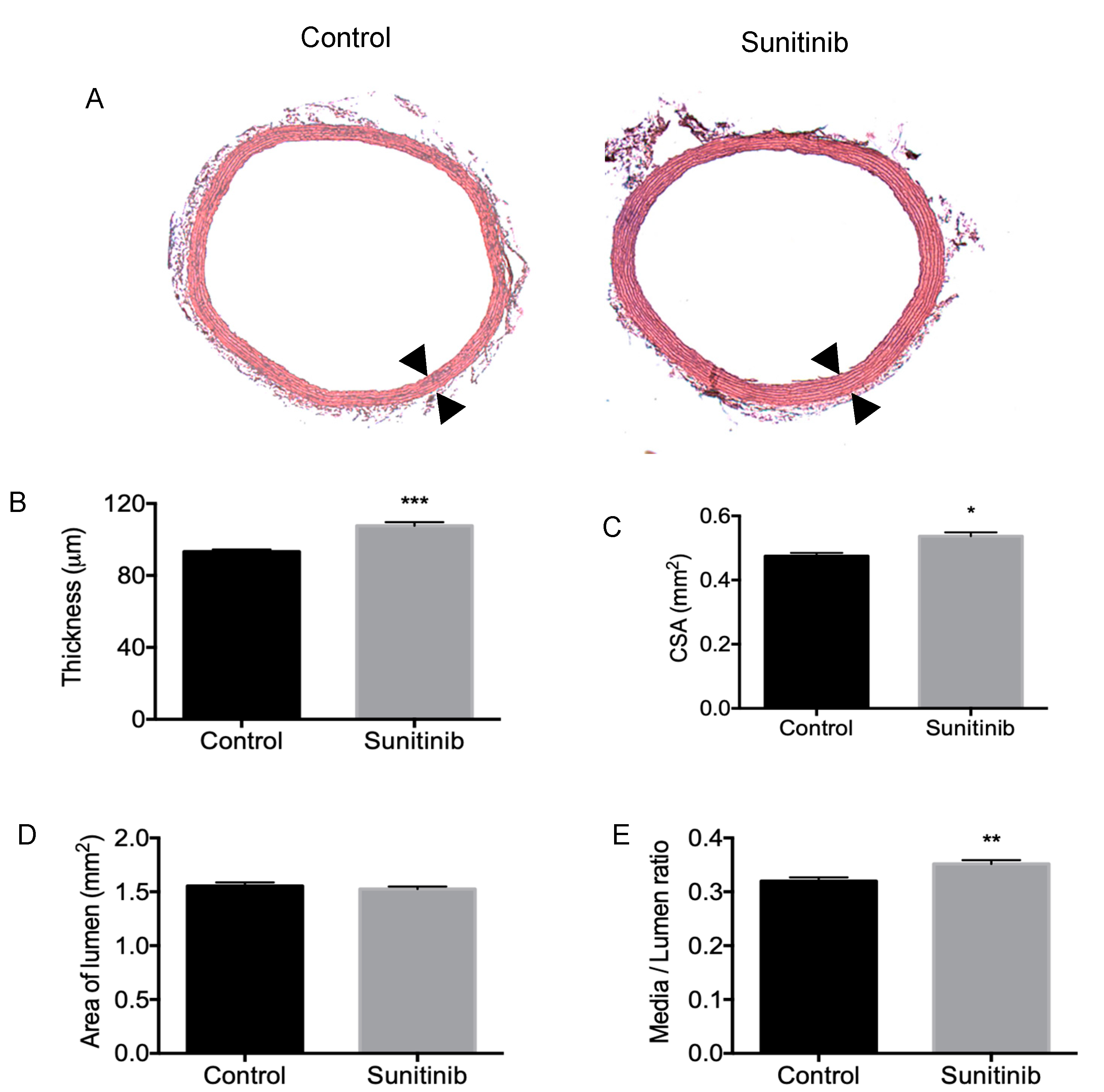
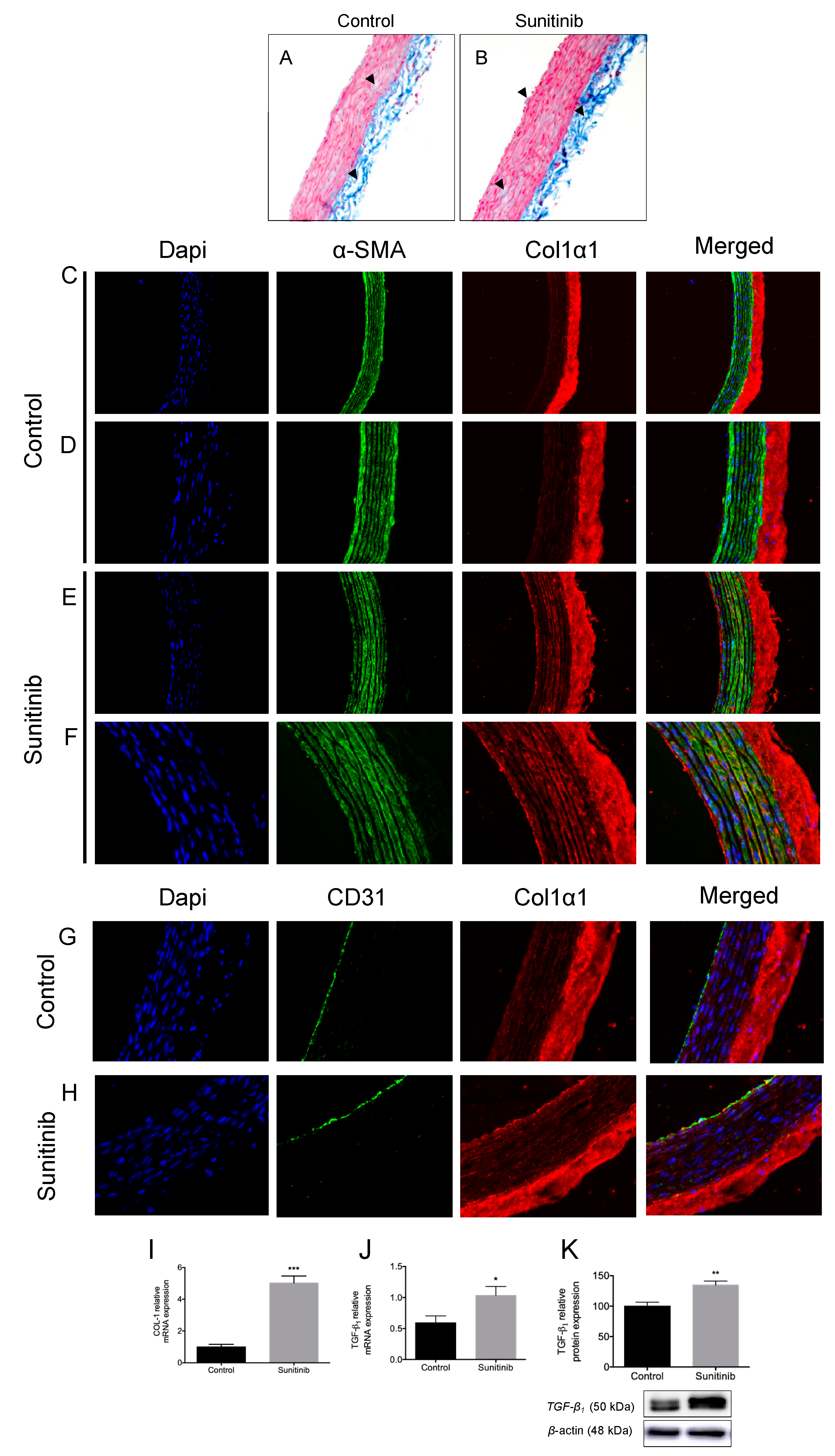
3.3. Vascular Remodeling and Fibrosis
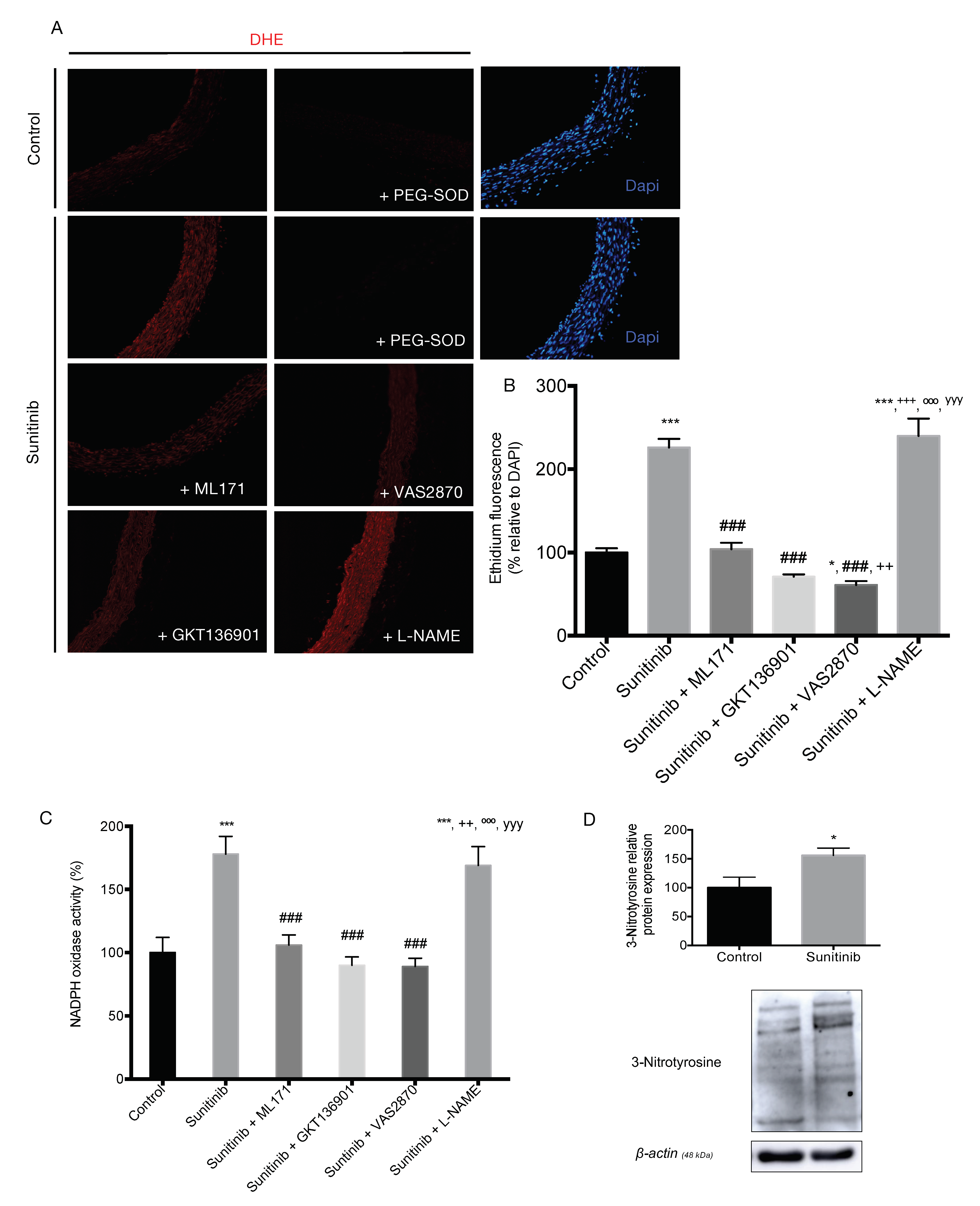
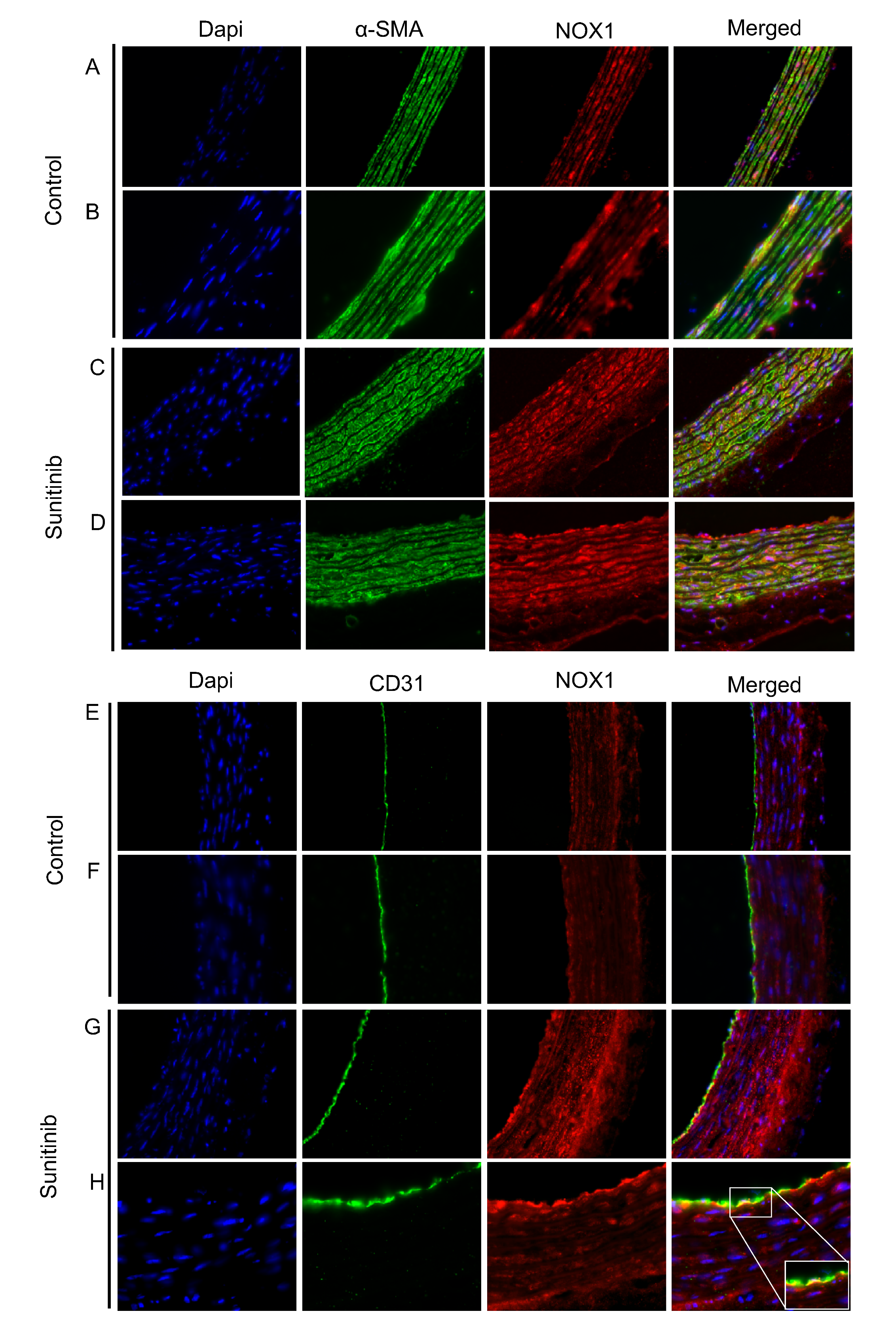
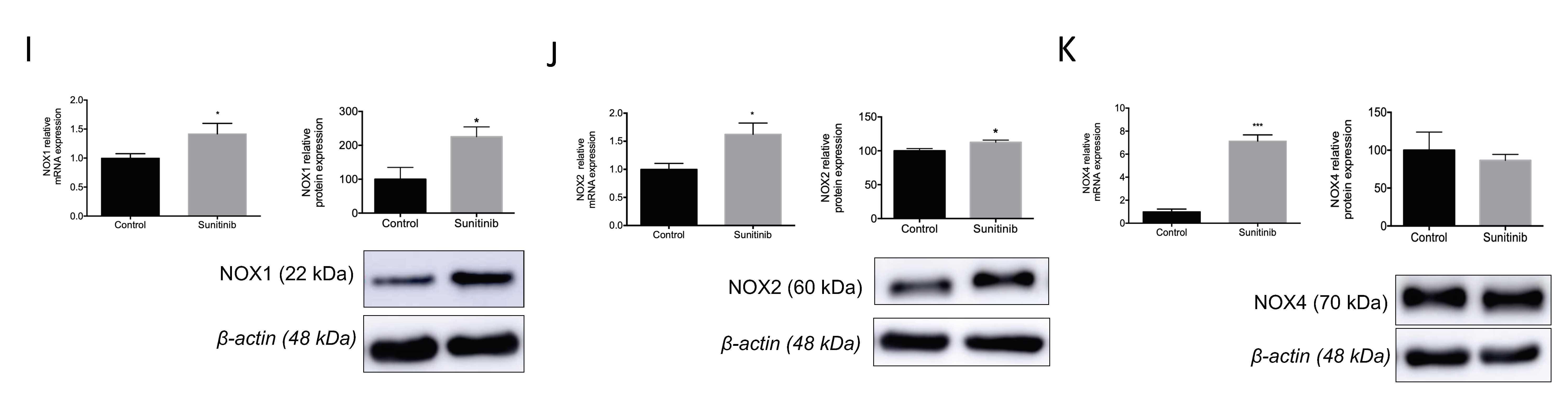
3.4. Assessment of Vascular Oxidative Stress
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Le Tourneau, C.; Faivre, S.; Raymond, E. New developments in multitargeted therapy for patients with solid tumours. Cancer Treat. Rev. 2008, 34, 37–48. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 1–12. [Google Scholar] [CrossRef]
- O’Farrell, A.M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W.H.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003. [Google Scholar] [CrossRef] [Green Version]
- Manir, K.S.; Banerjee, D.; Bhowmick, R.; Roy, C. Sunitinib-induced acute severe hypothyroidism in a case of metastatic gastrointestinal stromal tumor: A case report. J. Cancer Res. Ther. 2018. [Google Scholar] [CrossRef]
- Faivre, S.; Niccoli, P.; Castellano, D.; Valle, J.W.; Hammel, P.; Raoul, J.L.; Vinik, A.; Van Cutsem, E.; Bang, Y.J.; Lee, S.H.; et al. Sunitinib in pancreatic neuroendocrine tumors: Updated progression-free survival and final overall survival from a phase III randomized study. Ann. Oncol. 2017. [Google Scholar] [CrossRef]
