
Drug Repurposing Using Biological Networks


1
Department of Bioengineering, Carlos III University, Leganés, 28911 Madrid, Spain
2
Network Research on Rare Diseases (CIBERER), U714, 28029 Madrid, Spain
3
Regenerative Medicine and Tissue Engineering Group, Health Research Institute-Jimenez Diaz Foundation University Hospital (IIS-FJD), 28040 Madrid, Spain
*
Author to whom correspondence should be addressed.
Processes 2021, 9(6), 1057; https://doi.org/10.3390/pr9061057
Submission received: 29 April 2021
/
Revised: 25 May 2021
/
Accepted: 2 June 2021
/
Published: 17 June 2021
(This article belongs to the Section Biological Processes and Systems)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Drug repositioning is a strategy to identify new uses for existing, approved, or research drugs that are outside the scope of its original medical indication. Drug repurposing is based on the fact that one drug can act on multiple targets or that two diseases can have molecular similarities, among others. Currently, thanks to the rapid advancement of high-performance technologies, a massive amount of biological and biomedical data is being generated. This allows the use of computational methods and models based on biological networks to develop new possibilities for drug repurposing. Therefore, here, we provide an in-depth review of the main applications of drug repositioning that have been carried out using biological network models. The goal of this review is to show the usefulness of these computational methods to predict associations and to find candidate drugs for repositioning in new indications of certain diseases.
1. Introduction
Drug repurposing or drug repositioning is the process of finding new uses for already existing drugs. This is a challenging process but with a great potential both to reduce the cost of drug development [1], as well as to improve its security [2,3]. The traditional process of discovering new drugs is based on complex strategies that include five stages: discovery and preclinical studies, security validation, clinical research through phase trials I, II and III, review by the regulatory agency (FDA/EMA) and post-marketing safety-monitoring (pharmacovigilance) [4]. Drug repositioning uses strategies that simplify this process. In general, the drug repurposing process consists of four main steps.
a. Identification of a candidate molecule. This can be performed using either experimental or computational approaches [5]. Experimental approaches use disease related data and the understanding of drug phenotype modulation, while computational methods predict drug–protein interactions [6] or pharmacological secondary effects [7].
b. Acquisition of the candidate molecule.
c. Mechanistic evaluation of the drug effect in preclinical models followed by evaluation of drug efficacy in clinical trials. This step significantly reduces drug development costs, as it takes into account that there are enough data related to drug safety in phase I clinical trials, since they were already performed for the original indication.
d. Post-marketing safety monitoring (pharmacovigilance).
The contrast of traditional drug development and drug repositioning can be seen in Figure 1.
Historically, drug repositioning has been a serendipity. One of the best known examples of successful drug repositioning is that of sildenafil, which started as an antihypertensive drug, but was repurposed afterwards as a drug to treat pulmonary arterial hypertension and erectile dysfunction and was finally marketed as Viagra® [8]. Another classical example is the case of thalidomide, which was withdrawn from the market after its connection to severe fetal defects, but recent research has shown it to be effective in the treatment of leprosy and multiple myeloma [5]. These drug repositioning success stories have further inspired global pharmaceutical industries to explore the potential capacity of existing drugs. In fact, in the last ten years, governments, researchers, academics and pharmaceutical companies have encouraged activities to support studies related to drug repositioning [9].
Drug repositioning is based on the fact that any drug can act on multiple targets, that two different diseases may have cellular and molecular similarities and that a target can exhibit pleiotropic effects. With the help of current existing high-throughput technologies, the amount of data generated is rapidly increasing. These technologies foster the use of computational methodologies to find associations between drugs, diseases and targets and provide evidence to boost the drug repurposing process [10].
The rapid development of emerging information technologies, including cloud computing, social media and the Internet of Things, provide a large amount of data generated that is in continuous growth in numerous fields of research. However, there is an inherent complexity in the analysis of these data that arise from their huge variety, the speed at which they are obtained and their volume [11]. Recent advances in technologies, such as next-generation sequencing and high-performance biomedical data capture technologies, as well as the reduction of costs, allow researchers to generate large amounts of experimental data. These include data generated by powerful analytical technologies, such as DNA or RNA sequencing, and mass spectrometry for different applications, such as transcriptomics (gene expression and co-expression data), proteomics (protein profiles and interaction data of proteins), metabolomics (metabolic profiles) and epigenomics (methylation data of DNA), among others. Large amounts of clinical data available in electronic health records (EHR), clinical trials and biobanks are added to these already complex omic data. These data ares also stored in heterogeneous, normally unstructured formats, which makes data integration extremely complex and difficult. Even though several databases provide direct access to structured data, such as gene expression (e.g., EBI Expression atlas), there is still a large part of the genomic data that is only available in raw unstructured format (e.g., Sequence Read Archive). For these reasons, there is an urgent need for computational approaches that can integrate, analyze and interpret this type of datasets.
Computational techniques currently used to analyze these data are based on statistical approaches, machine learning, or, especially, biological networks-based models. These computational methods have already shown great possibilities to reduce the distance between the generation and interpretation of huge amounts of data in biomedical fields [12].
2. Biological Network Models
Networks are simple and versatile data structures that allow the discovery of different associations through statistical and computational approaches. The concept of biological networks has now been widely used to represent molecular associations and to model the interactions among biological entities. In addition, there has recently been considerable interest in investigating the structure of such networks and the relationship between networks and their basic biological properties. Networks provide an intuitive framework for integrating a wide variety of sources of information, capturing quantitative and qualitative relationships among entities, such as correlation of gene expression, or the presence or absence of a protein interaction [13].
In biological networks, nodes can represent various entities, not only genes, drugs, proteins or metabolites, but also complex phenotypes, such as biological functions or diseases. Moreover, network edges can be used to show different biological concepts that establish a relationship between nodes, such as the interactions among proteins, gene regulation, or the functional similarity between genes. In addition, both edges and nodes can be annotated with quantitative or qualitative information derived from high-throughput experiments to emphasize specific concepts [14,15].
