
Biomolecular Modifications Linked to Oxidative Stress in Amyotrophic Lateral Sclerosis: Determining Promising Biomarkers Related to Oxidative Stress

Abstract
:1. Introduction
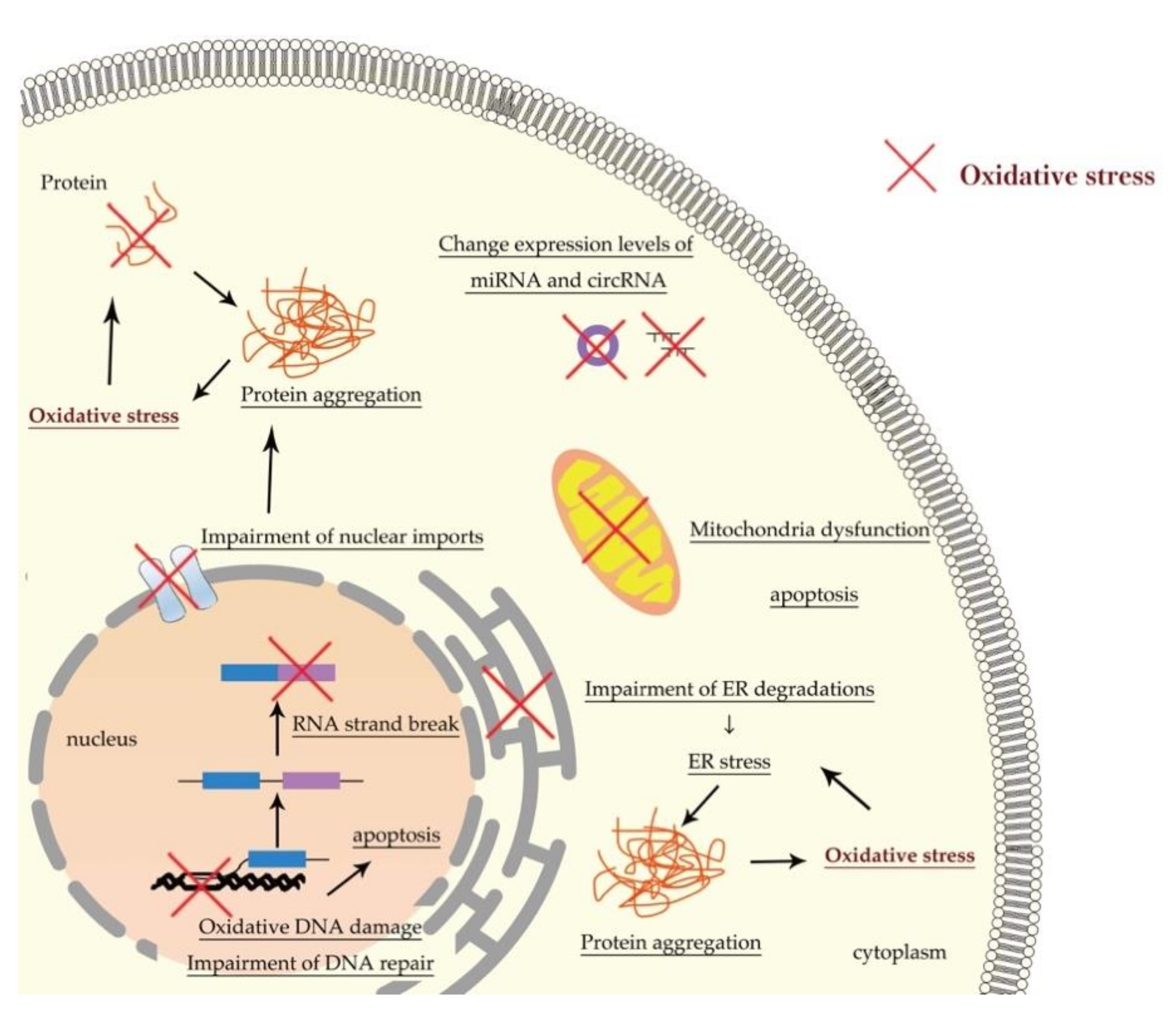
2. Oxidative Biomolecular Modification Leading to Neurodegeneration
2.1. Oxidative Modifications of Proteins Linked to Protein Aggregation
2.2. Oxidative DNA Damage Linked to Apoptosis
2.3. Oxidative RNA Damage
3. Biomolecular Modifications Associated with Oxidative Stress in ALS
3.1. Abnormal Protein Aggregations and Oxidative Stress in ALS
3.2. Oxidative DNA Damage and Impairment of DNA Repair in ALS
3.3. RNA Modifications Associated with Oxidative Stress in ALS
3.4. Treatment Linked to Oxidative Stress in ALS
4. Promising Biomarker Candidates Associated with Oxidative Stress in ALS
4.1. Protein
4.2. RNA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Carrera-Julia, S.; Moreno, M.L.; Barrios, C.; de la Rubia Orti, J.E.; Drehmer, E. Antioxidant Alternatives in the Treatment of Amyotrophic Lateral Sclerosis: A Comprehensive Review. Front. Physiol. 2020, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Cadenas, E. Oxidative stress: Damage to intact cells and organs. Philos. Trans. R. Soc. B Biol. Sci. 1985, 311, 617–631. [Google Scholar] [CrossRef]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in ALS: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated With Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Jagaraj, C.J.; Parakh, S.; Atkin, J.D. Emerging Evidence Highlighting the Importance of Redox Dysregulation in the Pathogenesis of Amyotrophic Lateral Sclerosis (ALS). Front. Cell. Neurosci. 2020, 14, 581950. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working, G. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Rappa, G.; Puglisi, C.; Santos, M.F.; Forte, S.; Memeo, L.; Lorico, A. Extracellular Vesicles from Thyroid Carcinoma: The New Frontier of Liquid Biopsy. Int. J. Mol. Sci. 2019, 20, 1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candelario, K.M.; Steindler, D.A. The role of extracellular vesicles in the progression of neurodegenerative disease and cancer. Trends Mol. Med. 2014, 20, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular Vesicle-Mediated Cell-Cell Communication in the Nervous System: Focus on Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, T.; Yamashita, T.; Tamaoka, A.; Kwak, S. Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and other Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int J. Mol. Sci 2018, 19, 3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.K. Oxidative stress in neurodegeneration: Cause or consequence? Nat. Med. 2004, 10, S18–S25. [Google Scholar] [CrossRef]
- Quinlan, S.; Kenny, A.; Medina, M.; Engel, T.; Jimenez-Mateos, E.M. MicroRNAs in Neurodegenerative Diseases. Int. Rev. Cell Mol. Biol. 2017, 334, 309–343. [Google Scholar] [CrossRef] [PubMed]
- Maniati, M.S.; Maniati, M.; Yousefi, T.; Ahmadi-Ahangar, A.; Tehrani, S.S. New insights into the role of microRNAs and long noncoding RNAs in most common neurodegenerative diseases. J. Cell. Biochem 2019. [Google Scholar] [CrossRef] [PubMed]
- Quek, C.; Hill, A.F. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun 2017, 483, 1178–1186. [Google Scholar] [CrossRef]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitia, R.; Molteni, S.N. Stress, protein (mis)folding, and signaling: The redox connection. Sci. STKE 2004, 2004, pe27. [Google Scholar] [CrossRef] [PubMed]
- Gianazza, E.; Crawford, J.; Miller, I. Detecting oxidative post-translational modifications in proteins. Amino Acids 2007, 33, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Weids, A.J.; Ibstedt, S.; Tamas, M.J.; Grant, C.M. Distinct stress conditions result in aggregation of proteins with similar properties. Sci. Rep. 2016, 6, 24554. [Google Scholar] [CrossRef] [Green Version]
- Serebryany, E.; Woodard, J.C.; Adkar, B.V.; Shabab, M.; King, J.A.; Shakhnovich, E.I. An Internal Disulfide Locks a Misfolded Aggregation-prone Intermediate in Cataract-linked Mutants of Human γD-Crystallin. J. Biol. Chem. 2016, 291, 19172–19183. [Google Scholar] [CrossRef] [Green Version]
- Oka, O.B.; Bulleid, N.J. Forming disulfides in the endoplasmic reticulum. Biochim. Biophys. Acta 2013, 1833, 2425–2429. [Google Scholar] [CrossRef] [Green Version]
