
Investigation of the Side Chain Effect on Gas and Water Vapor Transport Properties of Anthracene-Maleimide Based Polymers of Intrinsic Microporosity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
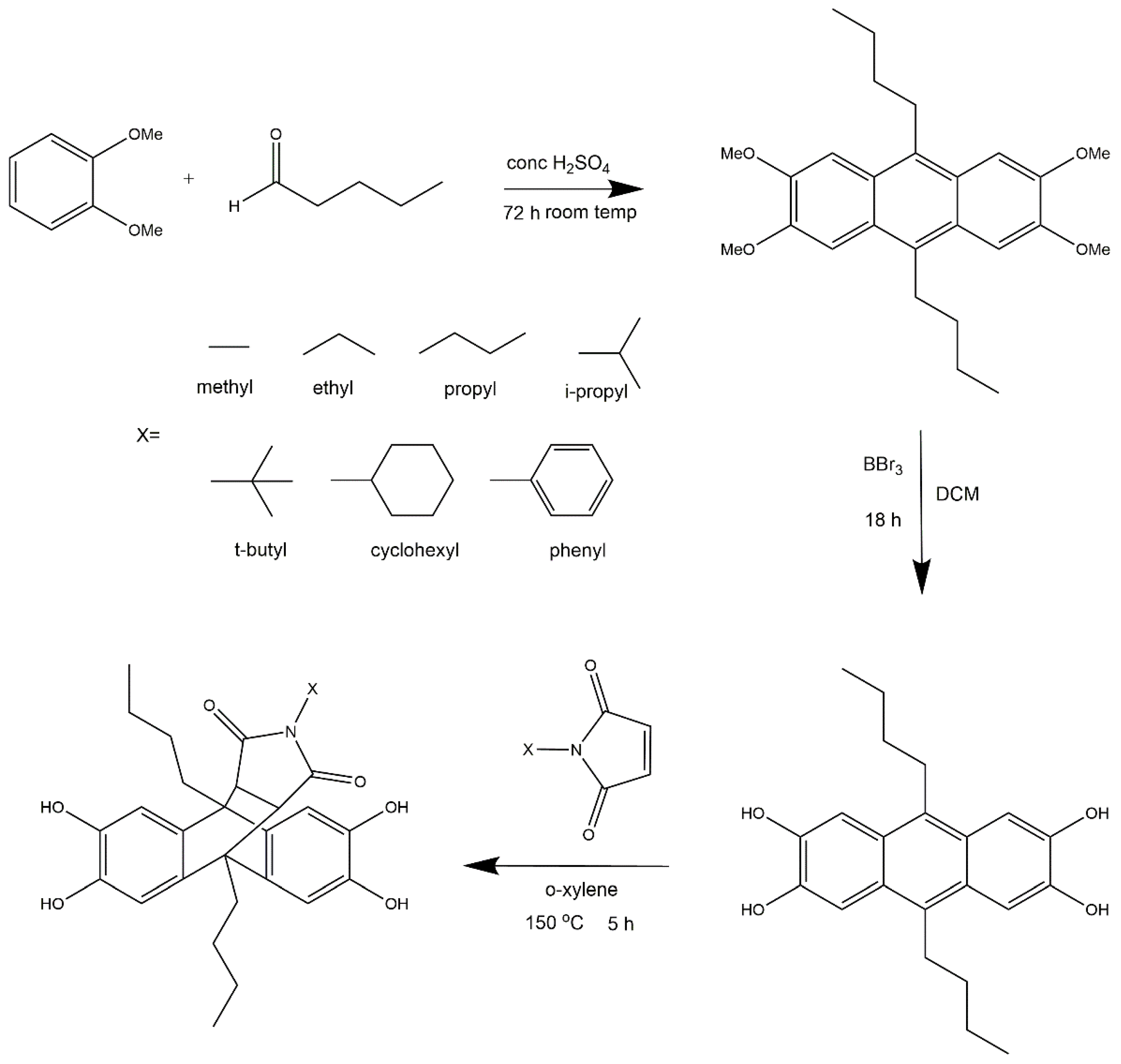
2.2. Monomer Synthesis
2.2.1. 2,3,6,7-Tetramethoxy-9,10-Dibutylanthracene
2.2.2. 2,3,6,7-Tetrahydroxy-9,10-Dibutylanthracene
2.2.3. Methyl-Comonomer
2.2.4. Ethyl-Comonomer
2.2.5. Propyl-Comonomer
2.2.6. i-Propyl-Comonomer
2.2.7. t-Butyl-Comonomer
2.2.8. Cyclohexyl-Comonomer
2.2.9. Phenyl-Comonomer
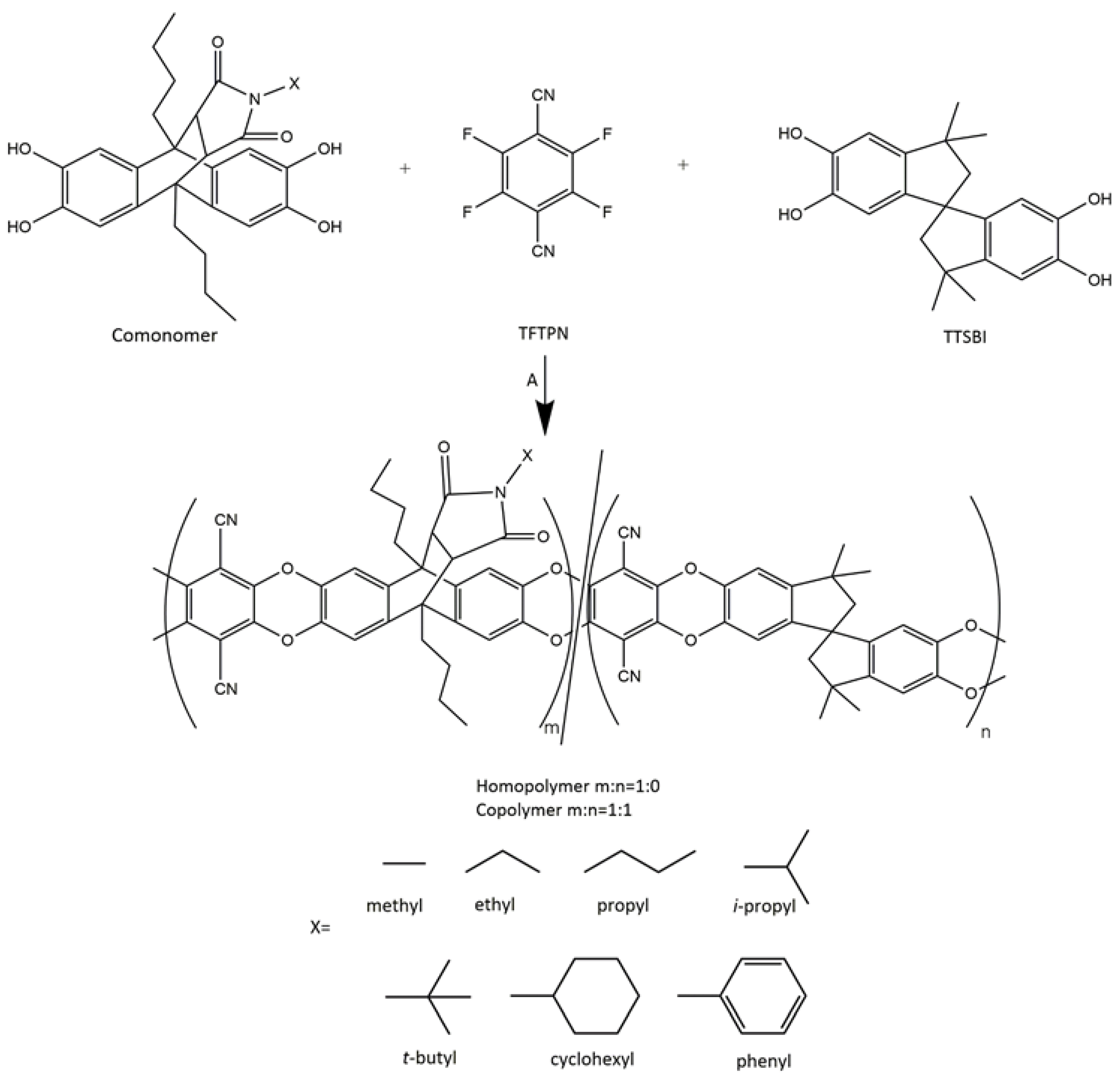
2.3. Polymer Synthesis
2.3.1. PIM-1 Synthesis
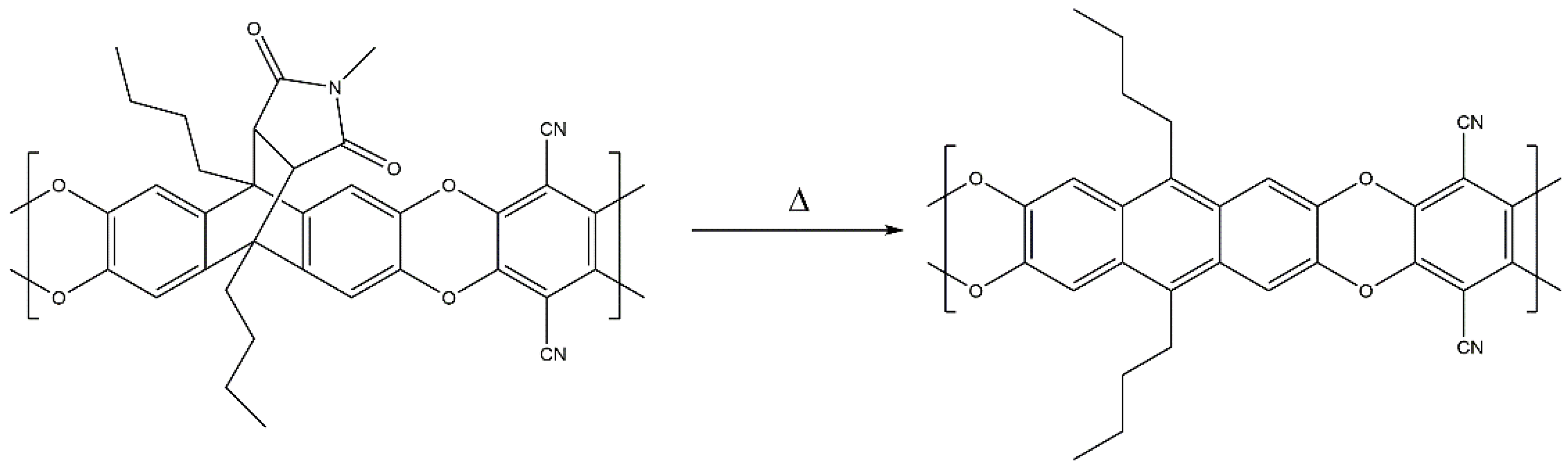
2.3.2. Synthesis of Homo- and Copolymers
2.4. Film Preparation
2.5. Methods
2.5.1. Size-Exclusion Chromatography (SEC)
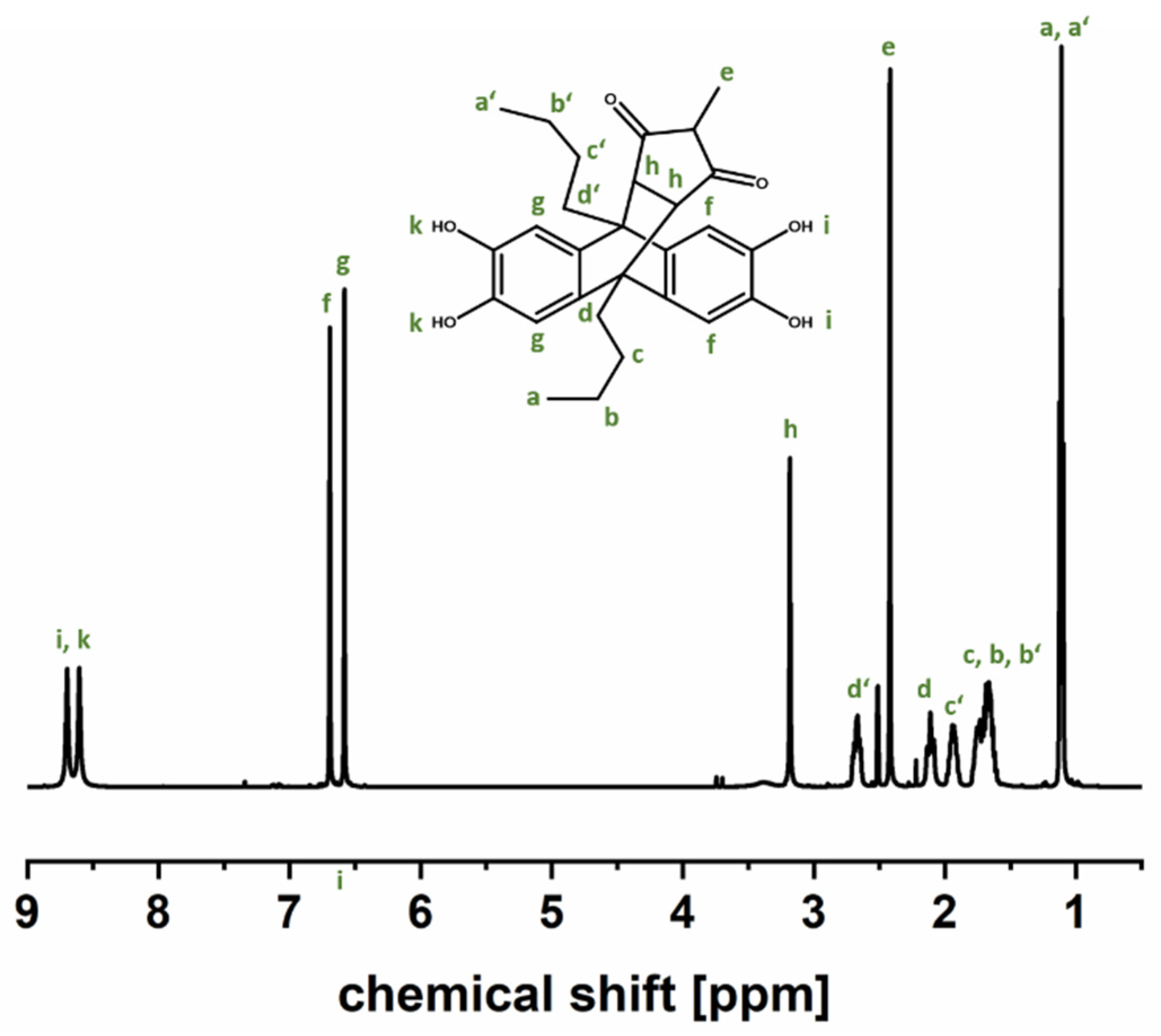
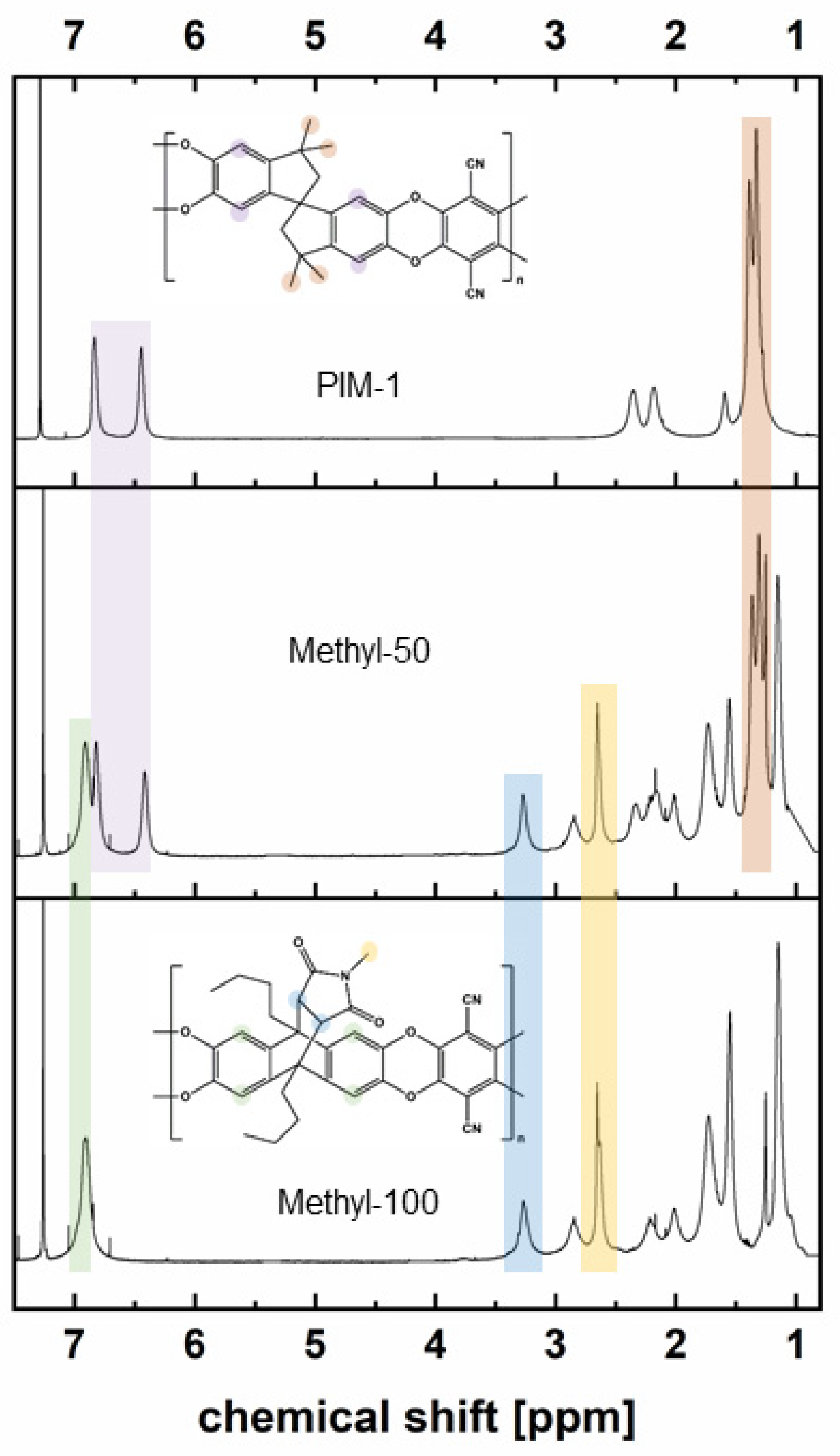
2.5.2. 1H Nuclear Magnetic Resonance (1H-NMR)
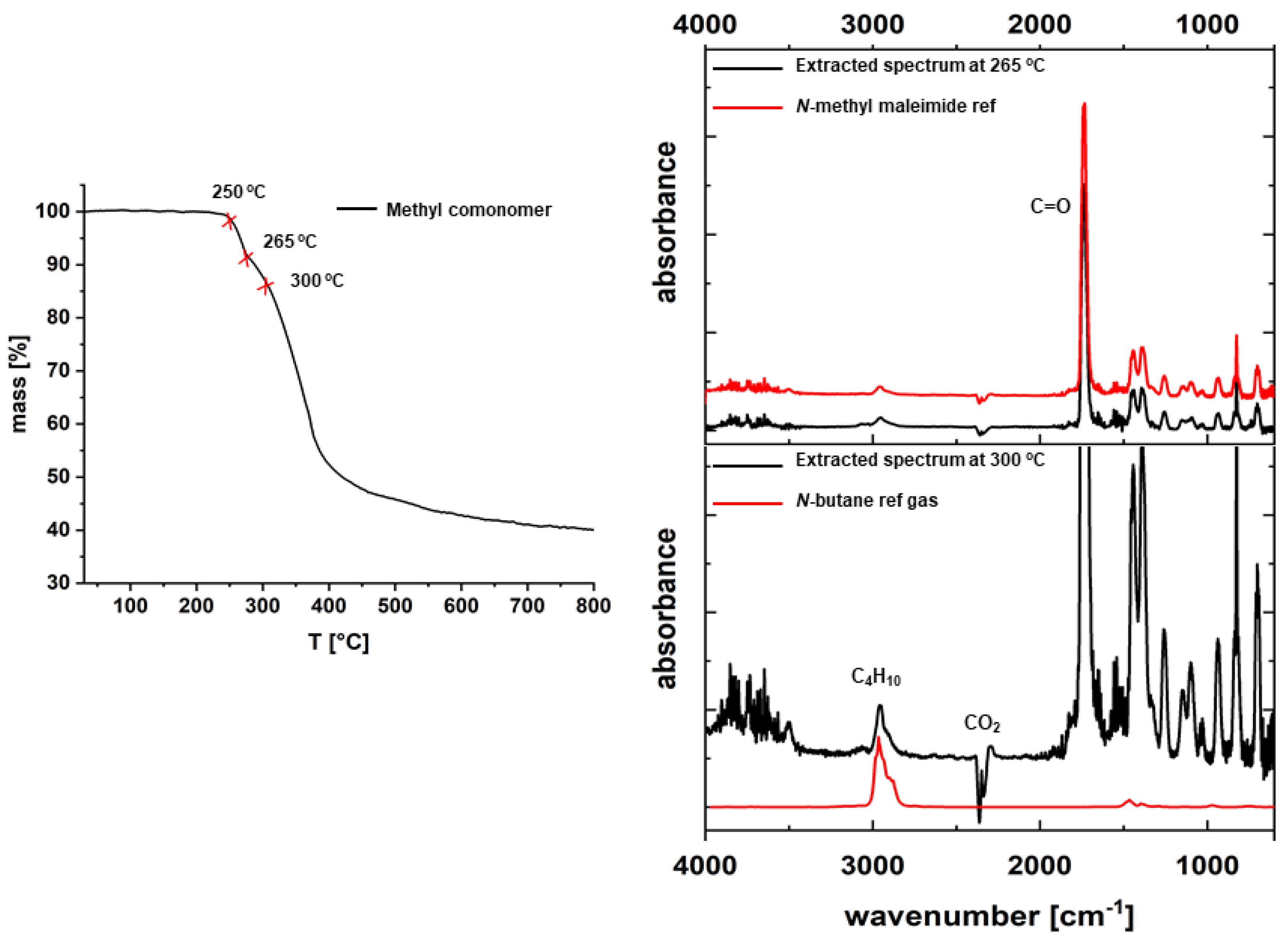
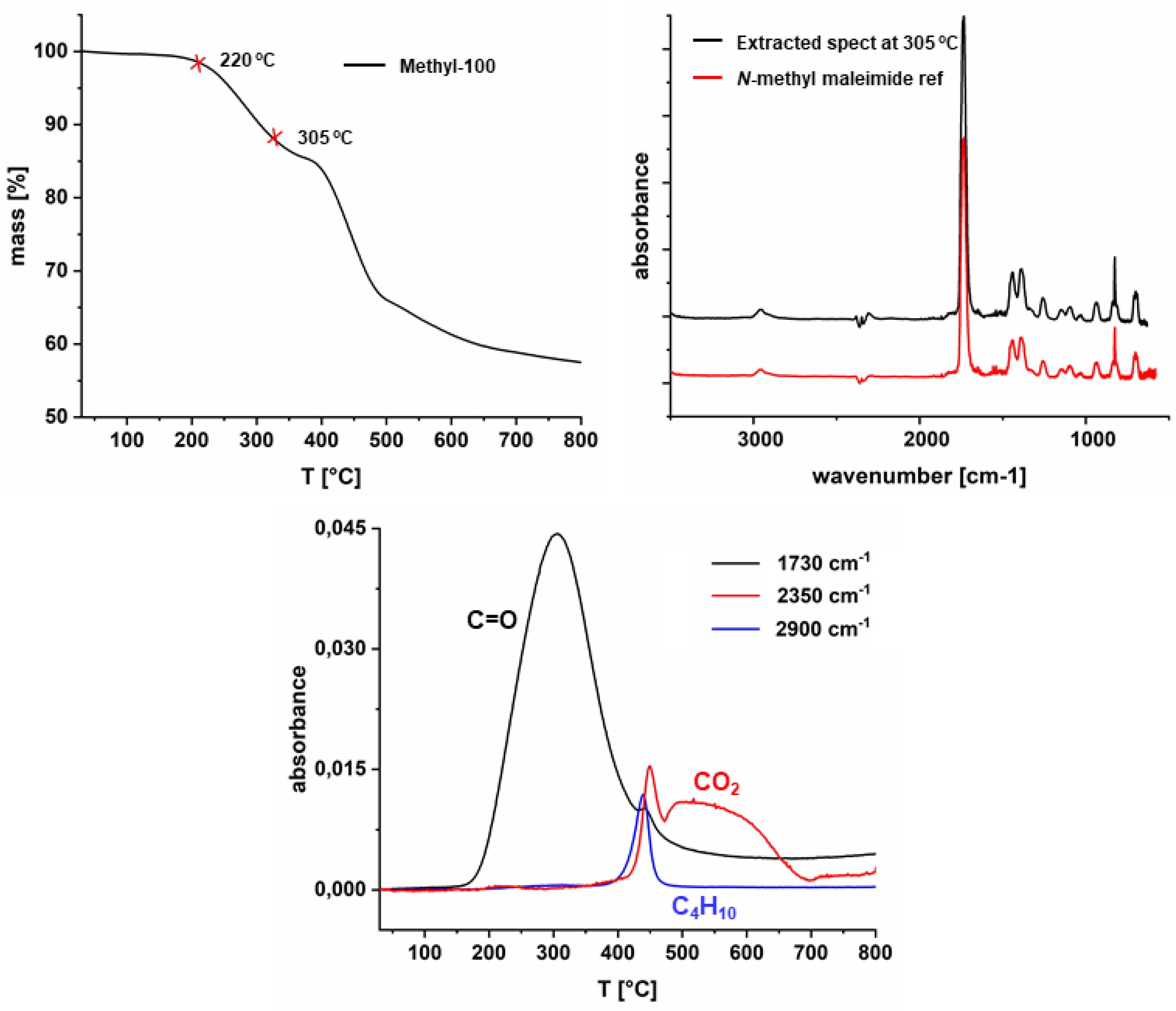
2.5.3. Thermal Gravimetric Analysis (TGA)
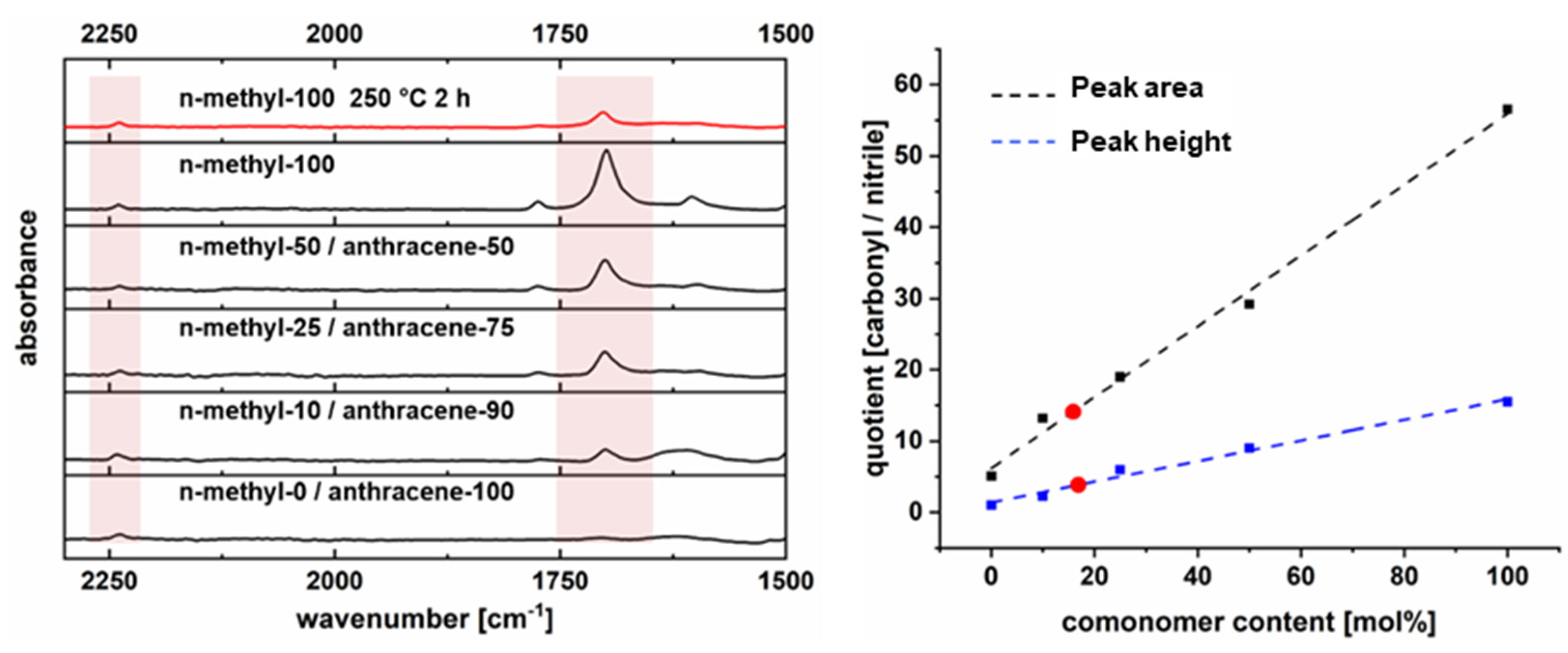
2.5.4. Thermogravimetric Analysis Coupled with Fourier Transform Infrared Spectroscopy (TG-FTIR)
2.5.5. Fourier Transform Infrared Spectroscopy (FTIR)
2.5.6. Density Measurement
2.5.7. Determination of Fractional Free Volume (FFV)
2.5.8. Gas Transport Properties Measurement