Networks can therefore be classified into categories based on the main source of biological data that they represent, such as gene regulatory networks or metabolic networks, among others. Regulatory gene networks built from genome-wide transcriptional profiles may represent intrinsic transcriptomic variation and can estimate causal relationships between molecules and identify the main disease drivers. Genome-scale metabolic network models built from sets of metabolic reactions can be used to simulate kinetic activities and enzymatic knockouts, as well as perform in silico knockouts that can help identify and prioritize new drug targets [13]. Biological networks may contain associations underlying physical protein–protein interactions, gene regulation by transcription factors, gene co-expression networks, which represent sets of genes regulated together, or metabolic signaling pathways [16]. Protein–protein interaction networks (PPI networks) are networks that model the protein binding and are derived from high-throughput experiments, such as, for example, the yeast two-hybrid system (Y2H), on the one hand and mass spectrometry-based methods, such as purification by tandem affinity, on the other [17,18]. Transcriptional regulatory networks (TR networks) are bipartite networks with a set of nodes representing genes and other representative transcription factors (TF). The data for such networks are normally obtained by chromatin immunoprecipitation (ChIP) methods. Metabolic networks are also bipartite networks that model the chemical interactions and reactions that take place in cells, as well as the substrates or metabolites involved in these reactions.
In recent years, network-based methods have become a major strategy for drug repositioning [4,9]. With the advances of bioinformatic software and high-throughput techniques, networks are a preferred computational approach to model interactions between molecules in biological systems [19]. Typically, in these network models for drug repurposing, nodes in the networks represent drugs, diseases, or gene products, while edges represent relationships between them [20]. These networks are built either on the basis of experimental knowledge, or through computational predictions using multiple data. Some examples are drug–target networks, drug–disease networks, drug–drug networks, regulatory networks, protein–protein interaction networks, or signaling networks, that are useful for the identification of drug targets or therapeutic approaches [19,21]. In a disease network, if a disease is a hub, it could indicate that it shares common elements with other multiple diseases, such as pathogenesis mechanisms or genetic mutations. On the contrary, outliers are defined as nodes that have few connections to other nodes. For example, in a protein–protein interaction network, a loosely connected target could be determined to a particular disease and, therefore, the corresponding drug might have low possibilities to be repositioned aiming at that target [22].
An important challenge related to network-based drug–target prediction is the integration of multiple interaction networks, as they mainly have to be constructed using heterogeneous information sources. Network integration approaches usually project various datasets into a single network representation. However, this projection process may result in a large loss of information. A number of web-based approaches exists to integrate multiple molecular networks from experimental data sources [23].
Network topological parameters can easily accomplish tasks, such as viewing multiple existing interactions, incorporating novel relationships and superimposing additional properties on the main nodes and their relationships [24]. For large networks, such as the ones normally involved in drug repurposing applications, these graph theory approaches are necessary to extract the most relevant information on the network structure and generate specific subnetworks that allow an easier visualization and analysis of data, by highlighting their fundamental properties [25]. Such findings are useful to reveal the connection between some topological features and their specific biological functions, such as molecular mechanisms or disease characteristics [26]. The identification of important nodes and other topological characteristics is crucial to understand how molecular interactions give rise to emerging biological processes and complex phenotypes in health and disease [27]. In fact, a better understanding of the implications of cellular interconnectivity in disease progression could lead to the identification of disease genes and disease pathways. This network medicine approach also proposes that disease-associated phenotypes are not only the outcome of single gene mutations, but are also originated by possible perturbations in the protein interaction network [28]. For this reason, the interpretation of disease mechanisms and the development of successful approaches for therapeutic effect requires a deep understanding of how networks of molecular interactions are pathologically dysregulated. In clinical practice, network analysis can become a relevant tool and a complementary approach to traditional enrichment analysis methods. This analysis would allow the discovery of better and more accurate biomarkers that monitor the functional integrity of the disease-disturbed network, to realize a better classification of diseases, paving the way to personalized therapies and treatments.
The nodes that are in the same module generally have similar biological properties. For example, if a drug module is enriched for a specific therapeutic category, all drugs in that module could, in principle, be applied for that therapeutic use. In contrast, nodes with high centrality of betweenness (the number of shortest paths that pass through a vertex) are typically the ones that join two different modules and, therefore, this measurement provides a quantifiable way to assess the relative importance of certain nodes in the network. In the drug network example, a drug with a high value of centrality of betweenness tends to be related to several therapeutic uses and has therefore a high potential value to be repositioned.
Computational strategies used to obtain relevant information from network-based models can be related to the construction of networks or to the analysis of network perturbations [4]. Methodologies based on network construction can be inference methods or modularity methods.
Network-based inference methods (NBI) use a priori known information about interactions, known as the “training set,” to predict new interactions and suggest new targets for drug repositioning [29]. Among the networks of greatest interest for inference-based methods are transcriptional regulation, gene, metabolic or protein–protein interaction networks. NBI methods can be formulated as a regression, where the individual response in terms of gene expression is considered as a function of the global expression of the rest of the genes, that act as predictors. For example, the TIGRESS algorithm [30] is applied to produce sparse patterns that can estimate partial correlation relationships between genes. Inferred networks are directional and weighted, due to the predefined settings of “predictor” and “response” [31]. On the other hand, the inference of possible pharmacodynamic interactions, based on pharmacological information on compounds and genomic information on proteins, can be carried out within the framework of supervised bipartite graphical inference methods. In this type of methods, there are two distinct classes of nodes and connections are only possible between nodes of different classes. Among the algorithms used for supervised bipartite graphical inference, the algorithm based on distance learning stands out, a method that works best in terms of precision, prediction and computational efficiency [32]. Inference-based methods show more robust and better performance on diverse datasets than other perturbation methods, such as propagation [33,34,35]. Although different inference methods may only represent individual partial lattice structures, they usually complement each other. Figure 2 shows a schematic representation of network-based inference based methods. For example, in network inference methods the response of the gene expression can be considered as the result of the expression of several other genes that act as predictors. The algorithm used for this approach is the Bayesian network (BN), a probabilistic graph model, which can also be used to estimate direct influences. BN algorithms can capture both linear and non-linear, as well as stochastic or combinatorial, relationships between variables and are one of the best alternatives to deal with noisy data due to their probabilistic nature [31]. Network-based methods are really interesting due to a huge capability for integration and analysis of multiple omic data and are expected to facilitate a more precise understanding of complex diseases and be very useful in drug repurposing approaches.