- Karri, S.; Singh, S.; Paripati, A.K.; Marada, A.; Krishnamoorthy, T.; Guruprasad, L.; Balasubramanian, D.; Sepuri, N.B.V. Adaptation of Mge1 to oxidative stress by local unfolding and altered Interaction with mitochondrial Hsp70 and Mxr2. Mitochondrion 2019, 46, 140–148. [Google Scholar] [CrossRef]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Kodiha, M.; Chu, A.; Matusiewicz, N.; Stochaj, U. Multiple mechanisms promote the inhibition of classical nuclear import upon exposure to severe oxidative stress. Cell Death Differ. 2004, 11, 862–874. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Sanchez de Groot, N.; Gomes, R.A.; Villar-Pique, A.; Babu, M.M.; Coelho, A.V.; Ventura, S. Proteome response at the edge of protein aggregation. Open Biol. 2015, 5, 140221. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Walker, C.; El-Khamisy, S.F. Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain 2018, 141, 1247–1262. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Radak, Z.; Zhao, Z.; Goto, S.; Koltai, E. Age-associated neurodegeneration and oxidative damage to lipids, proteins and DNA. Mol. Asp. Med. 2011, 32, 305–315. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrador, E.; Salvador-Palmer, R.; Lopez-Blanch, R.; Jihad-Jebbar, A.; Valles, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Coppede, F. An overview of DNA repair in amyotrophic lateral sclerosis. Sci. World J. 2011, 11, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gredilla, R.; Weissman, L.; Yang, J.L.; Bohr, V.A.; Stevnsner, T. Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 694–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Wilson, D.M., III; Bohr, V.A. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair 2007, 6, 544–559. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Chen, X.; Li, Z.; Ye, W.; Ding, H.; Li, P.; Aung, L.H.H. Role of RNA Oxidation in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 5022. [Google Scholar] [CrossRef]
- Nunomura, A.; Hofer, T.; Moreira, P.I.; Castellani, R.J.; Smith, M.A.; Perry, G. RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 2009, 118, 151–166. [Google Scholar] [CrossRef]
- Simms, C.L.; Zaher, H.S. Quality control of chemically damaged RNA. Cell. Mol. Life Sci. 2016, 73, 3639–3653. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, A.C.; Resendiz, M.J.; Greenberg, M.M. Direct strand scission from a nucleobase radical in RNA. J. Am. Chem. Soc. 2010, 132, 3668–3669. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Chock, P.B.; Stadtman, E.R. Oxidized messenger RNA induces translation errors. Proc. Natl. Acad. Sci. USA 2007, 104, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Dimayuga, E.; Keller, J.N. Oxidative stress alters neuronal RNA- and protein-synthesis: Implications for neural viability. Free Radic. Res. 2007, 41, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Shan, X.; Tashiro, H.; Lin, C.L. The identification and characterization of oxidized RNAs in Alzheimer’s disease. J. Neurosci. 2003, 23, 4913–4921. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Chang, Y.; Lin, C.L. Messenger RNA oxidation is an early event preceding cell death and causes reduced protein expression. FASEB J. 2007, 21, 2753–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From Oxidative Stress Damage to Pathways, Networks, and Autophagy via MicroRNAs. Oxid. Med. Cell. Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Gao, J.; Ding, S.L.; Wang, K.; Jiao, J.Q.; Wang, Y.; Sun, T.; Zhou, L.Y.; Long, B.; Zhang, X.J.; et al. Oxidative Modification of miR-184 Enables It to Target Bcl-xL and Bcl-w. Mol. Cell 2015, 59, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 Pathway in Age-Related Neurological Disorders: Insights into MicroRNAs. Cell. Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef]
- Li, L.; Ni, Z.; Si, X.; Jiang, L.; Sang, H.; Xia, W.; Chen, Z.; Huang, J.; Jin, J.; Shao, A.; et al. Emerging Clues of Regulatory Roles of Circular RNAs through Modulating Oxidative Stress: Focus on Neurological and Vascular Diseases. Oxid. Med. Cell. Longev. 2021, 2021, 6659908. [Google Scholar] [CrossRef]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Bogdanov, M.; Brown, R.H.; Matson, W.; Smart, R.; Hayden, D.; O’Donnell, H.; Flint Beal, M.; Cudkowicz, M. Increased oxidative damage to DNA in ALS patients. Free Radic. Biol. Med. 2000, 29, 652–658. [Google Scholar] [CrossRef]
- Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. [Google Scholar] [CrossRef] [Green Version]
