Ir-Ni Bimetallic OER Catalysts Prepared by Controlled Ni Electrodeposition on Irpoly and Ir(111)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Substrates
Ir(111) Single Crystal and Polycrystalline Ir (Irpoly)
2.2. Electrochemical Characterization
2.2.1. Ni Sweep Voltammetry
2.2.2. Controlled-Potential Technique
2.2.3. Preparation of Ni Overlayers (OLs) and Ni (near-) Surface Alloys (NSAs)
2.2.4. Electrochemical OER Protocol at RDE
2.2.5. CO Stripping Protocol
2.3. Theoretical Characterization
Density Functional Theory (DFT) Calculated H-Ir Binding Energies
2.4. Physicochemical Characterization
2.4.1. Inductively Coupled Plasma Mass Spectrometry (ICP-MS)
2.4.2. Atomic Force Microscopy (AFM)
2.4.3. X-ray Photoelectron Spectroscopy (XPS)
2.5. Mass Spectrometry
Differential Electrochemical Mass Spectrometry (DEMS)
3. Results and Discussion
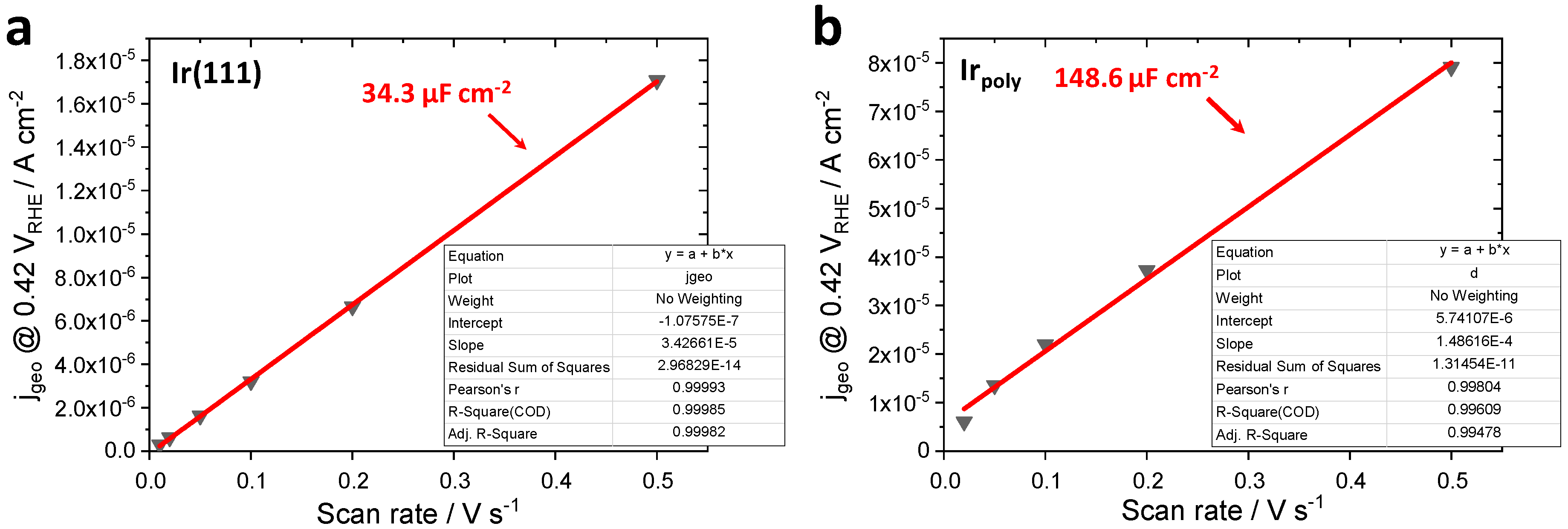
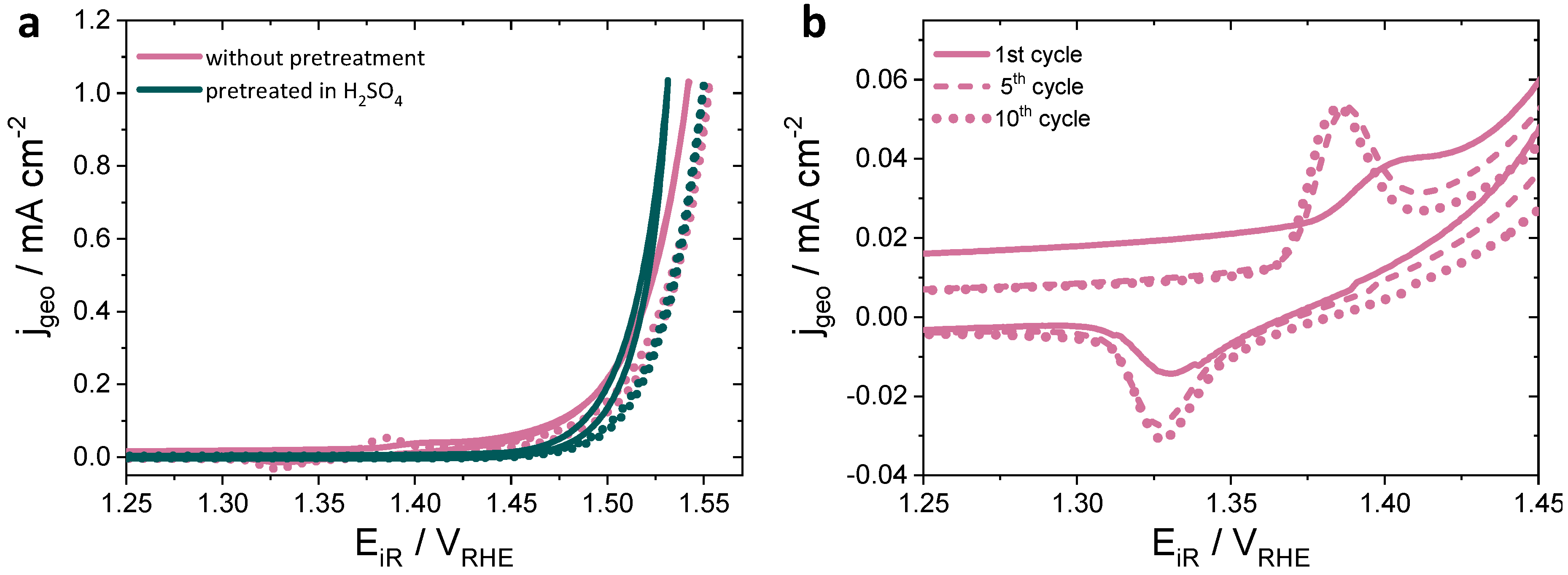
3.1. Roughness Factor (RF) of Irpoly
3.2. Surface Chemical Characterization
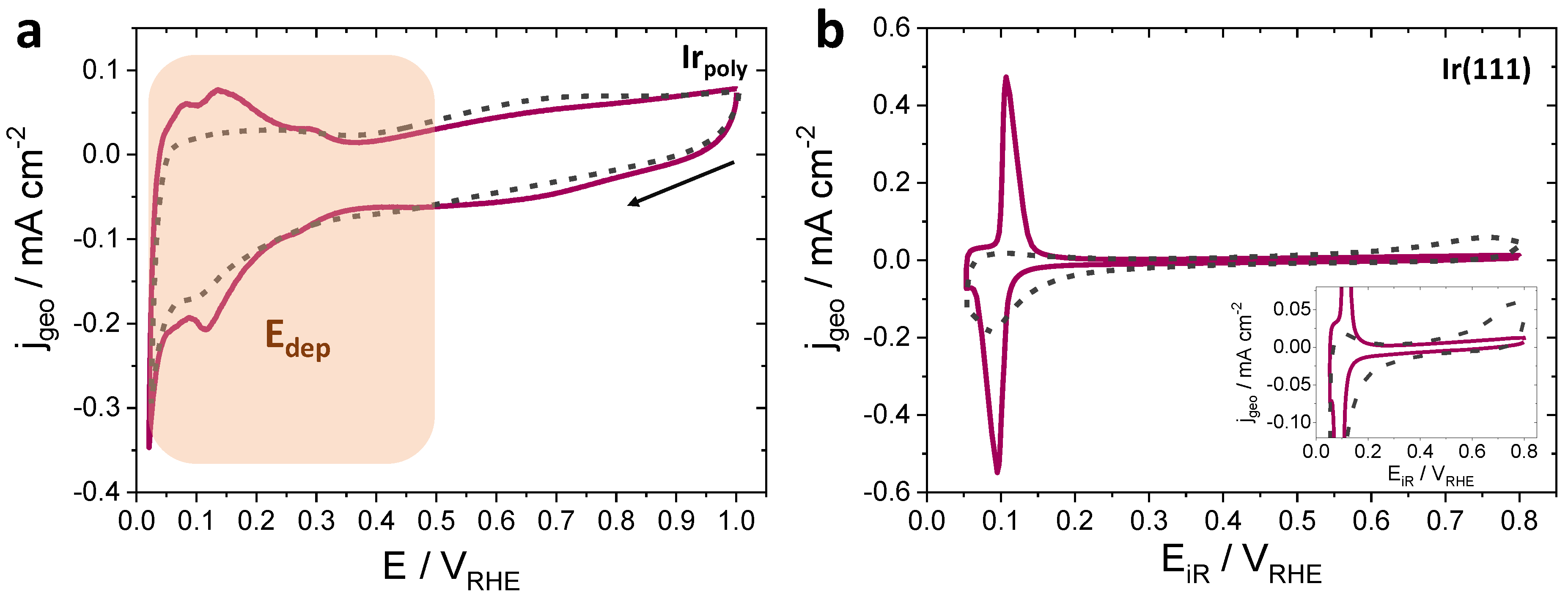
3.2.1. Ni Deposition on Irpoly and Ir(111) by Sweep Voltammetry
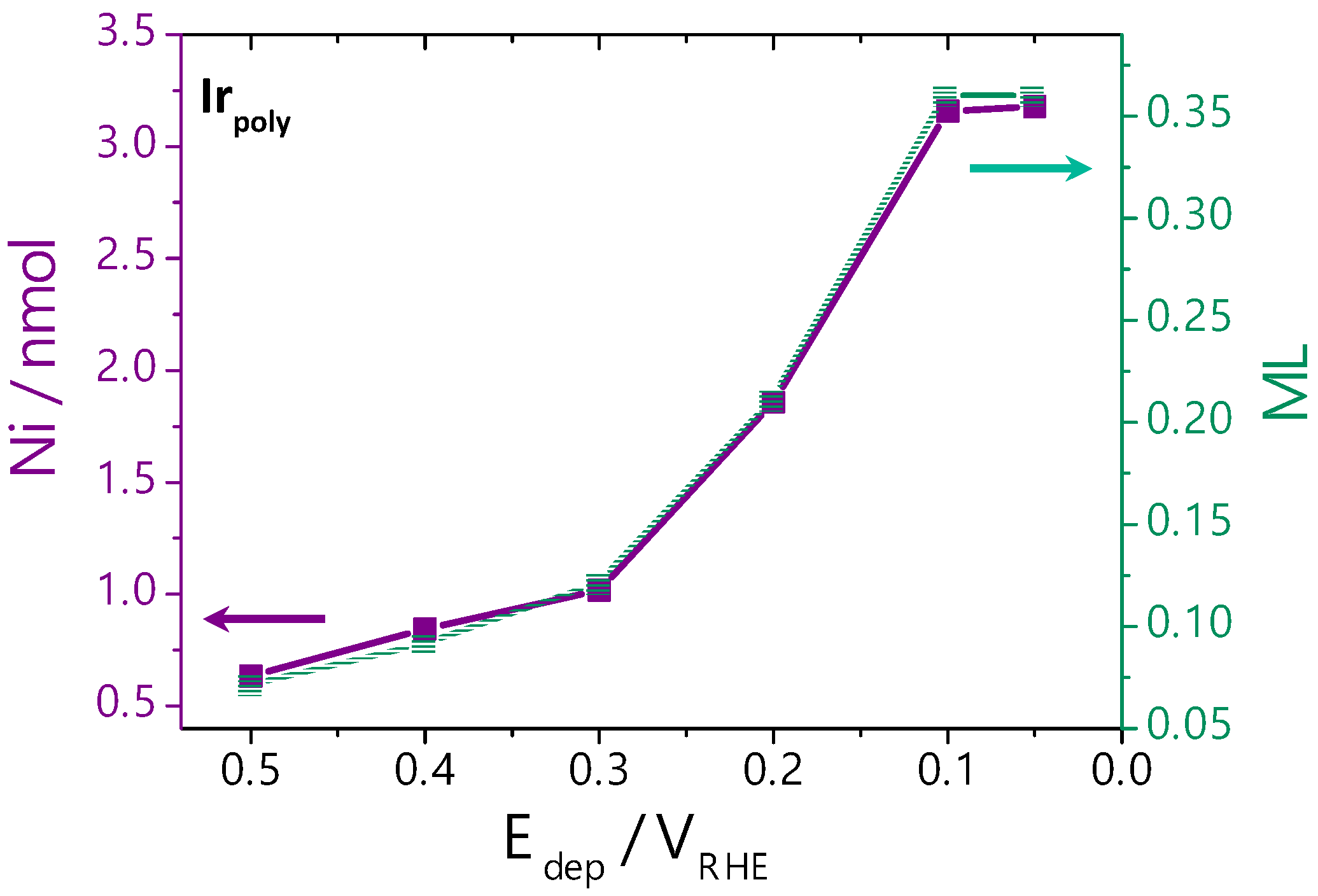
3.2.2. Ni Deposition on Irpoly by the Controlled-Potential Technique
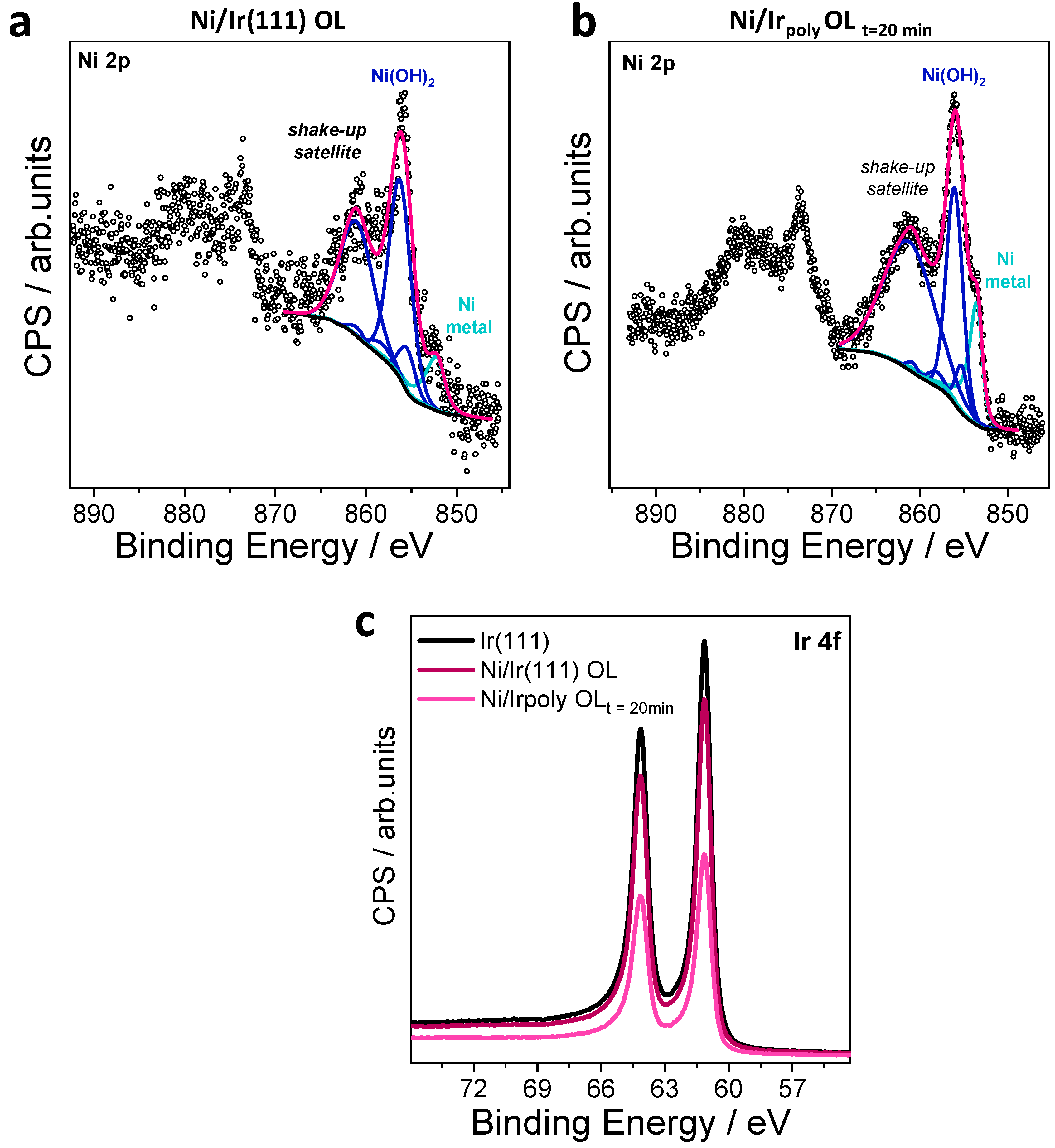
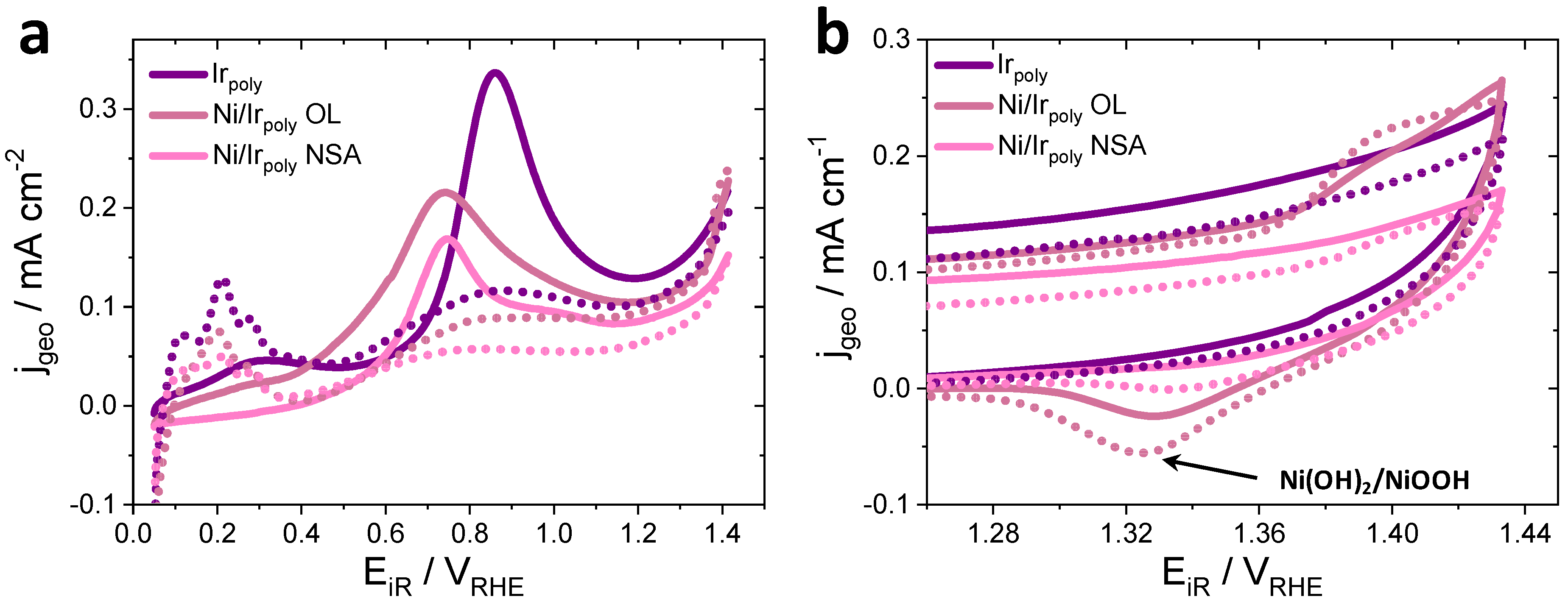
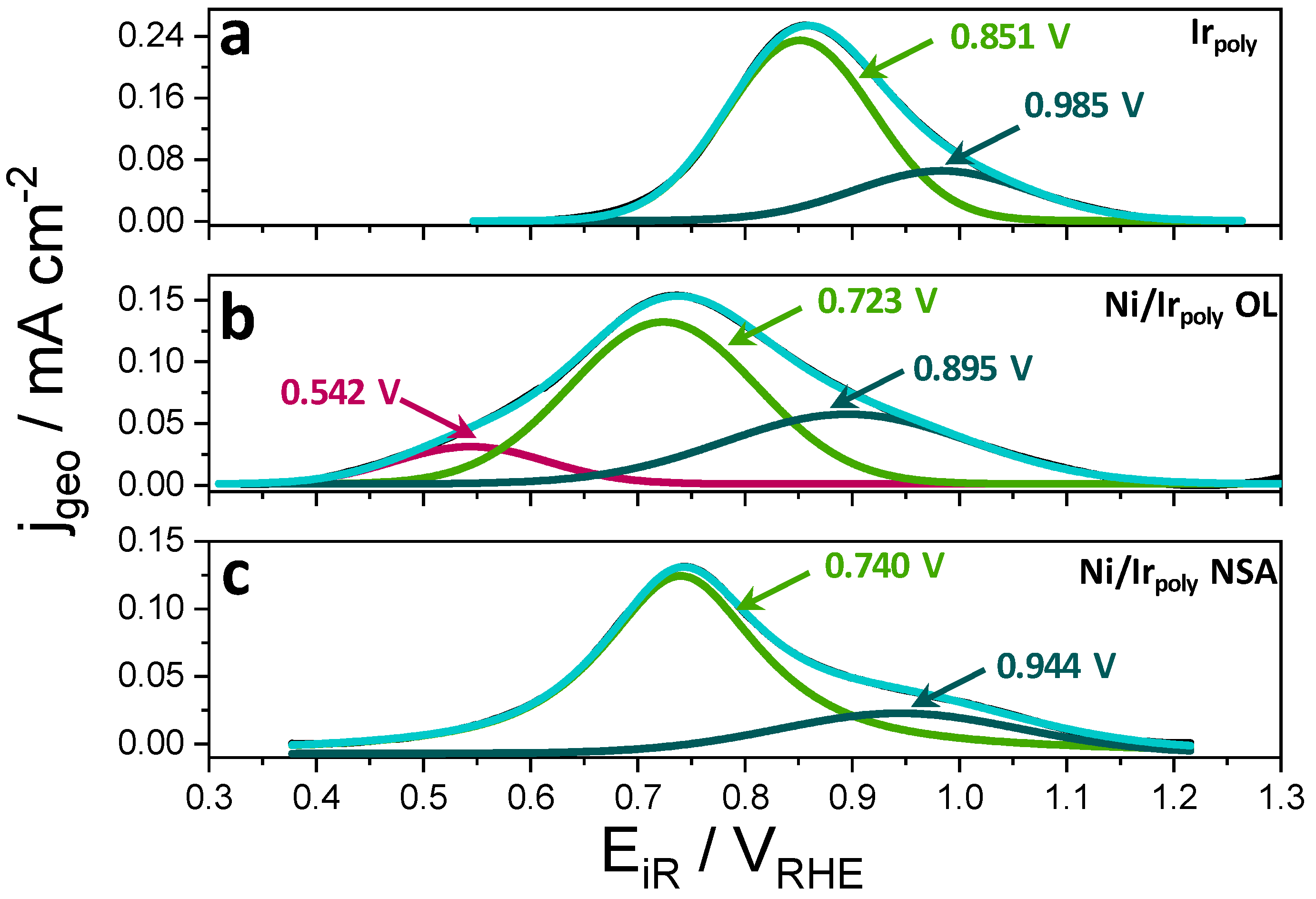
3.2.3. Surface Chemical State of Ni/Ir OL
3.3. Structure-Activity Correlations
3.3.1. Oxygen Evolution Reaction (OER) and Faraday Efficiencies (FE)
3.3.2. CO Stripping Voltammetry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cherevko, S.; Geiger, S.; Kasian, O.; Kulyk, N.; Grote, J.-P.; Savan, A.; Shrestha, B.R.; Merzlikin, S.; Breitbach, B.; Ludwig, A.; et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 2016, 262, 170–180. [Google Scholar] [CrossRef]