Modularity methods assume that all cellular components belonging to the same topological, functional, or disease module have a high probability of being involved in the same disease. Thus, these methods begin with the identification of disease modules and continue to screen their members for potential disease. Complex systems usually have clusters with a big number of internal connections, while they share only a few associations with the outside components of the network. The era of big data has motivated the development of clustering algorithms that deal with large datasets in a very efficient manner. General clustering algorithms have shown great potential to discover functional modules in the field of network biology. Furthermore, the use of graph clustering provides an interesting approach to the discovery of protein complexes in PPI networks due to their huge number of interactions [18]. For example, a study by Yu et al. proposed an approach to generate disease–protein–drug networks based on a symmetric conditional probability and the detection of modules in the network through the ClusterONE algorithm [36]. As a result, they found potential drug–disease associations, such as, for example, iloperidone, a known antipsychotic drug that could potentially be used to treat hypertension. These modularity methods use a wide variety of omic disease datasets, to rebuild disease-specific pathways that can give potential significant targets for the repositioned drugs. These methods provide the advantage of being able to reduce general signaling networks with a large number of proteins to a specific network with some proteins or targets [21].
Different perturbation analysis approaches can be taken to extract information that allows drug repositioning within each of these types of networks, such as propagation-based methods or random walk.
In network propagation methods, the information is propagated from a source node to all network nodes. According to the different forms of propagation, these approaches can be divided into two types, local and global [37]. Local propagation only takes into account limited network information and it could fail to make correct predictions in some cases [38]. Otherwise, global propagation uses information from the entire network and, therefore, is normally preferred for certain purposes. Network propagation uses the idea of resource flow within a network to define a score. For example, in drug–target networks, an initial score of 1 is defined for each of the targets corresponding to differentially expressed genes, while the targets of the remaining network are assigned a value of 0. Finally, an initial score is propagated through the network thanks to an iterative process, until the algorithm stabilizes. The result is an evaluation of the importance of nodes based on network connectivity. Scores represent knowledge of the disease and are smoothed through the network to prioritize candidates that are in the network vicinity of all differentially expressed genes [37]. Several studies have shown that network propagation-based methods work well for finding disease drug targets, disease genes and disease–drug relationships [39,40].
Finally, the random walk is a specific case of perturbation methods. It is an iterative process that describes the transition of a random walker through a network starting from a set of selected nodes (seed) and then calculating a score that gives a probability of being reached by this random walker to all other nodes, once the steady state is achieved [41]. This is considered a global method because the entire network structure is covered. In a typical random walk, the set of seed nodes in the network is purposely defined as those corresponding to differentially expressed genes. Random walk-based methods allow the identification of the shortest pathways to known disease genes. This methodology has been applied to detect disease genes related to a wide range of diseases, from diabetes mellitus to Alzheimer’s disease, or different cancers [28]. Several algorithms based on this method have been successfully implemented for drug repurposing applications [42,43].
3. Validation Strategies
The computational methods described above usually predict a large number of candidates for drug repositioning. However, the ultimate goal of drug repositioning is the prediction of one or two candidates who have the highest potential to be successful in clinical applications to benefit the patients. In this sense, the different algorithms must undergo a validation procedure to assess their ability to make relevant and accurate predictions [29]. This procedure requires reference datasets to which the algorithm can relate. These datasets are obtained from reliable sources, such as clinical trials (https://clinicaltrials.gov/), DrugBank (https://www.drugbank.ca/), or case studies specifically designed for that purpose. Biological validation based on successful studies on a single case are less rigorous, as they cannot be extrapolated to all predictions made by one method [44]. Currently, most in silico drug repositioning strategies are validated using in vitro or in vivo models.
The selection of an appropriate validation model is a critical step for the success of drug prediction, since their contexts may be different from those used to make the prediction, or they may not be reliable per se [19]. Some validation methods perform an analytical validation where computational results are compared with existing biomedical knowledge. Methods that identify known drug associations and diseases in a consistent manner are preferred. However, there is little agreement as to a set of best practices, when comparing studies and validating methods. The results are evaluated by using different parameters designed to measure the reliability and precision of the predictions. The performance algorithm can also be evaluated by calculating different characteristics, such as specificity and sensitivity, or using training datasets.
Sensitivity-only based validation alone provides analytical rigor by measuring the overlap between indications from currently approved or research drugs and indications predicted by a given repositioning method. The strategies that use this type of validation, evaluate the general capacity of the drug repurposing to make sensible assertions, instead of selecting one or several tentative predictions for a more in-depth test. This is an attractive strategy, because researchers only need to have a set of true positives in order to test their predictions.
Validations based on both sensitivity and specificity are the most common type of validations. These methods are those that report sensitivity and specificity, but also those that report the area below the characteristic receptor of operation (AUC-ROC). In contrast to sensitivity-only validation methods, these methods require information on false drug predictions (false positives). In these studies, researchers mark all drug-indication pairs not scored as false positives. However, this approach is problematic, since the annotation is derived from a variety of databases of drug information, which can substantially affect the sensitivity and estimated specificity. Furthermore, marking unnoticed pairs as false suggests that all new repositioning hypotheses are false positives. This is not very straight-forward, because computational methods for drug repurposing should predict new indications, for which there are currently no known associations, and this strategy creates a considerable disequilibrium between both true and false positives [44].
For methods such as inference and machine learning-based methods containing multiple parameters whose values must be set, the procedure of validation includes a first step called “training”, during which the algorithm uses a part of the reference dataset to find the values of the parameters used to optimize its performance. The goal of this validation is to evaluate the capability of the algorithm to generalize on different datasets with that exact fit of parameters. When the parameters are set, the validation itself is performed using the remaining datasets [29].
Finally, to implement a novel method or to add new characteristics to an already existing one, it is important to compare its performance with an already established one by using the same reference datasets. This step allows to understand to what extent, and in what circumstances, the novel method gives better predictions. When the validation provides satisfactory results, the algorithm can be used to discover new relationships among diseases, chemical compounds, or possible drugs for repositioning. Despite some limitations, in silico, in vitro and in vivo models have been employed to validate candidates through preclinical drug evaluation.
4. Applications in Diseases
There are several applications of high-potential drug repositioning computational methods in diseases or related therapeutic areas. An important application is the discovery of anticancer drugs. Due to the great demand that it has, the search for new cancer therapies for drugs already on the market has increased in recent years [19]. Drug repositioning also turns out to be very useful in various diseases, such as cardiovascular or neurological diseases, Alzheimer’s, but also a successful therapeutic alternative for rare diseases. Finally, another promising strategy is to use repositioning for the discovery of anti-infective drugs that can have more effective ways to deal with drug resistance, which can greatly reduce the effectiveness of the drug and have terrible consequences for humans.