- Singh, E.; Devasahayam, G. Neurodegeneration by oxidative stress: A review on prospective use of small molecules for neuroprotection. Mol. Biol. Rep. 2020, 47, 3133–3140. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar] [CrossRef]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (ALS): Immunohistochemical studies on sporadic ALS, superoxide dismutase 1 (SOD1)-mutated familial ALS, and SOD1-mutated ALS animal models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef]
- Mitsumoto, H.; Santella, R.M.; Liu, X.; Bogdanov, M.; Zipprich, J.; Wu, H.C.; Mahata, J.; Kilty, M.; Bednarz, K.; Bell, D.; et al. Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 2008, 9, 177–183. [Google Scholar] [CrossRef]
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765. [Google Scholar] [CrossRef]
- Walczak, J.; Debska-Vielhaber, G.; Vielhaber, S.; Szymanski, J.; Charzynska, A.; Duszynski, J.; Szczepanowska, J. Distinction of sporadic and familial forms of ALS based on mitochondrial characteristics. FASEB J. 2019, 33, 4388–4403. [Google Scholar] [CrossRef]
- Ikawa, M.; Okazawa, H.; Tsujikawa, T.; Matsunaga, A.; Yamamura, O.; Mori, T.; Hamano, T.; Kiyono, Y.; Nakamoto, Y.; Yoneda, M. Increased oxidative stress is related to disease severity in the ALS motor cortex. A PET study. Neurology 2015, 84, 2033–2039. [Google Scholar] [CrossRef]
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Pare, B.; Lehmann, M.; Beaudin, M.; Nordstrom, U.; Saikali, S.; Julien, J.P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 14223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuda, E.; Takei, Y.I.; Ohara, S.; Fujiwara, N.; Hozumi, I.; Furukawa, Y. Wild-type Cu/Zn-superoxide dismutase is misfolded in cerebrospinal fluid of sporadic amyotrophic lateral sclerosis. Mol. Neurodegener. 2019, 14, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Tak, Y.J.; Park, J.H.; Rhim, H.; Kang, S. ALS-Related Mutant SOD1 Aggregates Interfere with Mitophagy by Sequestering the Autophagy Receptor Optineurin. Int. J. Mol. Sci. 2020, 21, 7525. [Google Scholar] [CrossRef]
- Farrawell, N.E.; Lambert-Smith, I.; Mitchell, K.; McKenna, J.; McAlary, L.; Ciryam, P.; Vine, K.L.; Saunders, D.N.; Yerbury, J.J. SOD1A4V aggregation alters ubiquitin homeostasis in a cell model of ALS. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Beckman, J.S.; Estévez, A.G.; Crow, J.P.; Barbeito, L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001, 24, S15–S20. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell. Longev. 2020, 2020, 5021694. [Google Scholar] [CrossRef]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128, 1686–1706. [Google Scholar] [CrossRef]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [Green Version]
- Uchino, M.; Ando, Y.; Tanaka, Y.; Nakamura, T.; Uyama, E.; Mita, S.; Murakami, T.; Ando, M. Decrease in Cu/Zn- and Mn-superoxide dismutase activities in brain and spinal cord of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 127, 61–67. [Google Scholar] [CrossRef]
- O’Reilly, S.A.; Roedica, J.; Nagy, D.; Hallewell, R.A.; Alderson, K.; Marklund, S.L.; Kuby, J.; Kushner, P.D. Motor neuron-astrocyte interactions and levels of Cu,Zn superoxide dismutase in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 1995, 131, 203–210. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Toledo, J.B.; Robinson, J.L.; Irwin, D.J.; Grossman, M.; Suh, E.; Van Deerlin, V.M.; Wood, E.M.; Baek, Y.; et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yokoshi, M.; Okada, H.; Kawahara, Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat. Commun. 2015, 6, 6183. [Google Scholar] [CrossRef] [Green Version]
- Goh, C.W.; Lee, I.C.; Sundaram, J.R.; George, S.E.; Yusoff, P.; Brush, M.H.; Sze, N.S.K.; Shenolikar, S. Chronic oxidative stress promotes GADD34-mediated phosphorylation of the TAR DNA-binding protein TDP-43, a modification linked to neurodegeneration. J. Biol. Chem. 2018, 293, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Unger, T.; Trojanowski, J.Q.; Lee, V.M. Redox signalling directly regulates TDP-43 via cysteine oxidation and disulphide cross-linking. EMBO J. 2012, 31, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P., III; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moujalled, D.; Grubman, A.; Acevedo, K.; Yang, S.; Ke, Y.D.; Moujalled, D.M.; Duncan, C.; Caragounis, A.; Perera, N.D.; Turner, B.J.; et al. TDP-43 mutations causing amyotrophic lateral sclerosis are associated with altered expression of RNA-binding protein hnRNP K and affect the Nrf2 antioxidant pathway. Hum. Mol. Genet. 2017, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free Radic. Biol. Med. 2002, 32, 813–821. [Google Scholar] [CrossRef]