3. Results and Discussions
3.1. Comonomer Synthesis
3.2. Polymer Synthesis
3.3. Gas and Water Vapor Transport Properties
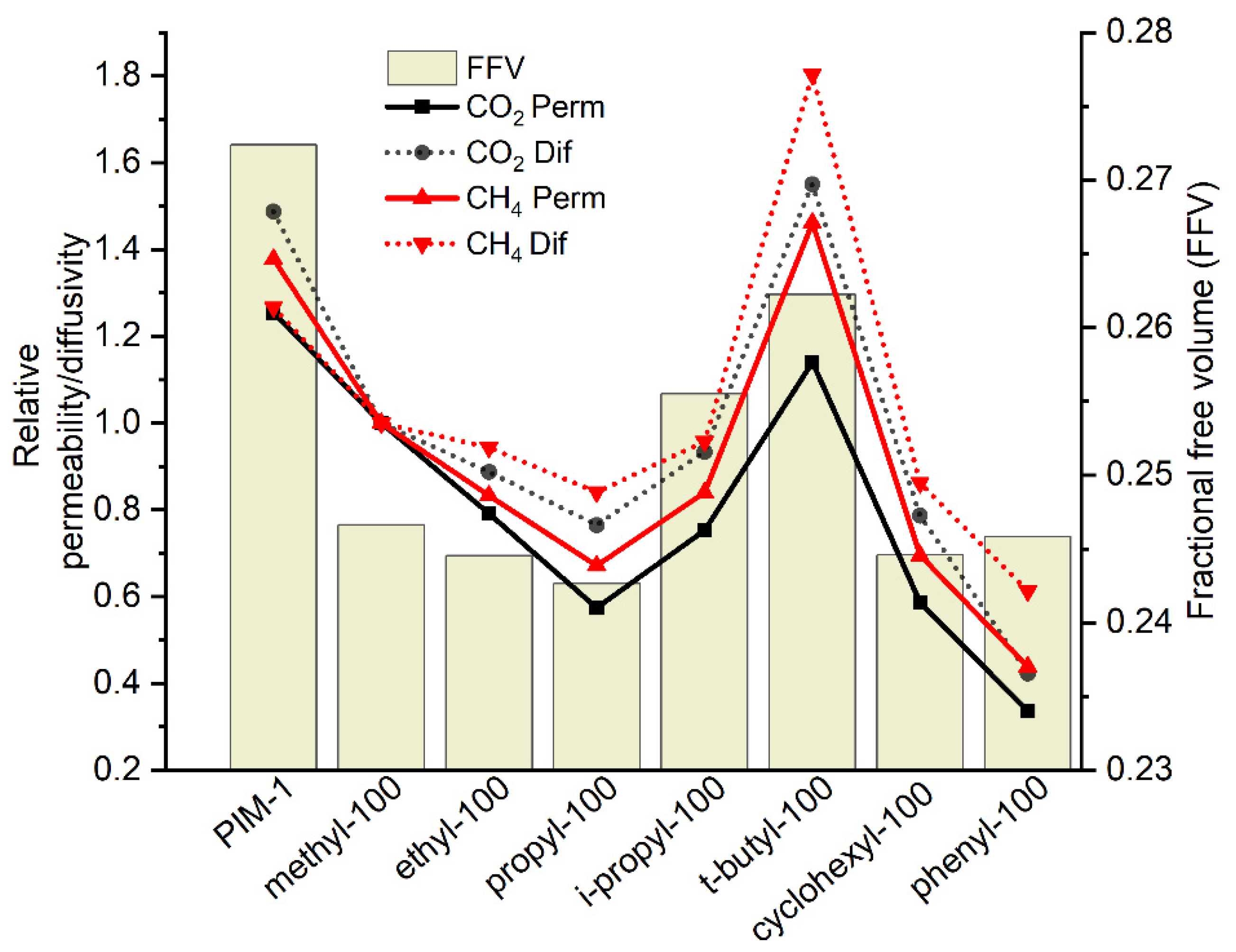
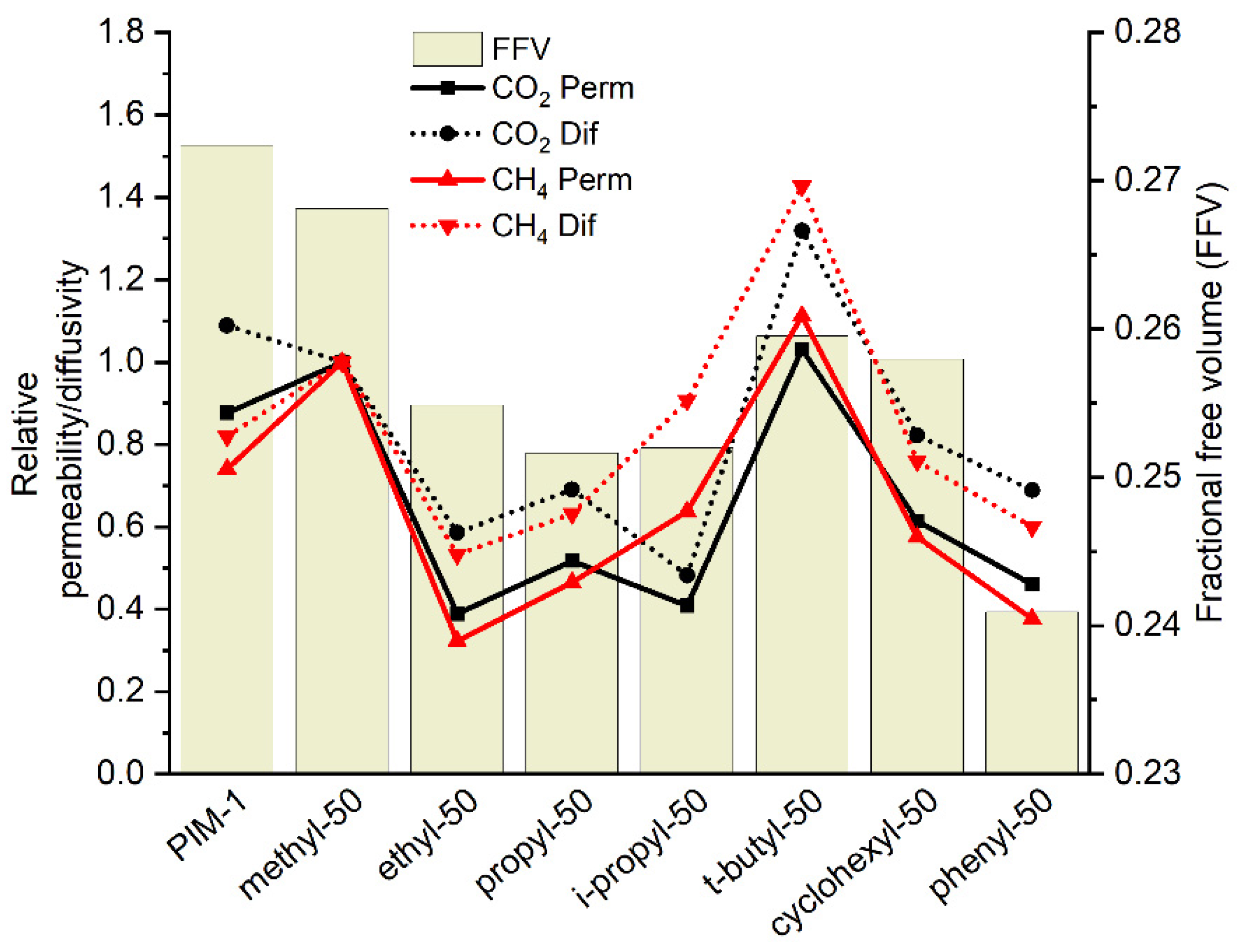
3.3.1. Gas Transport Properties of Homopolymers and Copolymers
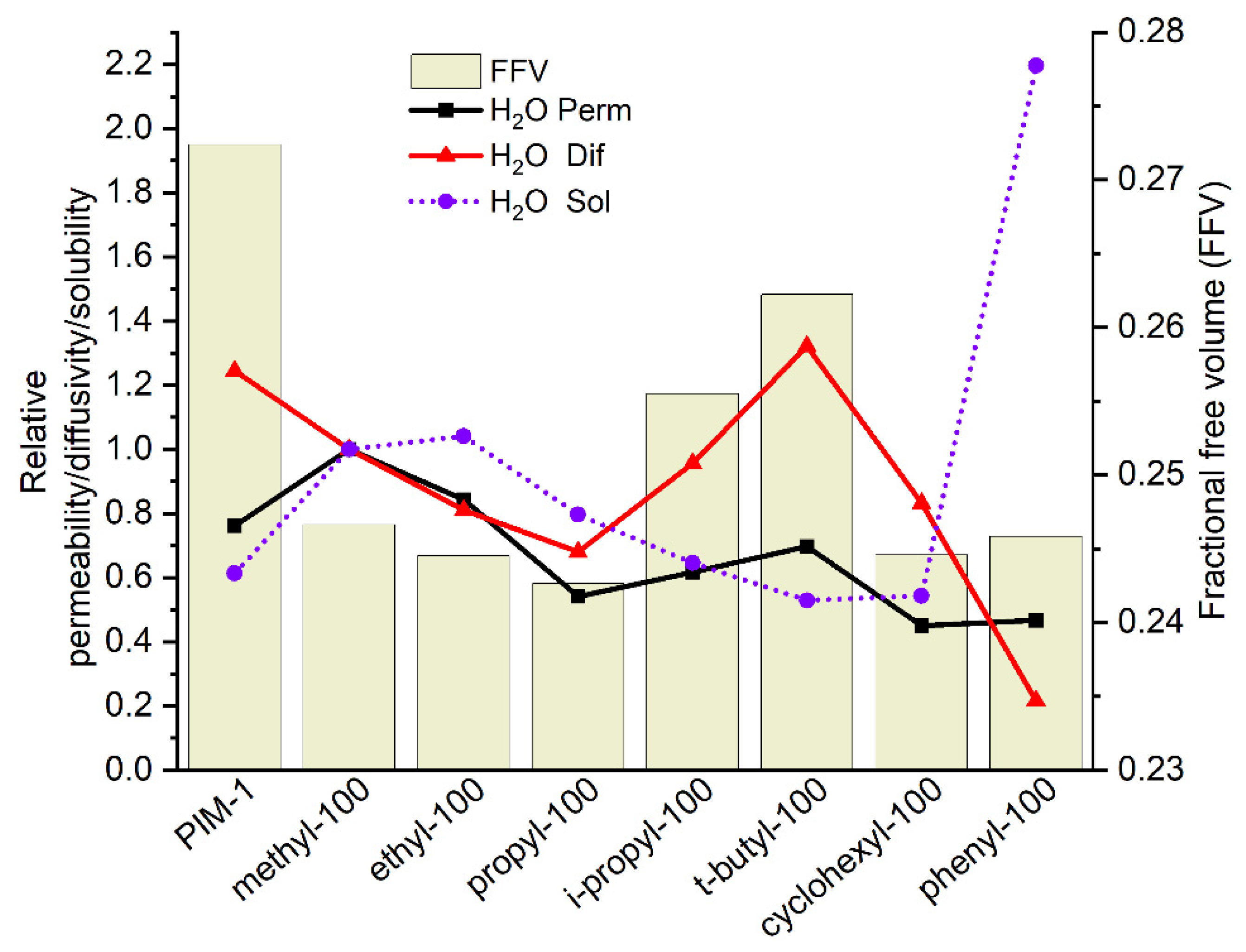
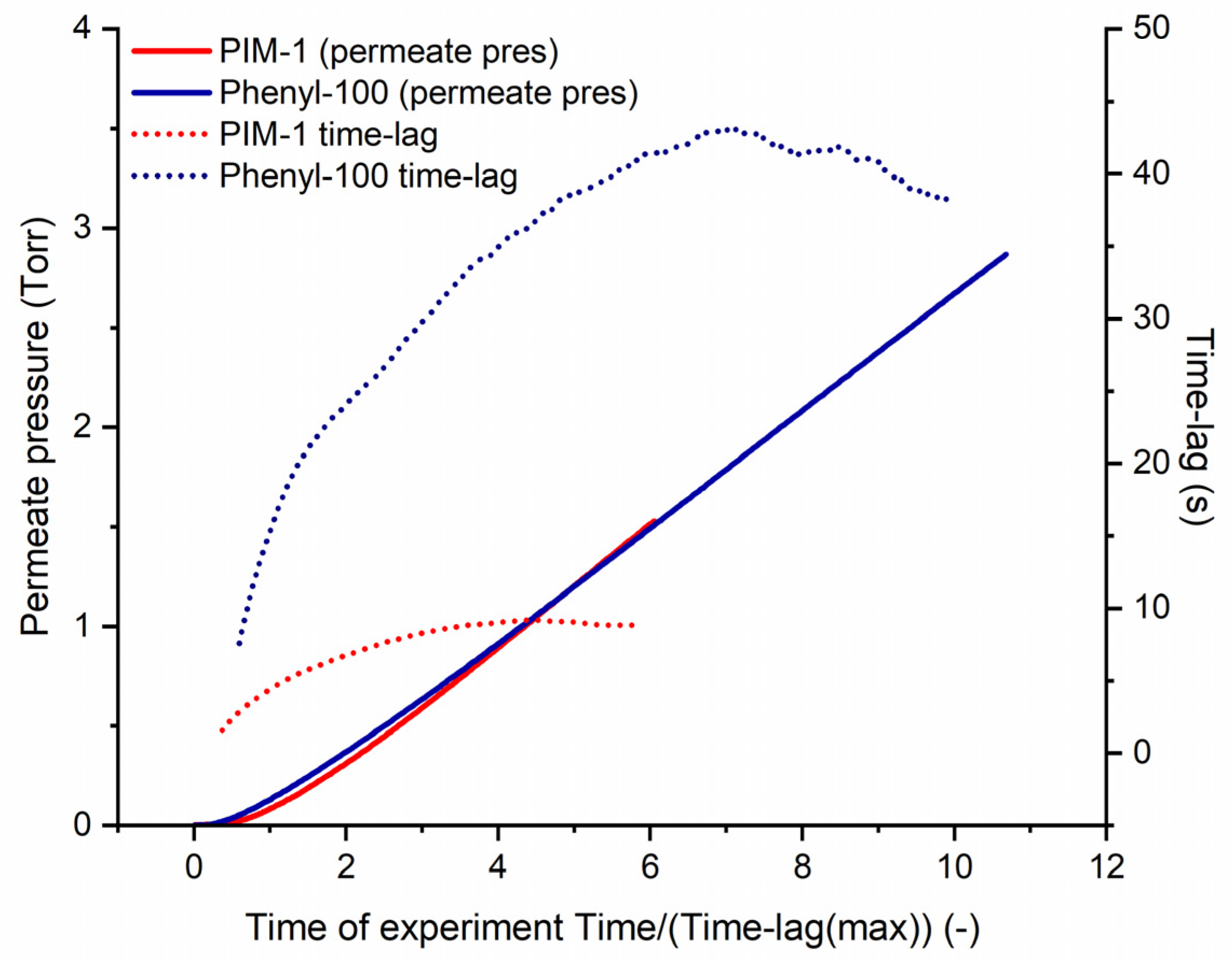
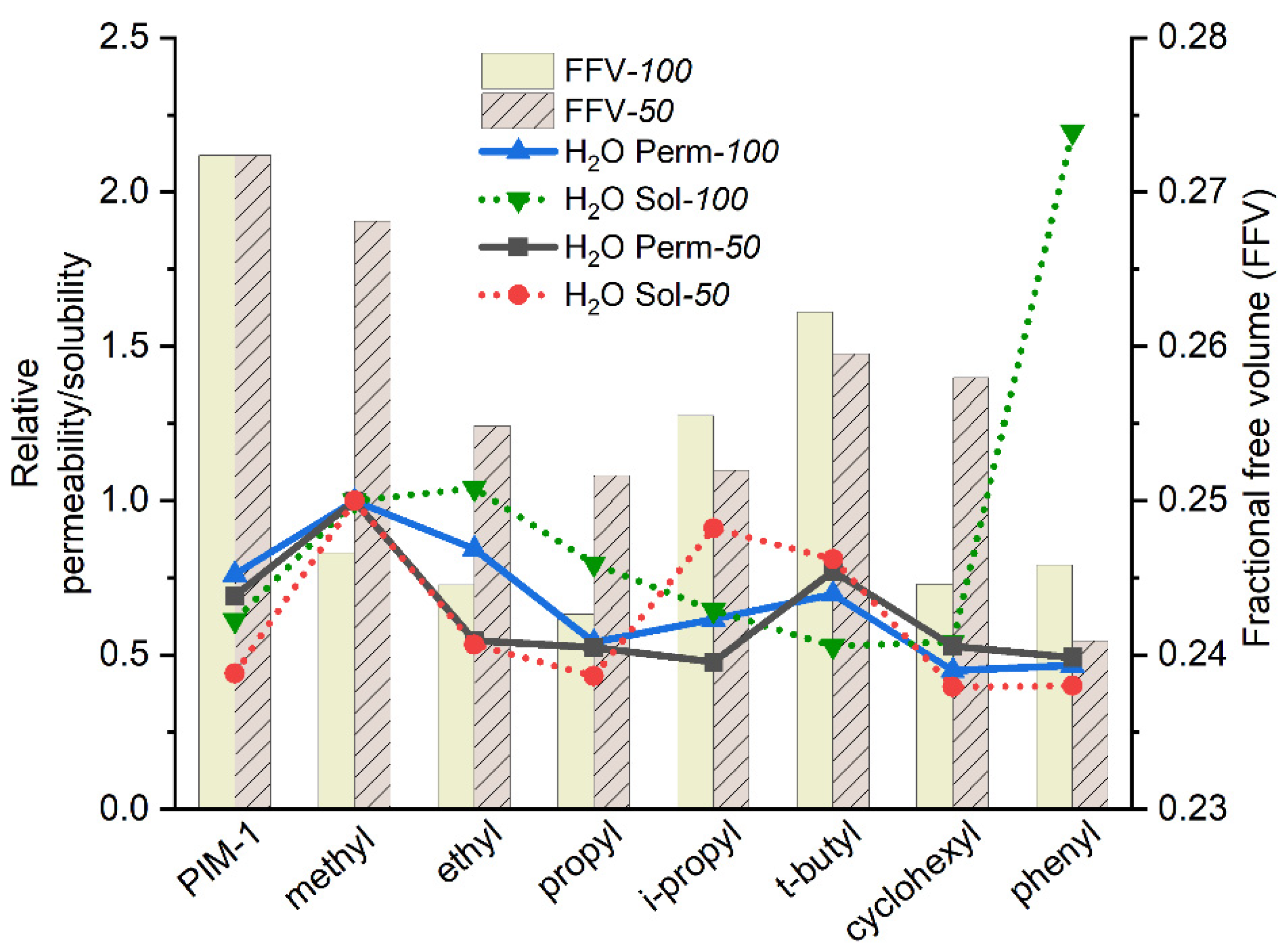
3.3.2. Water Vapor Transport Properties of Homo- and Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Budd, P.M.; McKeown, N.B. Highly permeable polymers for gas separation membranes. Polym. Chem. 2010, 1, 63–68. [Google Scholar] [CrossRef]
- Wijmans, J.G.; Baker, R.W. The solution-diffusion model: A review. J. Membr. Sci. 1995, 107, 1–21. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, L.; Xu, L.; Chung, T.-S. Molecular design of Tröger’s base-based polymers with intrinsic microporosity for gas separation. J. Membr. Sci. 2017, 521, 65–72. [Google Scholar] [CrossRef]
- Park, H.B.; Kamcev, J.; Robeson, L.M.; Elimelech, M.; Freeman, B.D. Maximizing the right stuff: The trade-off between membrane permeability and selectivity. Science 2017, 356, eaab0530. [Google Scholar] [CrossRef] [Green Version]
- Corrado, T.; Guo, R. Macromolecular design strategies toward tailoring free volume in glassy polymers for high performance gas separation membranes. Mol. Syst. Des. Eng. 2020, 5, 22–48. [Google Scholar] [CrossRef]