- Kasian, O.; Geiger, S.; Stock, P.; Polymeros, G.; Breitbach, B.; Savan, A.; Ludwig, A.; Cherevko, S.; Mayrhofer, K.J.J. On the Origin of the Improved Ruthenium Stability in RuO2–IrO2 Mixed Oxides. J. Electrochem. Soc. 2016, 163, F3099–F3104. [Google Scholar] [CrossRef]
- Schmeisser, D.; Jacobi, K. Photoelectron spectroscopy of nickel on zinc oxide in the monolayer and submonolayer range. Surf. Sci. 1979, 88, 138–152. [Google Scholar] [CrossRef]
- Gaebler, W.; Jacobi, K.; Ranke, W. The structure and electronic properties of thin palladium films on zinc oxide studied by AES and UPS. Surf. Sci. 1978, 75, 355–367. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Near-surface alloys for hydrogen fuel cell applications. Catal. Today 2006, 111, 52–58. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Alloy catalysts designed from first principles. Nat. Mater. 2004, 3, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Mavrikakis, M. Surface and Subsurface Hydrogen: Adsorption Properties on Transition Metals and Near-Surface Alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef] [PubMed]
- Herron, J.A.; Mavrikakis, M. On the composition of bimetallic near-surface alloys in the presence of oxygen and carbon monoxide. Catal. Commun. 2014, 52, 65–71. [Google Scholar] [CrossRef]
- Kandoi, S.; Ferrin, P.A.; Mavrikakis, M. Hydrogen on and in Selected Overlayer Near-Surface Alloys and the Effect of Subsurface Hydrogen on the Reactivity of Alloy Surfaces. Top. Catal. 2010, 53, 384–392. [Google Scholar] [CrossRef]
- Kim, C.; Möller, T.; Schmidt, J.; Thomas, A.; Strasser, P. Suppression of Competing Reaction Channels by Pb Adatom Decoration of Catalytically Active Cu Surfaces During CO2 Electroreduction. ACS Catal. under review.