- Radak, Z.; Boldogh, I. 8-Oxo-7,8-dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free Radic. Biol. Med. 2010, 49, 587–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Miyata, S.; Jono, T.; Horiuchi, S.; Hirano, A.; Kato, S.; Sasaki, S.; Asayama, K.; Kobayashi, M. Nonoxidative protein glycation is implicated in familial amyotrophic lateral sclerosis with superoxide dismutase-1 mutation. Acta Neuropathol. 2000, 100, 275–284. [Google Scholar] [CrossRef]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Warita, H.; Hayashi, T.; Murakami, T.; Manabe, Y.; Abe, K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Mol. Brain Res. 2001, 89, 147–152. [Google Scholar] [CrossRef]
- Aguirre, N.; Beal, M.F.; Matson, W.R.; Bogdanov, M.B. Increased oxidative damage to DNA in an animal model of amyotrophic lateral sclerosis. Free Radic. Res. 2005, 39, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef]
- Barbosa, L.F.; Cerqueira, F.M.; Macedo, A.F.; Garcia, C.C.; Angeli, J.P.; Schumacher, R.I.; Sogayar, M.C.; Augusto, O.; Carri, M.T.; Di Mascio, P.; et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 2010, 1802, 462–471. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Bradley, W.G.; Krasin, F. A new hypothesis of the etiology of amyotrophic lateral sclerosis. The DNA hypothesis. Arch. Neurol. 1982, 39, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Angkeow, P.; Deshpande, S.S.; Qi, B.; Liu, Y.X.; Park, Y.C.; Jeon, B.H.; Ozaki, M.; Irani, K. Redox factor-1: An extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ. 2002, 9, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Fishel, M.L.; Wu, X.; Devlin, C.M.; Logsdon, D.P.; Jiang, Y.; Luo, M.; He, Y.; Yu, Z.; Tong, Y.; Lipking, K.P.; et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2015, 290, 3057–3068. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, M.; Moh, S.; Kim, H.; Kim, D.H. Cytoplasmic Restriction of Mutated SOD1 Impairs the DNA Repair Process in Spinal Cord Neurons. Cells 2019, 8, 1502. [Google Scholar] [CrossRef] [Green Version]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.J.; Cooper-Knock, J.; Heath, P.R.; Highley, J.R. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kisby, G.E.; Milne, J.; Sweatt, C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. NeuroReport 1997, 8, 1337–1340. [Google Scholar] [CrossRef]
- Manabe, Y.; Warita, H.; Murakami, T.; Shiote, M.; Hayashi, T.; Nagano, I.; Shoji, M.; Abe, K. Early decrease of redox factor-1 in spinal motor neurons of presymptomatic transgenic mice with a mutant SOD1 gene. Brain Res. 2001, 915, 104–107. [Google Scholar] [CrossRef]
- Kikuchi, H.; Furuta, A.; Nishioka, K.; Suzuki, S.O.; Nakabeppu, Y.; Iwaki, T. Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol. 2002, 103, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Henkel, J.S.; Beers, D.R.; Sengun, I.S.; Simpson, E.P.; Goodman, J.C.; Engelhardt, J.I.; Siklós, L.; Appel, S.H. PARP expression is increased in astrocytes but decreased in motor neurons in the spinal cord of sporadic ALS patients. J. Neuropathol. Exp. Neurol 2003, 62, 88–103. [Google Scholar] [CrossRef] [Green Version]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef]
- Bozzo, F.; Mirra, A.; Carri, M.T. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Chang, Y.; Kong, Q.; Shan, X.; Tian, G.; Ilieva, H.; Cleveland, D.W.; Rothstein, J.D.; Borchelt, D.R.; Wong, P.C.; Lin, C.L. Messenger RNA oxidation occurs early in disease pathogenesis and promotes motor neuron degeneration in ALS. PLoS ONE 2008, 3, e2849. [Google Scholar] [CrossRef]
- Lenzken, S.C.; Romeo, V.; Zolezzi, F.; Cordero, F.; Lamorte, G.; Bonanno, D.; Biancolini, D.; Cozzolino, M.; Pesaresi, M.G.; Maracchioni, A.; et al. Mutant SOD1 and mitochondrial damage alter expression and splicing of genes controlling neuritogenesis in models of neurodegeneration. Hum. Mutat. 2011, 32, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Pham, J.; Keon, M.; Brennan, S.; Saksena, N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst A Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. Int. J. Mol. Sci. 2020, 21, 3464. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Dong, D.; Reece, E.A.; Wang, A.R.; Yang, P. Oxidative stress-induced miR-27a targets the redox gene nuclear factor erythroid 2-related factor 2 in diabetic embryopathy. Am. J. Obstet. Gynecol. 2018, 218, 131.e1–136.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Lu, Y.; Zhang, M.; Zhang, Z.; Wang, K.; Lv, J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed. Pharmacother. 2017, 89, 1187–1195. [Google Scholar] [CrossRef]