- Gorini, S.; De Angelis, A.; Berrino, L.; Malara, N.; Rosano, G.; Ferraro, E. Chemotherapeutic Drugs and Mitochondrial Dysfunction: Focus on Doxorubicin, Trastuzumab, and Sunitinib. Oxidative Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Robinson, E.S.; Khankin, E.V.; Choueiri, T.K.; Dhawan, M.S.; Rogers, M.J.; Karumanchi, S.A.; Humphreys, B.D. Suppression of the Nitric Oxide Pathway in Metastatic Renal Cell Carcinoma Patients Receiving Vascular Endothelial Growth Factor–Signaling Inhibitors. Hypertension 2010, 56, 1131–1136. [Google Scholar] [CrossRef] [Green Version]
- Belcik, J.T.; Qi, Y.; Kaufmann, B.A.; Xie, A.; Bullens, S.; Morgan, T.K.; Bagby, S.P.; Kolumam, G.; Kowalski, J.; Oyer, J.A.; et al. Cardiovascular and Systemic MicrovascularEffects of Anti-Vascular Endothelial Growth Factor Therapy for Cancer. J. Am. Coll. Cardiol. 2012, 60, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Kappers, M.H.W.; Van Esch, J.H.M.; Sluiter, W.; Sleijfer, S.; Danser, A.H.J.; Van Den Meiracker, A.H. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension 2010, 56, 675–681. [Google Scholar] [CrossRef] [Green Version]
- Lankhorst, S.; Kappers, M.H.W.; Van Esch, J.H.M.; Smedts, F.M.M.; Sleijfer, S.; Mathijssen, R.H.J.; Baelde, H.J.; Danser, A.H.J.; Van Den Meiracker, A.H. Treatment of hypertension and renal injury induced by the angiogenesis inhibitor sunitinib preclinical study. Hypertension 2014, 64, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.J.; Fretwell, L.V.; Woolard, J. Effects of 4 multitargeted receptor tyrosine kinase inhibitors on regional hemodynamics