- Özer, E.; Paul, B.; Spöri, C.; Strasser, P. Coupled Inductive Annealing-Electrochemical Setup for Controlled Preparation and Characterization of Alloy Crystal Surface Electrodes. Small Methods 2018, 1800232. [Google Scholar] [CrossRef]
- Özer, E.; Spöri, C.; Reier, T.; Strasser, P. Iridium(1 1 1), Iridium(1 1 0), and Ruthenium(0 0 0 1) Single Crystals as Model Catalysts for the Oxygen Evolution Reaction: Insights into the Electrochemical Oxide Formation and Electrocatalytic Activity. ChemCatChem 2016, 9, 597–603. [Google Scholar] [CrossRef]
- Trotochaud, L.; Young, S.L.; Ranney, J.K.; Boettcher, S.W. Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation. J. Am. Chem. Soc. 2014, 136, 6744–6753. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Jusys, Z.; Massong, H.; Baltruschat, H. A New Approach for Simultaneous DEMS and EQCM: Electro-oxidation of Adsorbed CO on Pt and Pt-Ru. J. Electrochem. Soc. 1999, 146, 1093–1098. [Google Scholar] [CrossRef]
- Wonders, A.H.; Housmans, T.H.M.; Rosca, V.; Koper, M.T.M. On-line mass spectrometry system for measurements at single-crystal electrodes in hanging meniscus configuration. J. Appl. Electrochem. 2006, 36, 1215–1221. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligaard, T.; Nørskov, J.K. Ligand effects in heterogeneous catalysis and electrochemistry. Electrochim. Acta 2007, 52, 5512–5516. [Google Scholar] [CrossRef]
- Reier, T.; Pawolek, Z.; Cherevko, S.; Bruns, M.; Jones, T.; Teschner, D.; Selve, S.; Bergmann, A.; Nong, H.N.; Schlögl, R.; et al. Molecular Insight in Structure and Activity of Highly Efficient, Low-Ir Ir–Ni Oxide Catalysts for Electrochemical Water Splitting (OER). J. Am. Chem. Soc. 2015, 137, 13031–13040. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Sanchez Casalongue Hernan, G.; Ng May, L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In Situ Observation of Surface Species on Iridium Oxide Nanoparticles during the Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2014, 53, 7169–7172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, M.; Valero-Vidal, C.; Eichhorn, J.; Toma, F.M.; Ross, P.N.; Yano, J.; Liu, Z.; Crumlin, E.J. Elucidating the alkaline oxygen evolution reaction mechanism on platinum. J. Mater. Chem. A 2017, 5, 11634–11643. [Google Scholar] [CrossRef] [Green Version]
- Doyle, R.L.; Lyons, M.E.G. The Oxygen Evolution Reaction: Mechanistic Concepts and Catalyst Design. In Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices, 1st ed.; Giménez, S., Bisquert, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 41–104. ISBN 978–3-319-29641-8. [Google Scholar]
- Gong, L.; Ren, D.; Deng, Y.; Yeo, B.S. Efficient and Stable Evolution of Oxygen Using Pulse-Electrodeposited Ir/Ni Oxide Catalyst in Fe-Spiked KOH Electrolyte. ACS Appl. Mater. Interfaces 2016, 8, 15985–15990. [Google Scholar] [CrossRef]
- Nong, H.N.; Oh, H.S.; Reier, T.; Willinger, E.; Willinger, M.G.; Petkov, V.; Teschner, D.; Strasser, P. Oxide-Supported IrNiOx Core–Shell Particles as Efficient, Cost-Effective, and Stable Catalysts for Electrochemical Water Splitting. Angew. Chem. 2015, 127, 3018–3022. [Google Scholar] [CrossRef]
- Wang, C.; Sui, Y.; Xu, M.; Liu, C.; Xiao, G.; Zou, B. Synthesis of Ni–Ir Nanocages with Improved Electrocatalytic Performance for the Oxygen Evolution Reaction. ACS Sustain. Chem. Eng. 2017, 5, 9787–9792. [Google Scholar] [CrossRef]
- Bode, H.; Dehmelt, K.; Witte, J. Zur kenntnis der nickelhydroxidelektrode—I.Über das nickel (II)-hydroxidhydrat. Electrochim. Acta 1966, 11, 1079–1087. [Google Scholar] [CrossRef]