4.1. Cancer
The use of powerful high-throughput technologies, such as mass spectrometry or next-generation sequencing, have been widely used to study the genomic environment in cancer. These technologies have identified more than 500 significant mutated genes for more than 20 types of cancer, in various projects. Nonetheless, the cost of cancer therapeutic novel drugs continues to rise spectacularly, making it necessary to employ innovative strategies to accelerate the discovery of cancer medicaments at a lower cost. Cancer drug repositioning provides several interesting advantages, such as, for example, the availability of drug safety studies, or others, such as pharmacokinetic and pharmacodynamic properties.
In a recent study, an in silico network approach focused on drug targets was used for the evaluation of drug repositioning in emerging cancer oncology development [45]. The comprehensive drug–target network was built by integrating drug and protein binding from various available data sources. Specifically, tools such as the DrugBank, BindingDB or canSAR databases were used to extract data on drug–protein or drug–gene interactions and HPRD (Human Protein Reference Database) to extract data on human protein–protein interaction. With these data, the authors built a support network that allowed the identification of failed drugs in clinical trials, as well as novel therapeutic targets for already marketed drugs, or new chemical possibilities. Once the network was developed, the researchers built predictive models based on weighted or unweighted networks through inference algorithms and assessed their performance through cross-validation and external validation. Finally, they experimentally verified new predictions using in vitro or in vivo experimental trials, or electronically available patient data in medical records or other healthcare databases.
In another example, a protein–protein interaction network constructed by the combined information from triple negative breast cancer experiments, obtained from repositories and databases, allowed the identification of promising multi-target drugs, as later validated with in vitro experiments. The application of graph-based algorithms highlighted the most interesting combination of drug targets and a data fusion approach based on matrix tri-factorization was used together with known drug mechanisms of action to identify the repurposed candidates [46].
4.2. Cardiovascular Diseases
Cardiovascular diseases (CVD) cover a wide range of disorders that affect different parts of the cardiovascular system and include coronary heart disease, carotid disease, peripheral arterial disease and aneurysm, among others. The etiology of cardiovascular diseases is not simple, as most are complex diseases that occur as a result of the interaction between multiple genes. Therefore, different approaches have been used in CVD research, focusing on the mathematical concepts of system biology networks, as they accurately capture the inner workings of complex biological systems. As in cancer, new advances related to high-throughput techniques have provided a large amount of biological datasets related to CVD. Therefore, numerous molecular interaction databases, such as BioGRID and DRYGIN, have been used. These databases provide biological information related to protein–protein interactions (PPI), genetic interactions or enzyme–substrate relationships. Data stored also include functional gene annotations, data related to genetic disorders, metabolic pathway circuits, or disease associations.
As explained above, network representation provides the chance to reduce the complexity of the biological data needed for computational analysis. Network analysis provides accurate information about the interconnectedness of data describing different processes within a living cell. There have been several attempts to create biological networks relevant to various cardiovascular disorders. One of them was based on building a cardiac transcription network, integrating DNA-binding and mRNA profiles related to interesting cardiac transcription factors [47]. In this network, target genes relevant to the cardiovascular system were selected based on their biological functions, such as muscle contractility and cardiac growth. The network described common regulatory mechanisms related to several transcription factors and the result of miRNA post-transcriptional modulation of expression levels. The quality of the biological data is crucial to building a reliable CVD network. Sarajlic et al. studied different methods that used biological network topology in CVD research and found that only a few approaches were able to identify new genes relevant to CVD that are based solely on the topological properties of the entire PPI network [26]. Among these, it is worth highlighting a computational method based on six topological characteristics (grade, count of disease gene neighbors, proportion of disease genes among neighbors, centrality of intermediation, clustering coefficient and mean length of the shortest path to the disease gene), whose constructed classifier was used in the PPI network to predict candidate genes for coronary artery disease [48]. The enrichment of various CVD networks in disease-relevant biological functions was verified and functional modules were identified in the networks with the help of topological calculations. However, the topological evaluation did not investigate the context of a larger and more complete network and was generally restricted to the disease-specific subnetwork. Nevertheless, this led to predictions of new modules, pathways and genes.
Another example in a recent study analyzed the topological features of a miRNA–gene–drug network and identified miRNA–gene–drug triplets in cardiac hypertrophy and acute myocardial infarction; it later used the miRNA dysregulated pathways to identify novel drug repurposing candidates [49].
4.3. Neurological Disorders: Alzheimer
The biomedical big data accumulated to date warrant extensive research to better understand the pathogenesis of Alzheimer’s disease and facilitate the process of repositioning anti-Alzheimer’s disease (anti-AD) drugs. Zhang et al. generated a list of anti-AD tentative protein targets by evaluating publicly available genomic, epigenomic, proteomic and metabolomic data [50]. The information related to the pathogenesis of Alzheimer’s came from both PubMed and OMIM databases and the drug target data were extracted from the drug target database and the drug bank by searching the NHGRI-EBI Catalog. The genome wide association studies (GWAS) catalog was used to extract genetic variations associated with Alzheimer’s disease and the Human Metabolome Database (HMDB) to extract AD-related metabolites. In total, 98 proteins, 14 epigenetic modifications, 244 genetic mutations and 86 metabolites associated with AD were obtained from OMIM, PubMed and the GWAS Catalog. In addition, 1179 metabolite–protein interactions from the HMDB database were extracted and 200 proteins linked to more than two metabolites associated with AD were found. In total, they selected 524 Alzheimer’s relevant proteins, out of which eight were revealed by two alternative ‘omics’ technologies. With the aid of DrugBank and TTD databases, they obtained information from drugs, targets and drug mechanisms. Considering the pathogenesis of AD together with the known drug mechanisms, they discovered 19 targets out of 92 drugs with an anti-AD tentative sign that could be potentially repositioned. After this, a score was given to these targets based on a weighted sum model that used three parameters: level of change in AD related proteins, number of citations and number of articles that reported the relationship of the target with AD based on a PubMed search. This algorithm found that CD33 and MIF, which are related to microglial activation and neuroinflammation, were the two best ranked targets and that they were associated with seven existing drugs. Inhibitors or antibodies that targeted MIF and CD33 had already been evaluated in clinical trials for the treatment of acute myelogenous leukemia or solid tumors. These results suggested that they could also be good candidates for treating Alzheimer’s-related neuroinflammation.