in conscious, freely moving rats. FASEB J. 2017, 31, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Alivon, M.; Giroux, J.; Briet, M.; Goldwasser, F.; Laurent, S.; Boutouyrie, P. Large artery stiffness and hypertension after antiangiogenic drugs: Influence on cancer progression. J. Hypertens. 2015, 33, 1310–1317. [Google Scholar] [CrossRef]
- Svilaas, T.; Lefrandt, J.D.; Gietema, J.A.; Kamphuisen, P.W. Long-term arterial complications of chemotherapy in patients with cancer. Thromb. Res. 2016, 140, S109–S118. [Google Scholar] [CrossRef]
- Blanca, A.J.; Ruiz-Armenta, M.V.; Zambrano, S.; Miguel-Carrasco, J.L.; Arias, J.L.; Arévalo, M.; Mate, A.; Aramburu, O.; Vázquez, C.M. Inflammatory and fibrotic processes are involved in the cardiotoxic effect of sunitinib: Protective role of l -carnitine. Toxicol. Lett. 2016, 241, 9–18. [Google Scholar] [CrossRef]
- Neves, K.B.; Rios, F.J.; Van Der Mey, L.; Alves-Lopes, R.; Cameron, A.C.; Volpe, M.; Montezano, A.C.; Savoia, C.; Touyz, R.M. VEGFR (vascular endothelial growth factor receptor) inhibition induces cardiovascular damage via redox-sensitive processes. Hypertension 2018, 71, 638–647. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, V.; Thomas, C.J.; Cao, M.; Melnyk, S.B.; Pavliv, O.; Joseph, J.; Singh, S.P.; Sharma, S.; Moros, E.G.; Boerma, M. Effects of local irradiation combined with sunitinib on early remodeling, mitochondria, and oxidative stress in the rat heart. Radiother. Oncol. 2016, 119, 259–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976. [Google Scholar] [CrossRef]