- Budd, P.M.; Elabas, E.S.; Ghanem, B.S.; Makhseed, S.; McKeown, N.B.; Msayib, K.J.; Tattershall, C.E.; Wang, D. Solution-Processed, Organophilic Membrane Derived from a Polymer of Intrinsic Microporosity. Adv. Mater. 2004, 16, 456–459. [Google Scholar] [CrossRef]
- Rose, I.; Bezzu, C.G.; Carta, M.; Comesana-Gandara, B.; Lasseuguette, E.; Ferrari, M.C.; Bernardo, P.; Clarizia, G.; Fuoco, A.; Jansen, J.C.; et al. Polymer ultrapermeability from the inefficient packing of 2D chains. Nat. Mater. 2017, 16, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Guiver, M.D.; Lee, Y.M. Polymer Rigidity Improves Microporous Membranes. Science 2013, 339, 284–285. [Google Scholar] [CrossRef] [Green Version]
- McKeown, N.B.; Budd, P.M. Exploitation of Intrinsic Microporosity in Polymer-Based Materials. Macromolecules 2010, 43, 5163–5176. [Google Scholar] [CrossRef]
- Budd, P.M.; McKeown, N.B.; Fritsch, D. Free volume and intrinsic microporosity in polymers. J. Mater. Chem. 2005, 15, 1977–1986. [Google Scholar] [CrossRef]
- Budd, P.M.; Ghanem, B.S.; Makhseed, S.; McKeown, N.B.; Msayib, K.J.; Tattershall, C.E. Polymers of intrinsic microporosity (PIMs): Robust, solution-processable, organic nanoporous materials. Chem. Commun. 2004, 230–231. [Google Scholar] [CrossRef]