- Pegoraro, V.; Merico, A.; Angelini, C. Micro-RNAs in ALS muscle: Differences in gender, age at onset and disease duration. J. Neurol. Sci. 2017, 380, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Aschrafi, A.; Kar, A.N.; Natera-Naranjo, O.; MacGibeny, M.A.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell. Mol. Life Sci. 2012, 69, 4017–4027. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Manfellotto, F.; Fiorentino, G.; Annunziata, A.; Biffali, E.; Pannone, R.; Federico, A. Wide-Ranging Analysis of MicroRNA Profiles in Sporadic Amyotrophic Lateral Sclerosis Using Next-Generation Sequencing. Front. Genet. 2018, 9, 310. [Google Scholar] [CrossRef]
- Li, C.; Wei, Q.; Gu, X.; Chen, Y.; Chen, X.; Cao, B.; Ou, R.; Shang, H. Decreased Glycogenolysis by miR-338-3p Promotes Regional Glycogen Accumulation Within the Spinal Cord of Amyotrophic Lateral Sclerosis Mice. Front. Mol. Neurosci. 2019, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- De Felice, B.; Annunziata, A.; Fiorentino, G.; Borra, M.; Biffali, E.; Coppola, C.; Cotrufo, R.; Brettschneider, J.; Giordana, M.L.; Dalmay, T.; et al. miR-338-3p is over-expressed in blood, CFS, serum and spinal cord from sporadic amyotrophic lateral sclerosis patients. Neurogenetics 2014, 15, 243–253. [Google Scholar] [CrossRef]
- Li, N.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech. Ageing Dev. 2011, 132, 75–85. [Google Scholar] [CrossRef]
- Zhou, F.; Zhang, C.; Guan, Y.; Chen, Y.; Lu, Q.; Jie, L.; Gao, H.; Du, H.; Zhang, H.; Liu, Y.; et al. Screening the expression characteristics of several miRNAs in G93A-SOD1 transgenic mouse: Altered expression of miRNA-124 is associated with astrocyte differentiation by targeting Sox2 and Sox9. J. Neurochem. 2018, 145, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.P.; Barabino, S.; Nizzardo, M.; et al. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 2018, 8, 10105. [Google Scholar] [CrossRef]
- Di Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef] [PubMed]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Siddiqui, S.; Gabriely, G.; Lanser, A.J.; Dake, B.; Murugaiyan, G.; Doykan, C.E.; Wu, P.M.; Gali, R.R.; Iyer, L.K.; et al. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J. Clin. Investig. 2012, 122, 3063–3087. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastiao, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar] [CrossRef]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Song, Y.; Li, M.; Li, J.C.; Wei, E.Q. Edaravone protects PC12 cells from ischemic-like injury via attenuating the damage to mitochondria. J. Zhejiang Univ. Sci. B 2006, 7, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Shoge, M.; Oda, E.; Yamamoto, Y.; Giddings, J.C.; Kashiwagi, S.; Suematsu, M.; Yamamoto, J. The free-radical scavenger, edaravone, augments NO release from vascular cells and platelets after laser-induced, acute endothelial injury in vivo. Platelets 2006, 17, 201–206. [Google Scholar] [CrossRef]
- Qi, X.; Okuma, Y.; Hosoi, T.; Nomura, Y. Edaravone protects against hypoxia/ischemia-induced endoplasmic reticulum dysfunction. J. Pharmacol. Exp. Ther. 2004, 311, 388–393. [Google Scholar] [CrossRef]
- Luo, L.; Song, Z.; Li, X.; Zeng, Y.; He, J. Efficacy and safety of edaravone in treatment of amyotrophic lateral sclerosis-a systematic review and meta-analysis. Neurol. Sci. 2019, 40, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Yamashita, S.; Ueyama, H.; Ishizaki, M.; Maeda, Y.; Ando, Y. Long-Term effects of edaravone on survival of patients with amyotrophic lateral sclerosis. ENeurologicalSci 2018, 11, 11–14. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, S.Y.; Park, D.; Park, J.S. Effect of edaravone therapy in Korean amyotrophic lateral sclerosis (ALS) patients. Neurol. Sci. 2020, 41, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Kimura, A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph. Lateral Scler. 2006, 7, 241–245. [Google Scholar] [CrossRef]