- Wei, J.; Xu, L.; Du, Y.; Tang, X.; Ye, M.; Wu, Y.; Han, W.; Gao, P. Membrane raft redox signalling contributes to endothelial dysfunction and vascular remodelling of thoracic aorta in angiotensin II-infused rats. Exp. Physiol. 2019, 104, 946–956. [Google Scholar] [CrossRef]
- Marzinzig, M.; Nussler, A.K.; Stadler, J.; Marzinzig, E.; Barthlen, W.; Nussler, N.C.; Beger, H.G.; Morris, S.M.; Brückner, U.B. Improved methods to measure end products of nitric oxide in biological fluids: Nitrite, nitrate, and S-nitrosothiols. Nitric Oxide 1997, 1, 177–189. [Google Scholar] [CrossRef]
- Blanca, A.J.; Ruiz-Armenta, M.V.; Zambrano, S.; Miguel-Carrasco, J.L.; González-Roncero, F.M.; Fortuño, A.; Revilla, E.; Mate, A.; Vázquez, C.M. l-Carnitine ameliorates the oxidative stress response to angiotensin II by modulating NADPH oxidase through a reduction in protein kinase c activity and NF-κB translocation to the nucleus. Food Chem. 2017, 228, 356–366. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Alvarez de Sotomayor, M.; Perez-Guerrero, C.; Gomez-Amores, L.; Vazquez, C.M.; Herrera, M.D. L-carnitine and propionyl-L-carnitine improve endothelial dysfunction in spontaneously hypertensive rats: Different participation of NO and COX-products. Life Sci. 2005, 77, 2082–2097. [Google Scholar] [CrossRef]