- Corrigan, D.A.; Knight, S.L. Electrochemical and Spectroscopic Evidence on the Participation of Quadrivalent Nickel in the Nickel Hydroxide Redox Reaction. J. Electrochem. Soc. 1989, 136, 613–619. [Google Scholar] [CrossRef]
- Pfeifer, V.; Jones, T.E.; Velasco Velez, J.J.; Arrigo, R.; Piccinin, S.; Havecker, M.; Knop-Gericke, A.; Schlögl, R. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surfaces. Chem. Sci. 2017, 8, 2143–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, V.; Jones, T.E.; Velasco Velez, J.J.; Massue, C.; Greiner, M.T.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Allan, J.; et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 2016, 18, 2292–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nong, H.N.; Reier, T.; Oh, H.-S.; Gliech, M.; Paciok, P.; Vu, T.H.T.; Teschner, D.; Heggen, M.; Petkov, V.; Schlögl, R.; et al. A unique oxygen ligand environment facilitates water oxidation in hole-doped IrNiOx core–shell electrocatalysts. Nat. Catal. 2018. [Google Scholar] [CrossRef]
- Seitz, L.C.; Dickens, C.F.; Nishio, K.; Hikita, Y.; Montoya, J.; Doyle, A.; Kirk, C.; Vojvodic, A.; Hwang, H.Y.; Norskov, J.K.; et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 2016, 353, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Ahmadi, M.; Behafarid, F.; Gan, L.; Neumann, M.; Heggen, M.; Cuenya, B.R.; Strasser, P. Shape-selected bimetallic nanoparticle electrocatalysts: Evolution of their atomic-scale structure, chemical composition, and electrochemical reactivity under various chemical environments. Faraday Discuss. 2013, 162, 91–112. [Google Scholar] [CrossRef] [PubMed]