Another example of the prediction of anti-Alzheimer’s disease drugs calculated a drug repositioning perturbation score using a network constructed with data from RNA-seq, microarray and proteomic information collected from the Synapse database and the CMAP database. They found 31 potential candidates with high score and, from these drugs, only four were classified in the nervous system group of anatomical therapeutic chemicals [51].
4.4. Rare Diseases: Adrenocortical Carcinoma
Adrenocortical carcinoma (ACC) is a rare disease for which few effective treatment options are available. The drugs used in the treatment of this disease are highly toxic and new therapeutic options are urgently needed. A recent publication proposed a drug repurposing model for ACC by predicting links through a heterogeneous network consisting of drugs, diseases, drug targets and their relationships, called Heter-LP [52]. This model consists on a machine learning method that uses the propagation of labels in non-homogeneous networks in three principal steps: data modeling, projection and label propagation. In the first step, the imported data are collected and incorporated into a heterogeneous network, which is formed by three homogeneous subnetworks (drugs, diseases and drug targets) and three heterogeneous subnetworks (drug–disease, drug–target and disease–target). During the second stage, the calculation of topological similarity between pairs of entities is achieved, with the goal of determining the relationships between different nodes, as well as their own similarity. Finally, the third step of label propagation uses these topological similarities to integrate them with homogeneous subnetworks. The purpose of this new method is to predict relationships that do not yet exist in the input data. The results obtained with Heter-LP, regarding novel predicted drugs potentially related to ACC, are weighted using the probability of the existence of the found relationship. The plausibility of several predicted links was supported by the literature and datasets not used in the actual prediction of the links. Furthermore, compared to another computational drug repositioning method, Heter-LP showed great success in suggesting new therapeutic pathways for rare diseases.
The importance of drug repurposing methods for orphan diseases is huge, due to the importance of the rapid advancement of novel therapies in this field. As a final remark of this importance, the FDA has developed a Rare Disease Repurposing Database (RDRD) to include the FDA-approved orphan drug or designation. This database can be used as a standard library for the development of computational models to help in drug repurposing [53].
4.5. Infectious Diseases: COVID-19
Human coronaviruses (HCoV) are a family that includes severe acute respiratory syndrome coronavirus (SARS-CoV) and, specifically, the 2019 novel coronavirus (2019-nCoV, also known as SARS-CoV-2), which is, nowadays, leading a global pandemic characterized by its high morbidity and mortality. In a recent study carried out by Zhou et al., an integrative methodology for repositioning antiviral drugs was presented [54]. This methodology used a network medicine platform based on systems pharmacology that quantified the interaction between the interactome virus–host and drug targets in the human PPI network (Figure 3).
This network is based on the idea that proteins that are functionally associated with viral infection are located in the corresponding subnetwork within the comprehensive network of human PPI. In addition, the model is based on the notion that proteins that serve as drug targets for a disease-specific drug can also be suitable drug targets for possible antiviral infection due to common virus–host-protein, protein–protein interactions and functional pathways elucidated by the human interactome. Using proximity analysis of the drug target network and HCoV–host interactions in the human interactome, they prioritized 16 potential repositioned anti-HCoV drugs (melatonin, mercaptopurine and sirolimus, among others), that were further validated by enrichment analysis of drug genetic signatures and HCoV, induced by transcriptomic data in human cell lines. However, although most of the predictions were validated by various data from the literature, before using them in patients, all drugs that can be repositioned and drug combinations predicted by the network need to be validated in several SARS-CoV2 clinical trials. In addition, they identified two possible drug combinations (sirolimus plus dactinomycin, mercaptopurine plus melatonin) using a complementary exposure pattern method, where drug targets reach the host SARS-CoV2 subnetwork, but target separate vicinities in the human protein–protein interaction network. However, the network data are not yet complete and some of the considered drug–target relationships can only be associated functionally, rather than having actual physical links. The network strategies used in this study, if widely implemented, could potentially aid in the development of additional treatment approaches for other infectious diseases and novel viral challenges.
In this line, Fiscon et al. developed a new network-based algorithm for drug repositioning, called SAveRUNNER (Searching off-lAbel dRUg aNd NEtwoRk) [55]. This algorithm quantifies the relationship between drug targets and disease-specific proteins in the human interactome and uses this to predict drug–disease associations via a network-based similarity measurement. In the paper, they applied SAveRUNNER to a panel of 14 diseases related to COVID-19, based on genetic similarity or comorbidity, and found 282 repositioned drugs, including some widely used nowadays for COVID-19 treatments.
5. Conclusions
Drug repositioning is a strategy to identify new uses for existing, approved or investigational drugs that are outside the scope of the original medical indication. This process is a rapidly evolving challenge in the area of drug development, as it has great potential to reduce both the cost of drug development and the time it takes for these drugs to reach the market [1], as well as reducing safety risks [2,3]. However, drug repositioning is a complex process that involves multiple factors, such as technology, patents, business models, investments and market demand [4]. With current modern approaches, such as omic technologies, computational methodologies and screening platforms, a wide array of associations between drugs, diseases and potential targets can be predicted [10]. In this sense, network-based approaches have become a widely used strategy and popular tool for computational drug repositioning [4,9], providing clues about causal interactions between components that perform certain functionalities. Networks give us an interesting and not very complex structure to study the integration of a huge amount of information sources and to represent data from both qualitative and quantitative interactions between molecules, such as the correlation of gene expression, or the absence or presence of an existing relationship [13]. Different algorithms, such as network propagation, random walk, or network inference, allow to extract meaningful information from these network models, posing a promising tool for their application in fields such as biomedical drug repurposing.