- Ohta, Y.; Yamashita, T.; Nomura, E.; Hishikawa, N.; Ikegami, K.; Osakada, Y.; Matsumoto, N.; Kawahara, Y.; Yunoki, T.; Takahashi, Y.; et al. Improvement of a decreased anti-oxidative activity by edaravone in amyotrophic lateral sclerosis patients. J. Neurol. Sci. 2020, 415, 116906. [Google Scholar] [CrossRef]
- Fortuna, A.; Gizzi, M.; Bello, L.; Martinelli, I.; Bertolin, C.; Pegoraro, E.; Corbetta, M.; Soraru, G.; Edaravone Study, G. Safety and efficacy of edaravone compared to historical controls in patients with amyotrophic lateral sclerosis from North-Eastern Italy. J. Neurol. Sci. 2019, 404, 47–51. [Google Scholar] [CrossRef]
- Lunetta, C.; Moglia, C.; Lizio, A.; Caponnetto, C.; Dubbioso, R.; Giannini, F.; Mata, S.; Mazzini, L.; Sabatelli, M.; Siciliano, G.; et al. The Italian multicenter experience with edaravone in amyotrophic lateral sclerosis. J. Neurol. 2020, 267, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Desnuelle, C.; Dib, M.; Garrel, C.; Favier, A. A double-blind, placebo-controlled randomized clinical trial of α-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph. Lateral Scler. Motor Neuron Disord. 2001, 2, 9–18. [Google Scholar] [CrossRef]
- Fitzgerald, K.C.; O’Reilly, E.J.; Fondell, E.; Falcone, G.J.; McCullough, M.L.; Park, Y.; Kolonel, L.N.; Ascherio, A. Intakes of vitamin C and carotenoids and risk of amyotrophic lateral sclerosis: Pooled results from 5 cohort studies. Ann. Neurol. 2013, 73, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, K.L.; Shefner, J.; Zhang, H.; Betensky, R.; O’Brien, M.; Yu, H.; Fantasia, M.; Taft, J.; Beal, M.F.; Traynor, B.; et al. Tolerance of high-dose (3000 mg/day) coenzyme Q10 in ALS. Neurology 2005, 65, 1834–1836. [Google Scholar] [CrossRef]
- Ahmadi, M.; Agah, E.; Nafissi, S.; Jaafari, M.R.; Harirchian, M.H.; Sarraf, P.; Faghihi-Kashani, S.; Hosseini, S.J.; Ghoreishi, A.; Aghamollaii, V.; et al. Safety and Efficacy of Nanocurcumin as Add-On Therapy to Riluzole in Patients With Amyotrophic Lateral Sclerosis: A Pilot Randomized Clinical Trial. Neurotherapeutics 2018, 15, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chico, L.; Ienco, E.C.; Bisordi, C.; Lo Gerfo, A.; Petrozzi, L.; Petrucci, A.; Mancuso, M.; Siciliano, G. Amyotrophic Lateral Sclerosis and Oxidative Stress: A Double-Blind Therapeutic Trial After Curcumin Supplementation. CNS Neurol Disord Drug Targets 2018, 17, 767–779. [Google Scholar] [CrossRef] [PubMed]
- de la Rubia, J.E.; Drehmer, E.; Platero, J.L.; Benlloch, M.; Caplliure-Llopis, J.; Villaron-Casales, C.; de Bernardo, N.; AlarcOn, J.; Fuente, C.; Carrera, S.; et al. Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: A randomized, double-blind, placebo-controlled human pilot study. Amyotroph. Lateral Scler. Frontotemporal Degener. 2019, 20, 115–122. [Google Scholar] [CrossRef]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in Oxidative Stress Markers in Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Oxid. Med. Cell. Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Ihara, Y.; Nobukuni, K.; Takata, H.; Hayabara, T. Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol. Res. 2005, 27, 105–108. [Google Scholar] [CrossRef]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. 2017, 44, 90–95. [Google Scholar] [CrossRef] [Green Version]
- Moumen, R.; Nouvelot, A.; Duval, D.; Lechevalier, B.; Viader, F. Plasma superoxide dismutase and glutathione peroxidase activity in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1997, 151, 35–39. [Google Scholar] [CrossRef]
- Baillet, A.; Chanteperdrix, V.; Trocme, C.; Casez, P.; Garrel, C.; Besson, G. The role of oxidative stress in amyotrophic lateral sclerosis and Parkinson’s disease. Neurochem. Res. 2010, 35, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Tohgi, H.; Abe, T.; Yamazaki, K.; Murata, T.; Ishizaki, E.; Isobe, C. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateral sclerosis. Ann. Neurol. 1999, 46, 129–131. [Google Scholar] [CrossRef]
- Ryberg, H.; Soderling, A.S.; Davidsson, P.; Blennow, K.; Caidahl, K.; Persson, L.I. Cerebrospinal fluid levels of free 3-nitrotyrosine are not elevated in the majority of patients with amyotrophic lateral sclerosis or Alzheimer’s disease. Neurochem. Int. 2004, 45, 57–62. [Google Scholar] [CrossRef]
- Ihara, Y.; Mori, A.; Hayabara, T.; Kawai, M.; Namba, R.; Nobukuni, K.; Sato, K.; Kibata, M. Superoxide dismutase and free radicals in sporadic amyotrophic lateral sclerosis: Relationship to clinical data. J. Neurol. Sci. 1995, 134, 51–56. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Lacomblez, L.; Jaudon, M.; Lepage, S.; Salachas, F.; Bensimon, G.; Bizard, C.; Doppler, V.; Delattre, J.; Meininger, V. Blood oxidative stress in amyotrophic lateral sclerosis. J. Neurol. Sci. 2000, 178, 57–62. [Google Scholar] [CrossRef]