- Rudi, S.; Cui, C.; Gan, L.; Strasser, P. Comparative study of the electrocatalytically active surface areas (ECSAs) of Pt alloy nanoparticles evaluated by Hupd and CO-stripping voltammetry. Electrocatalysis 2014, 5, 408–418. [Google Scholar] [CrossRef]
- Beermann, V.; Gocyla, M.; Kühl, S.; Padgett, E.; Schmies, H.; Goerlin, M.; Erini, N.; Shviro, M.; Heggen, M.; Dunin-Borkowski, R.E.; et al. Tuning the Electrocatalytic Oxygen Reduction Reaction Activity and Stability of Shape-Controlled Pt–Ni Nanoparticles by Thermal Annealing—Elucidating the Surface Atomic Structural and Compositional Changes. J. Am. Chem. Soc. 2017, 139, 16536–16547. [Google Scholar] [CrossRef]
- Chatenet, M.; Soldo-Olivier, Y.; Chaînet, E.; Faure, R. Understanding CO-stripping mechanism from NiUPD/Pt(110) in view of the measured nickel formal partial charge number upon underpotential deposition on platinum surfaces in sulphate media. Electrochim. Acta 2007, 53, 369–376. [Google Scholar] [CrossRef]
- Spendelow, J.S.; Goodpaster, J.D.; Kenis, P.J.A.; Wieckowski, A. Mechanism of CO Oxidation on Pt(111) in Alkaline Media. J. Phys. Chem. B 2006, 110, 9545–9555. [Google Scholar] [CrossRef]
- García, G.; Koper, M.T.M. Stripping voltammetry of carbon monoxide oxidation on stepped platinum single-crystal electrodes in alkaline solution. Phys. Chem. Chem. Phys. 2008, 10, 3802–3811. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.P.; Rodes, A.; Feliu, J.M.; Koper, M.T.M.; van Santen, R.A. Role of Crystalline Defects in Electrocatalysis: CO Adsorption and Oxidation on Stepped Platinum Electrodes As Studied by in situ Infrared Spectroscopy. J. Phys. Chem. B 2002, 106, 9863–9872. [Google Scholar] [CrossRef]