- Budd, P.M.; McKeown, N.B.; Ghanem, B.S.; Msayib, K.J.; Fritsch, D.; Starannikova, L.; Belov, N.; Sanfirova, O.; Yampolskii, Y.; Shantarovich, V. Gas permeation parameters and other physicochemical properties of a polymer of intrinsic microporosity: Polybenzodioxane PIM-1. J. Membr. Sci. 2008, 325, 851–860. [Google Scholar] [CrossRef]
- Alberto, M.; Bhavsar, R.; Luque-Alled, J.M.; Vijayaraghavan, A.; Budd, P.M.; Gorgojo, P. Impeded physical aging in PIM-1 membranes containing graphene-like fillers. J. Membr. Sci. 2018, 563, 513–520. [Google Scholar] [CrossRef] [Green Version]
- Bengtson, G.; Neumann, S.; Filiz, V. Membranes of Polymers of Intrinsic Microporosity (PIM-1) Modified by Poly(ethylene glycol). Membranes 2017, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Bezzu, C.G.; Carta, M.; Tonkins, A.; Jansen, J.C.; Bernardo, P.; Bazzarelli, F.; McKeown, N.B. A Spirobifluorene-Based Polymer of Intrinsic Microporosity with Improved Performance for Gas Separation. Adv. Mater. 2012, 24, 5930–5933. [Google Scholar] [CrossRef]
- Tian, M.; Rochatz, S.; Fawcett, H.; Burrows, A.D.; Bowen, C.R.; Mays, T.J. Chemical modification of the polymer of intrinsic microporosity PIM-1 for enhanced hydrogen storage, (in English). Adsorpt.-J. Int. Adsorpt. Soc. 2020, 26, 1083–1091. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, D.G.; Lee, K.; Baek, Y.; Yoo, Y.; Kim, Y.S.; Kim, B.G.; Lee, J.C. A Carbonaceous Membrane based on a Polymer of Intrinsic Microporosity (PIM-1) for Water Treatment. Sci. Rep. 2016, 6, 36078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, E.; Cottis, P.; Rong, Y.; Rogers, A.T.; Stone, J.M.; Malpass-Evans, R.; Carta, M.; McKeown, N.B.; Marken, F. Water desalination concept using an ionic rectifier based on a polymer of intrinsic microporosity (PIM). J. Mater. Chem. A 2015, 3, 15849–15853. [Google Scholar] [CrossRef] [Green Version]
- Du, N.; Cin, M.M.; Pinnau, I.; Nicalek, A.; Robertson, G.P.; Guiver, M.D. Azide-based cross-linking of polymers of intrinsic microporosity (PIMs) for condensable gas separation. Macromol. Rapid Commun. 2011, 32, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Mason, C.R.; Maynard-Atem, L.; Heard, K.W.J.; Satilmis, B.; Budd, P.M.; Friess, K.; Lanč, M.; Bernardo, P.; Clarizia, G.; Jansen, J.C. Enhancement of CO2 Affinity in a Polymer of Intrinsic Microporosity by Amine Modification. Macromolecules 2014, 47, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Song, J.; Pinnau, I.; Guiver, M.D. High-Performance Carboxylated Polymers of Intrinsic Microporosity (PIMs) with Tunable Gas Transport Properties†. Macromolecules 2009, 42, 6038–6043. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Swaidan, R.; Belmabkhout, Y.; Zhu, Y.; Litwiller, E.; Jouiad, M.; Pinnau, I.; Han, Y. Synthesis and Gas Transport Properties of Hydroxyl-Functionalized Polyimides with Intrinsic Microporosity. Macromolecules 2012, 45, 3841–3849. [Google Scholar] [CrossRef]