- León-Mateos, L.; Mosquera, J.; Antón Aparicio, L. Treatment of sunitinib-induced hypertension in solid tumor by nitric oxide donors. Redox Biol. 2015, 6, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Miller, J.D.; Li, J.Z.; Russell, M.W.; Charbonneau, C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): A literature review. Cancer Treat. Rev. 2008, 34, 193–205. [Google Scholar] [CrossRef]
- Speed, B.; Bu, H.Z.; Pool, W.F.; Peng, G.W.; Wu, E.Y.; Patyna, S.; Bello, C.; Kang, P. Pharmacokinetics, distribution, and metabolism of [14C] sunitinib in rats, monkeys, and humans. Drug Metab. Dispos. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijs, A.M.J.; Van Herpen, C.M.L.; Verweij, V.; Pertijs, J.; Van Den Broek, P.H.H.; Van Der Graaf, W.T.A.; Rongen, G.A. Impaired endothelium-dependent vasodilation does not initiate the development of sunitinib-associated hypertension. J. Hypertens. 2015, 33, 2075–2082. [Google Scholar] [CrossRef] [PubMed]
- Goodman, V.L.; Rock, E.P.; Dagher, R.; Ramchandani, R.P.; Abraham, S.; Gobburu, J.V.S.; Booth, B.P.; Verbois, S.L.; Morse, D.E.; Liang, C.Y.; et al. Approval Summary: Sunitinib for the Treatment of Imatinib Refractory or Intolerant Gastrointestinal Stromal Tumors and Advanced Renal Cell Carcinoma. Clin. Cancer Res. 2007, 13, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Blasi, E.; Heyen, J.; Patyna, S.; Hemkens, M.; Ramirez, D.; John-Baptiste, A.; Steidl-Nichols, J.; McHarg, A. Sunitinib, a Receptor Tyrosine Kinase Inhibitor, Increases Blood Pressure in Rats without Associated Changes in Cardiac Structure and Function. Cardiovasc. Ther. 2012, 30, 287–294. [Google Scholar] [CrossRef]