- López-Cudero, A.; Cuesta, A.; Gutiérrez, C. Potential dependence of the saturation CO coverage of Pt electrodes: The origin of the pre-peak in CO-stripping voltammograms. Part 1: Pt(111). J. Electroanal. Chem. 2005, 579, 1–12. [Google Scholar] [CrossRef]
- Garrick, T.R.; Moylan, T.E.; Carpenter, M.K.; Kongkanand, A. Editors’ Choice—Electrochemically Active Surface Area Measurement of Aged Pt Alloy Catalysts in PEM Fuel Cells by CO Stripping. J. Electrochem. Soc. 2017, 164, F55–F59. [Google Scholar] [CrossRef]
- Hasché, F.; Oezaslan, M.; Strasser, P. Activity, stability and degradation of multi walled carbon nanotube (MWCNT) supported Pt fuel cell electrocatalysts. Phys. Chem. Chem. Phys. 2010, 12, 15251–15258. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Anderson, L.; Chen, Y.; Pan, M.; Abel Chuang, P.-Y. New insights into evaluating catalyst activity and stability for oxygen evolution reactions in alkaline media. Sustain. Energy Fuels 2018, 2, 237–251. [Google Scholar] [CrossRef]
- Bandarenka Aliaksandr, S.; Varela Ana, S.; Karamad, M.; Calle-Vallejo, F.; Bech, L.; Perez-Alonso Francisco, J.; Rossmeisl, J.; Stephens Ifan, E.L.; Chorkendorff, I. Design of an Active Site towards Optimal Electrocatalysis: Overlayers, Surface Alloys and Near-Surface Alloys of Cu/Pt(111). Angew. Chem. Int. Ed. 2012, 51, 11845–11848. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen adsorption, absorption and diffusion on and in transition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Hagedorn, C.J.; Weiss, M.J.; Weinberg, W.H. Dissociative chemisorption of hydrogen on Ir(111): Evidence for terminal site adsorption. Phys. Rev. B 1999, 60, R14016–R14018. [Google Scholar] [CrossRef]
- Krekelberg, W.P.; Greeley, J.; Mavrikakis, M. Atomic and Molecular Adsorption on Ir(111). J. Phys. Chem. B 2004, 108, 987–994. [Google Scholar] [CrossRef]