- Du, N.; Park, H.B.; Robertson, G.P.; Dal-Cin, M.M.; Visser, T.; Scoles, L.; Guiver, M.D. Polymer nanosieve membranes for CO2-capture applications. Nat. Mater. 2011, 10, 372–375. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Al-Harbi, N.M.; Budd, P.M.; Bernardo, P.; Bazzarelli, F.; Clarizia, G.; Jansen, J.C. Polymer of Intrinsic Microporosity Incorporating Thioamide Functionality: Preparation and Gas Transport Properties. Macromolecules 2011, 44, 6471–6479. [Google Scholar] [CrossRef]
- Halder, K.; Neumann, S.; Bengtson, G.; Khan, M.M.; Filiz, V.; Abetz, V. Polymers of Intrinsic Microporosity Postmodified by Vinyl Groups for Membrane Applications. Macromolecules 2018, 51, 7309–7319. [Google Scholar] [CrossRef]
- Khan, M.M.; Filiz, V.; Bengtson, G.; Shishatskiy, S.; Rahman, M.; Abetz, V. Functionalized carbon nanotubes mixed matrix membranes of polymers of intrinsic microporosity for gas separation. Nanoscale Res. Lett. 2012, 7, 504. [Google Scholar] [CrossRef] [Green Version]
- Aliyev, E.M.; Khan, M.M.; Nabiyev, A.M.; Alosmanov, R.M.; Bunyad-zadeh, I.A.; Shishatskiy, S.; Filiz, V. Covalently Modified Graphene Oxide and Polymer of Intrinsic Microporosity (PIM-1) in Mixed Matrix Thin-Film Composite Membranes. Nanoscale Res. Lett. 2018, 13, 359. [Google Scholar] [CrossRef] [PubMed]