- Przedborski, S.; Donaldson, D.M.; Murphy, P.L.; Hirsch, O.; Lange, D.; Naini, A.B.; McKenna-Yasek, D.; Brown, R.H., Jr. Blood superoxide dismutase, catalase and glutathione peroxidase activities in familial and sporadic amyotrophic lateral sclerosis. Neurodegeneration 1996, 5, 57–64. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Ikeda, K.; Kinoshita, M. Decreased cerebrospinal-fluid superoxide dismutase in amyotrophic lateral sclerosis. Lancet 1993, 342, 1118. [Google Scholar] [CrossRef]
- Robberecht, W.; Sapp, P.; Viaene, M.K.; Rosen, D.; McKenna-Yasek, D.; Haines, J.; Horvitz, R.; Theys, P.; Brown, R., Jr. Cu/Zn superoxide dismutase activity in familial and sporadic amyotrophic lateral sclerosis. J. Neurochem. 1994, 62, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Puymirat, J.; Cossette, L.; Gosselin, F.; Bouchard, J.P. Red blood cell Cu/Zn superoxide dismutase activity in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 1994, 127, 121–123. [Google Scholar] [CrossRef]
- Tórsdóttir, G.; Kristinsson, J.; Gudmundsson, G.; Snaedal, J.; Jóhannesson, T. Copper, ceruloplasmin and superoxide dismutase (SOD) in amyotrophic lateral sclerosis. Pharmacol. Toxicol. 2000, 87, 126–130. [Google Scholar] [CrossRef]
- Mitchell, J.D.; Gatt, J.A.; Phillips, T.M.; Houghton, E.; Rostron, G.; Wignall, C.; Whittington, J.; Shaw, I.C. Cu/Zn superoxide dismutase free radicals, and motoneuron disease. Lancet 1993, 342, 1051–1052. [Google Scholar] [CrossRef]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef] [Green Version]
- Oteiza, P.I.; Uchitel, O.D.; Carrasquedo, F.; Dubrovski, A.L.; Roma, J.C.; Fraga, C.G. Evaluation of antioxidants, protein, and lipid oxidation products in blood from sporadic amyotrophic lateral sclerosis patients. Neurochem. Res. 1997, 22, 535–539. [Google Scholar] [CrossRef]
- Molina, J.A.; de Bustos, F.; Jiménez-Jiménez, F.J.; Gómez-Escalonilla, C.; García-Redondo, A.; Esteban, J.; Guerrero-Sola, A.; del Hoyo, P.; Martínez-Salio, A.; Ramírez-Ramos, C.; et al. Serum levels of coenzyme Q10 in patients with amyotrophic lateral sclerosis. J. Neural. Transm. 2000, 107, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Miyazaki, Y.; Yoshino, H. Increased oxidative stress in patients with amyotrophic lateral sclerosis and the effect of edaravone administration. Redox Rep. 2016, 21, 104–112. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Acimovic, J.; Lovgren-Sandblom, A.; Parini, P.; Andersen, P.M.; Bjorkhem, I. Cholesterol, oxysterol, triglyceride, and coenzyme Q homeostasis in ALS. Evidence against the hypothesis that elevated 27-hydroxycholesterol is a pathogenic factor. PLoS ONE 2014, 9, e113619. [Google Scholar] [CrossRef]
- Murata, T.; Ohtsuka, C.; Terayama, Y. Increased mitochondrial oxidative damage and oxidative DNA damage contributes to the neurodegenerative process in sporadic amyotrophic lateral sclerosis. Free Radic. Res. 2008, 42, 221–225. [Google Scholar] [CrossRef]
- Sohmiya, M.; Tanaka, M.; Suzuki, Y.; Tanino, Y.; Okamoto, K.; Yamamoto, Y. An increase of oxidized coenzyme Q-10 occurs in the plasma of sporadic ALS patients. J. Neurol. Sci. 2005, 228, 49–53. [Google Scholar] [CrossRef]
- Keizman, D.; Ish-Shalom, M.; Berliner, S.; Maimon, N.; Vered, Y.; Artamonov, I.; Tsehori, J.; Nefussy, B.; Drory, V.E. Low uric acid levels in serum of patients with ALS: Further evidence for oxidative stress? J. Neurol. Sci. 2009, 285, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kawabe, K.; Iwasaki, Y. Do serum uric acid levels reflect oxidative stress in the progression of ALS? J. Neurol. Sci. 2009, 287, P294. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Hirayama, T.; Takazawa, T.; Kawabe, K.; Iwasaki, Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: A cross-sectional study. Intern. Med. 2012, 51, 1501–1508. [Google Scholar] [CrossRef] [Green Version]
- Zoccolella, S.; Simone, I.L.; Capozzo, R.; Tortelli, R.; Leo, A.; D’Errico, E.; Logroscino, G. An exploratory study of serum urate levels in patients with amyotrophic lateral sclerosis. J. Neurol. 2011, 258, 238–243. [Google Scholar] [CrossRef]