- Lerch, D.; Klein, A.; Schmidt, A.; Müller, S.; Hammer, L.; Heinz, K.; Weinert, M. Unusual adsorption site of hydrogen on the unreconstructed Ir(100) surface. Phys. Rev. B 2006, 73, 075430. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Steps | Calculation |
|---|---|
| surface area calculation | |
| surface area/10−15 cm2 | 1.277 |
| atoms per (111) face in unit cell | 2 |
| atoms per unit area/1015 cm−2 | 1.566 |
| amount of substance per surface area/nmol cm−2 | 2.6 |
| amount of substance for monolayer/nmol ML−1 | 2.04 |
| mass of Ni per monolayer/µg ML−1 | 0.12 |
| mass of Ni per monolayer for Irpoly/µg ML−1 | 0.52 |
| Face | CN | Site | H BE/eV | H-Ir Bond Distance/pm |
|---|---|---|---|---|
| (100) | 6 | bridging | −2.91 | 2 × 181 |
| (111) | 9 | atop | −2.73 | 159 |
| (110) | 7 | bridging | −2.74 | 2 × 181 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özer, E.; Sinev, I.; Mingers, A.M.; Araujo, J.; Kropp, T.; Mavrikakis, M.; Mayrhofer, K.J.J.; Cuenya, B.R.; Strasser, P. Ir-Ni Bimetallic OER Catalysts Prepared by Controlled Ni Electrodeposition on Irpoly and Ir(111). Surfaces 2018, 1, 165-186. https://doi.org/10.3390/surfaces1010013
Özer E, Sinev I, Mingers AM, Araujo J, Kropp T, Mavrikakis M, Mayrhofer KJJ, Cuenya BR, Strasser P. Ir-Ni Bimetallic OER Catalysts Prepared by Controlled Ni Electrodeposition on Irpoly and Ir(111). Surfaces. 2018; 1(1):165-186. https://doi.org/10.3390/surfaces1010013
Chicago/Turabian StyleÖzer, Ebru, Ilya Sinev, Andrea M. Mingers, Jorge Araujo, Thomas Kropp, Manos Mavrikakis, Karl J. J. Mayrhofer, Beatriz Roldan Cuenya, and Peter Strasser. 2018. "Ir-Ni Bimetallic OER Catalysts Prepared by Controlled Ni Electrodeposition on Irpoly and Ir(111)" Surfaces 1, no. 1: 165-186. https://doi.org/10.3390/surfaces1010013