- Kojonazarov, B.; Sydykov, A.; Pullamsetti, S.S.; Luitel, H.; Dahal, B.K.; Kosanovic, D.; Tian, X.; Majewski, M.; Baumann, C.; Evans, S.; et al. Effects of multikinase inhibitors on pressure overload-induced right ventricular remodeling. Int. J. Cardiol. 2013, 167, 2630–2637. [Google Scholar] [CrossRef]
- Zhu, X.; Stergiopoulos, K.; Wu, S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: Systematic review and meta-analysis. Acta Oncol. 2009, 48, 9–17. [Google Scholar] [CrossRef]
- Henderson, K.A.; Borders, R.B.; Ross, J.B.; Huwar, T.B.; Travis, C.O.; Wood, B.J.; Ma, Z.J.; Hong, S.P.; Vinci, T.M.; Roche, B.M. Effects of tyrosine kinase inhibitors on rat isolated heart function and protein biomarkers indicative of toxicity. J. Pharmacol. Toxicol. Methods 2013, 68, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, H.; Cooper, S.; Hussain, A.; Mee, C.; Maddock, H. Attenuation of Sunitinib-induced cardiotoxicity through the A3 adenosine receptor activation. Eur. J. Pharmacol. 2017, 814, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Eechoute, K.; Van Der Veldt, A.A.M.; Oosting, S.; Kappers, M.H.W.; Wessels, J.A.M.; Gelderblom, H.; Guchelaar, H.J.; Reyners, A.K.L.; Van Herpen, C.M.L.; Haanen, J.B.; et al. Polymorphisms in endothelial