- Oh, S.I.; Baek, S.; Park, J.S.; Piao, L.; Oh, K.W.; Kim, S.H. Prognostic Role of Serum Levels of Uric Acid in Amyotrophic Lateral Sclerosis. J. Clin. Neurol. 2015, 11, 376–382. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Guo, X.; Wei, Q.; Song, W.; Cao, B.; Huang, R.; Ou, R.; Chen, X.; Shang, H. Serum uric acid level is associated with the prevalence but not with survival of amyotrophic lateral sclerosis in a Chinese population. Metab. Brain Dis. 2014, 29, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Lv, Q.; Wang, Z.; Zhong, Z.; Huang, W. Role of Long Noncoding RNAs in Parkinson’s Disease: Putative Biomarkers and Therapeutic Targets. Parkinson’s Dis. 2020, 2020, 5374307. [Google Scholar] [CrossRef]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as Biomarkers in Amyotrophic Lateral Sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [Green Version]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, Y.; Zhou, X.; Luan, J.; Cui, Y.; Han, J. Comparison of the extraction and determination of serum exosome and miRNA in serum and the detection of miR-27a-3p in serum exosome of ALS patients. Intractable Rare Dis. Res. 2018, 7, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed miRNAs Related to Neural and Glial Activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Proteins | |||||
|---|---|---|---|---|---|
| Related to Redox System | Name | Kinds of Body Fluids | Changes | Related to Parameter of ALS | Reference |
| Products of oxidative stress | 8-OHdG | CSF Plasma Urine | Increase | 8-OHdG levels in urine were negatively correlated with both the rate of change of the ALSFRS-R and the FVC | [69,74,165,166] |
| MDA | Plasma | Increase | Not described | [166,167,168] | |
| HNE | Plasma CSF | Increase | HNE levels in serum were positively correlated with the extent of disease | [75] | |
| 3-NT | CSF | Increase or no change (inconsistent result) | Not described | [169,170] | |
| Antioxidant enzyme | Activity of SOD | RBC Plasma CSF | Increase or reduce or no change (inconsistent result) | The activity in bulbar onset sporadic ALS was significantly higher than in spinal onset sporadic ALS | [165,166,167,168,171,172,173,174,175,176,177] |
| activity of GPX | Whole blood RBC Plasma | Reduce or no change (inconsistent result) | Not described | [166,167,168,172,173,178] | |
| GSH | Whole blood Plasma | Reduce or no change ( inconsistent result) | No significant temporal change during 6 months was observed in sporadic ALS | [166,168,179,180] | |
| GSSG | Whole blood | Increase | Not described | [166] | |
| CoQ10 | Plasma Serum | No change | CoQ10 levels were not influenced by clinical form, age at onset, and duration of disease | [181,182,183] | |
| Proportion of oxidized forms of CoQ10 | Serum CSF | Increase | Proportion of oxidized forms of CoQ10 correlated with duration of disease | [184,185] | |
| UA | Serum Plasma | Reduce or no change (inconsistent result) | The levels of UA in bulbar onset sporadic ALS were significantly decreased compared with spinal onset sporadic ALS patients. The baseline levels of UA were negatively correlated with the annual decline of ALSFRS-R. | [182,186,187,188,189,190,191] | |
| RNAs | |||||
| Related to Redox System | Name | Kinds of Body Fluids | Changes | Related to the Parameter of ALS | Reference |
| The NRF2-ARE pathway | miR-27a | Serum | Decrease | Downregulation of miR-27a was correlated with the degree of muscle atrophy | [194,195] |
| miR-142-5p | CSF | Decrease | Not described | [145,196] | |
| Mitochondrial dysfunction and neuroinflammation | miR-155 | Peripheral monocyte | Increase | Not described | [145,196] |
| Mitochondrial dysfunction | miR-338-3p | Serum PBL CSF | Increase | Not described | [138,197] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosaka, T.; Tsuji, H.; Tamaoka, A. Biomolecular Modifications Linked to Oxidative Stress in Amyotrophic Lateral Sclerosis: Determining Promising Biomarkers Related to Oxidative Stress. Processes 2021, 9, 1667. https://doi.org/10.3390/pr9091667
Hosaka T, Tsuji H, Tamaoka A. Biomolecular Modifications Linked to Oxidative Stress in Amyotrophic Lateral Sclerosis: Determining Promising Biomarkers Related to Oxidative Stress. Processes. 2021; 9(9):1667. https://doi.org/10.3390/pr9091667
Chicago/Turabian StyleHosaka, Takashi, Hiroshi Tsuji, and Akira Tamaoka. 2021. "Biomolecular Modifications Linked to Oxidative Stress in Amyotrophic Lateral Sclerosis: Determining Promising Biomarkers Related to Oxidative Stress" Processes 9, no. 9: 1667. https://doi.org/10.3390/pr9091667