- Aliyev, E.; Warfsmann, J.; Tokay, B.; Shishatskiy, S.; Lee, Y.J.; Lillepaerg, J.; Champness, N.R.; Filiz, V. Gas Transport Properties of the Metal–Organic Framework (MOF)-Assisted Polymer of Intrinsic Microporosity (PIM-1) Thin-Film Composite Membranes. ACS Sustain. Chem. Eng. 2020, 9, 684–694. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Wakimoto, K.; Gibbons, A.H.; Isfahani, A.P.; Kusuda, H.; Sivaniah, E.; Ghalei, B. Enhanced PIM-1 membrane gas separation selectivity through efficient dispersion of functionalized POSS fillers. J. Membr. Sci. 2017, 539, 178–186. [Google Scholar] [CrossRef]
- Lee, W.H.; Seong, J.G.; Hu, X.; Lee, Y.M. Recent progress in microporous polymers from thermally rearranged polymers and polymers of intrinsic microporosity for membrane gas separation: Pushing performance limits and revisiting trade-off lines. J. Polym. Sci. 2020, 58, 2450–2466. [Google Scholar] [CrossRef]
- Low, Z.-X.; Budd, P.M.; McKeown, N.B.; Patterson, D.A. Gas Permeation Properties, Physical Aging, and Its Mitigation in High Free Volume Glassy Polymers. Chem. Rev. 2018, 118, 5871–5911. [Google Scholar] [CrossRef]
- Hart, K.E.; Colina, C.M. Estimating gas permeability and permselectivity of microporous polymers. J. Membr. Sci. 2014, 468, 259–268. [Google Scholar] [CrossRef]