nitric oxide synthase (eNOS) and vascular endothelial growth factor (VEGF) predict sunitinib-induced hypertension. Clin. Pharmacol. Ther. 2012, 92, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Renna, N.F.; de las Heras, N.; Miatello, R.M. Pathophysiology of Vascular Remodeling in Hypertension. Int. J. Hypertens. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Lakatta, E.G. The reality of aging viewed from the arterial wall. Artery Res. 2013, 7, 73. [Google Scholar] [CrossRef] [Green Version]
- Coen, M.; Gabbiani, G.; Bochaton-Piallat, M.-L. ATVB in Focus Vascular Cell Lineage Determination and Differentiation Myofibroblast-Mediated Adventitial Remodeling An Underestimated Player in Arterial Pathology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2391–2396. [Google Scholar] [CrossRef] [Green Version]
- Tsamis, A.; Krawiec, J.T.; Vorp, D.A. Elastin and collagen fibre microstructure of the human aorta in ageing and disease: A review. J. R. Soc. Interface 2013, 10. [Google Scholar] [CrossRef]
- Zhang, M.-J.; Zhou, Y.; Chen, L.; Wang, Y.-Q.; Wang, X.; Pi, Y.; Gao, C.-Y.; Li, J.-C.; Zhang, L.-L. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem. Cell Biol. 2016, 145, 119–130. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y.; Ali, F. Atherosclerotic cardiovascular disease: A review of initiators and protective factors. Inflammopharmacology 2016, 24, 1–10. [Google Scholar] [CrossRef]