Author Contributions
Conceptualization, C.L.; methodology, F.J.S.; writing—original draft preparation, F.J.S.; writing—review and editing, C.L. and S.G.-A.; supervision, C.L.; visualization: S.G.-A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mullen, J.; Cockell, S.J.; Tipney, H.; Woollard, P.M.; Wipat, A. Mining integrated semantic networks for drug repositioning opportunities. PeerJ 2016, 4, e1558. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.-W. Drug Repositioning Approaches for the Discovery of New Therapeutics for Alzheimer’s Disease. Neurotherapeutics 2014, 12, 132–142. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, H.; Xi, Z.; Rogaeva, E. Drug Repositioning for Diabetes Based on ’Omics’ Data Mining. PLoS ONE 2015, 10, e0126082. [Google Scholar] [CrossRef]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chen, J.; Shi, L.; Mikailov, M.; Zhu, H.; Wang, K.; He, L.; Yang, L. DRAR-CPI: A server for identifying drug reposi-tioning potential and adverse drug reactions via the chemical–protein interactome. Nucleic Acids Res. 2011, 39, W492–W498. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Agarwal, P. Systematic Drug Repositioning Based on Clinical Side-Effects. PLoS ONE 2011, 6, e28025. [Google Scholar] [CrossRef]
- Beachy, S.H.; Johnson, S.G.; Olson, S.; Berger, A.C. Roundtable on Translating Genomic-Based Research for, Policy, Board on Health Sciences, and Institute of Medicine; Drug Repurposing and Repositioning; National Academies Press: Washington, DC, USA, 2014. [Google Scholar] [CrossRef]
- Luo, H.; Li, M.; Yang, M.; Wu, F.X.; Li, Y.; Wang, J. Biomedical Data and Computational Models for Drug Repositioning: A Comprehensive Review. Brief. Bioinform. 2020, 22, 1604–1619. [Google Scholar] [CrossRef]
- Polamreddy, P.; Gattu, N. The drug repurposing landscape from 2012 to 2017: Evolution, challenges, and possible solutions. Drug Discov. Today 2019, 24, 789–795. [Google Scholar] [CrossRef]
- Kankanhalli, A.; Hahn, J.; Tan, S.; Gao, G. Big data and analytics in healthcare: Introduction to the special section. Inf. Syst. Front. 2016, 18, 233–235. [Google Scholar] [CrossRef] [Green Version]
- Gligorijević, V.; Malod-Dognin, N.; Pržulj, N. Integrative methods for analyzing big data in precision medicine. Proteomic 2016, 16, 741–758. [Google Scholar] [CrossRef]
- Hodos, R.A.; Kidd, B.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silicomethods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [Green Version]
- Alaimo, S.; Pulvirenti, A. Network-Based Drug Repositioning: Approaches, Resources, and Research Directions. Methods Mol. Biol. 2019, 1903, 97–113. [Google Scholar]
- Shahreza, M.L.; Ghadiri, N.; Mousavi, S.R.; Varshosaz, J.; Green, J.R. Heter-LP: A heterogeneous label propagation algorithm and its application in drug repositioning. J. Biomed. Inform. 2017, 68, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.M.; Gupta, S.; Wolkenhauer, O. Integrative workflows for network analysis. Essays Biochem. 2018, 62, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Rivas, J.D.L.; Fontanillo, C. Protein–Protein Interactions Essentials: Key Concepts to Building and Analyzing Interactome Networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [Green Version]
- Winterbach, W.; Van Mieghem, P.; Reinders, M.; Wang, H.; De Ridder, D. Topology of molecular interaction networks. BMC Syst. Biol. 2013, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2016, 17, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yella, J.K.; Yaddanapudi, S.; Wang, Y.; Jegga, A.G. Changing Trends in Computational Drug Repositioning. Pharmaceuticals 2018, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning—What we need to know. Drug Discov. Today 2013, 18, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Deng, L. Integrating Multiple Interaction Networks for Gene Function Inference. Molecules 2018, 24, 30. [Google Scholar] [CrossRef] [Green Version]
- Arrell, D.K.; Terzic, A. Network Systems Biology for Drug Discovery. Clin. Pharmacol. Ther. 2010, 88, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, S.; Xia, J. Network-Based Approaches for Multi-Omics Integration. Methods Mol. Biol. 2020, 2104, 469–487. [Google Scholar] [CrossRef]
- Sarajlić, A.; Pržulj, N. Survey of Network-Based Approaches to Research of Cardiovascular Diseases. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using biological networks to integrate, visualize and analyze genomics data. Genet. Sel. Evol. 2016, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaelen, Q.; Mamoshina, P.; Aliper, A.M.; Artemov, A.; Lezhnina, K.; Ozerov, I.; Labat, I.; Zhavoronkov, A. Design of efficient computational workflows for in silico drug repurposing. Drug Discov. Today 2017, 22, 210–222. [Google Scholar] [CrossRef]
- Haury, A.-C.; Mordelet, F.; Vera-Licona, P.; Vert, J.-P. TIGRESS: Trustful Inference of Gene REgulation using Stability Selec-tion. BMC Syst. Biol. 2012, 6, 145. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Risacher, S.L.; Shen, L.; Saykin, A.J. Network approaches to systems biology analysis of complex disease: Integrative methods for multi-omics data. Brief. Bioinform. 2017, 19, 1370–1381. [Google Scholar] [CrossRef] [Green Version]