- Wang, Q.; Zou, M.H. Measurement of reactive oxygen species (ROS) and mitochondrial ROS in AMPK knockout mice blood vessels. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Col1 | TCAGGGGCGAAGGCAACAGT | TTGGGATGGAGGGAGTTTACACGA |
| TGF-β1 | GCCCTGGATACCAACTACTGCT | AGGCTCCAAATGTAGGGGCAGG |
| NOX1 | TTCACCAATTCCCAGGATTGAAGTGGATGGTC | GACCTGTCACGATGTCAGTGGCCTTGTCAA |
| NOX2 | CCCTTTGGTACAGCCAGTGAAGAT | CAATCCCAGCTCCCACTAACATCA |
| NOX4 | TTGCTTTTGTATCTTC | CTTACCTTCGTCACAG |
| eNOS | GGGCCAGGGTGATGAGCTCTG | CCCTCCTGGCTTCCAGTGTCC |
| GAPDH | GCCAAAAGGGTCATCATCTCCGC | GGATGACCTTGCCCACAGCCTTG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Goya, C.; Santana-Garrido, Á.; Soto-Astacio, E.; Aramburu, Ó.; Zambrano, S.; Mate, A.; Vázquez, C.M. Mechanism of Vascular Toxicity in Rats Subjected to Treatment with a Tyrosine Kinase Inhibitor. Toxics 2020, 8, 49. https://doi.org/10.3390/toxics8030049
Reyes-Goya C, Santana-Garrido Á, Soto-Astacio E, Aramburu Ó, Zambrano S, Mate A, Vázquez CM. Mechanism of Vascular Toxicity in Rats Subjected to Treatment with a Tyrosine Kinase Inhibitor. Toxics. 2020; 8(3):49. https://doi.org/10.3390/toxics8030049
Chicago/Turabian StyleReyes-Goya, Claudia, Álvaro Santana-Garrido, Estefanía Soto-Astacio, Óscar Aramburu, Sonia Zambrano, Alfonso Mate, and Carmen M. Vázquez. 2020. "Mechanism of Vascular Toxicity in Rats Subjected to Treatment with a Tyrosine Kinase Inhibitor" Toxics 8, no. 3: 49. https://doi.org/10.3390/toxics8030049