- Yamanishi, Y.; Kotera, M.; Kanehisa, M.; Goto, S. Drug-target interaction prediction from chemical, genomic and pharmaco-logical data in an integrated framework. Bioinformatics 2010, 26, i246–i254. [Google Scholar] [CrossRef]
- Alaimo, S.; Pulvirenti, A.; Giugno, R.; Ferro, A. Drug–target interaction prediction through domain-tuned network-based inference. Bioinformatics 2013, 29, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.-X.; Huang, J.; Tang, Y. Prediction of Drug-Target Interactions and Drug Repositioning via Network-Based Inference. PLoS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, S.; Li, J. Drug Target Predictions Based on Heterogeneous Graph Inference. Biocomputing 2013, 2013, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Huang, J.; Ma, Z.; Zhang, J.; Zou, Y.; Gao, L. Inferring drug-disease associations based on known protein complexes. BMC Med. Genom. 2015, 8, S2. [Google Scholar] [CrossRef] [Green Version]
- Emig, D.; Ivliev, A.; Pustovalova, O.; Lancashire, L.; Bureeva, S.; Nikolsky, Y.; Bessarabova, M. Drug Target Prediction and Repositioning Using an Integrated Network-Based Approach. PLoS ONE 2013, 8, e60618. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.-P.; Kwoh, C.-K.; Yang, P.; Li, X.-L.; Zheng, J. Drug–target interaction prediction by learning from local information and neighbors. Bioinformatics 2013, 29, 238–245. [Google Scholar] [CrossRef]
- Martínez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug–disease prioritization by inte-grating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Yeh, S.-H.; Yeh, H.-Y.; Soo, V.-W. A network flow approach to predict drug targets from microarray data, disease genes and interactome network—Case study on prostate cancer. J. Clin. Bioinform. 2012, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Li, M.; Wang, S.; Liu, Q.; Li, Y.; Wang, J. Computational drug repositioning using low-rank matrix approximation and randomized algorithms. Bioinformatics 2018, 34, 1904–1912. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. Drug–target interaction prediction by random walk on the heterogeneous network. Mol. Bio-Syst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.-X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A review of validation strategies for computational drug repositioning. Brief. Bioinform. 2016, 19, 174–177. [Google Scholar] [CrossRef]
- Cheng, F. In Silico Oncology Drug Repositioning and Polypharmacology. In Methods in Molecular Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2019; Volume 1878, pp. 243–261. [Google Scholar]
- Vitali, F.; Cohen, L.D.; DeMartini, A.; Amato, A.; Eterno, V.; Zambelli, A.; Bellazzi, R. Correction: A Network-Based Data Integration Approach to Support Drug Repurposing and Multi-Target Therapies in Triple Negative Breast Cancer. PLoS ONE 2017, 12, e0170363. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, J.; Schueler, M.; Grunert, M.; Fischer, J.J.; Zhang, Q.; Krueger, T.; Lange, M.; Tönjes, M.; Dunkel, I.; Sperling, S.R. The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs. PLoS Genet. 2011, 7, e1001313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, X.; Tai, J.; Li, W.; Chen, L. Predicting Candidate Genes Based on Combined Network Topological Features: A Case Study in Coronary Artery Disease. PLoS ONE 2012, 7, e39542. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Yang, J.; Chi, J.; Ding, X.; Lv, N. Identification of drug repurposing candidates based on a miRNA-mediated drug and pathway network for cardiac hypertrophy and acute myocardial infarction. Hum. Genom. 2018, 12, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Schmitt-Ulms, G.; Sato, C.; Xi, Z.; Zhang, Y.; Zhou, Y.; George-Hyslop, P.S.; Rogaeva, E. Drug Repositioning for Alzheimer’s Disease Based on Systematic ‘omics’ Data Mining. PLoS ONE 2016, 11, e0168812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Song, M.-Y.; Kim, D.; Park, C.; Park, D.K.; Kim, D.G.; Yoo, J.S.; Kim, Y.H. A Proteotranscriptomic-Based Computa-tional Drug-Repositioning Method for Alzheimer’s Disease. Front. Pharmacol. 2020, 10, 1653. [Google Scholar] [CrossRef] [Green Version]
- Shahreza, M.L.; Ghadiri, N.; Green, J.R. A computational drug repositioning method applied to rare diseases: Adrenocortical carcinoma. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Xu, K.; Coté, T.R. Database identifies FDA-approved drugs with potential to be repurposed for treatment of orphan diseases. Brief. Bioinform. 2011, 12, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. SAveRUNNER: A network-based algorithm for drug repurposing and its application to COVID-19. PLoS Comput. Biol. 2021, 17, e1008686. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differences between traditional (A) and reposition (B) drug development. From [4], under open access (CC BY-NC).
Figure 1.
Differences between traditional (A) and reposition (B) drug development. From [4], under open access (CC BY-NC).

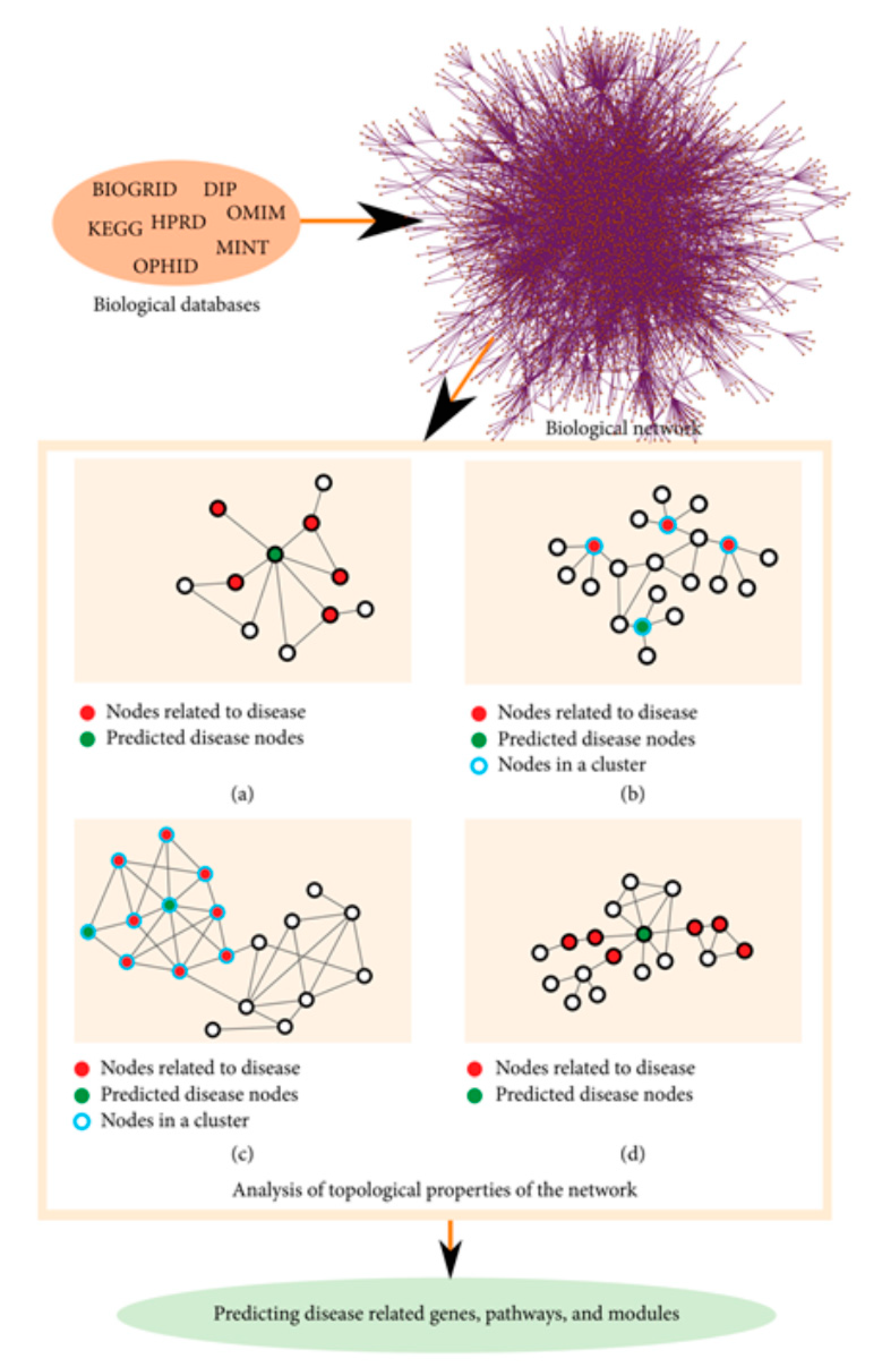
Figure 2.
Schematic representation of network topology methods to infer elements involved in disease. (a) Neighboring method. (b) Cluster enrichment in disease nodes. (c) Community enrichment. (d) Shortest path. From [26], under open access (CC BY-NC).
Figure 2.
Schematic representation of network topology methods to infer elements involved in disease. (a) Neighboring method. (b) Cluster enrichment in disease nodes. (c) Community enrichment. (d) Shortest path. From [26], under open access (CC BY-NC).

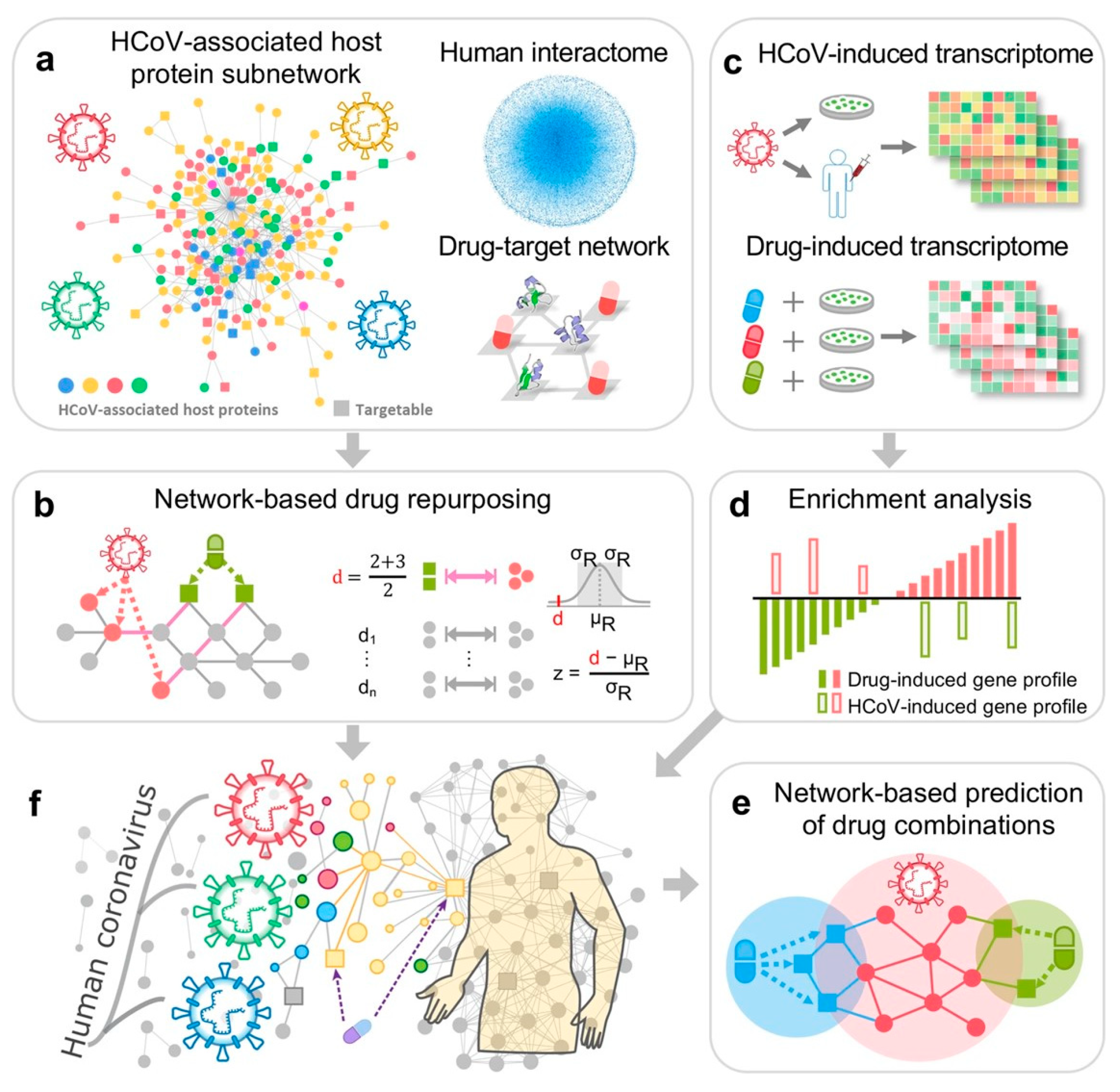
Figure 3.
Network-based methodology for COVID-19 drug repurposing. (a) Human coronavirus (HCoV)-associated host proteins. (b) Network proximity between drug targets and HCoV-associated proteins. (c,d) Gene set enrichment analysis to validate the network-based prediction. (e) Drug combinations using network-based method. (f) Overall hypothesis of the network-based methodology. From [54], under open access (CC BY-NC).
Figure 3.
Network-based methodology for COVID-19 drug repurposing. (a) Human coronavirus (HCoV)-associated host proteins. (b) Network proximity between drug targets and HCoV-associated proteins. (c,d) Gene set enrichment analysis to validate the network-based prediction. (e) Drug combinations using network-based method. (f) Overall hypothesis of the network-based methodology. From [54], under open access (CC BY-NC).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Somolinos, F.J.; León, C.; Guerrero-Aspizua, S. Drug Repurposing Using Biological Networks. Processes 2021, 9, 1057. https://doi.org/10.3390/pr9061057
AMA Style
Somolinos FJ, León C, Guerrero-Aspizua S. Drug Repurposing Using Biological Networks. Processes. 2021; 9(6):1057. https://doi.org/10.3390/pr9061057
Chicago/Turabian StyleSomolinos, Francisco Javier, Carlos León, and Sara Guerrero-Aspizua. 2021. "Drug Repurposing Using Biological Networks" Processes 9, no. 6: 1057. https://doi.org/10.3390/pr9061057
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.