- Carta, M.; Malpass-Evans, R.; Croad, M.; Rogan, Y.; Jansen, J.C.; Bernardo, P.; Bazzarelli, F.; McKeown, N.B. An Efficient Polymer Molecular Sieve for Membrane Gas Separations. Science 2013, 339, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Rose, I.; Carta, M.; Malpass-Evans, R.; Ferrari, M.C.; Bernardo, P.; Clarizia, G.; Jansen, J.C.; McKeown, N.B. Highly Permeable Benzotriptycene-Based Polymer of Intrinsic Microporosity. ACS Macro Lett. 2015, 4, 912–915. [Google Scholar] [CrossRef]
- Carta, M.; Croad, M.; Malpass-Evans, R.; Jansen, J.C.; Bernardo, P.; Clariza, G.; Friess, K.; Lanc, M.; McKeown, N.B. Triptycene induced enhancement of membrane gas selectivity for microporous Troger’s base polymers. Adv. Mater. 2014, 26, 3526–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calle, M.; Lozano, A.E.; de Abajo, J.; de la Campa, J.G.; Álvarez, C. Design of gas separation membranes derived of rigid aromatic polyimides. 1. Polymers from diamines containing di-tert-butyl side groups. J. Membr. Sci. 2010, 365, 145–153. [Google Scholar] [CrossRef]
- Rodriguez-Gonzalez, F.E.; Perez, G.; Niebla, V.; Jessop, I.; Martin-Trasanco, R.; Coll, D.; Ortiz, P.; Aguilar-Vega, M.; Tagle, L.H.; Terraza, C.A.; et al. New Poly(imide)s Bearing Alkyl Side-Chains: A Study on the Impact of Size and Shape of Lateral Groups on Thermal, Mechanical, and Gas Transport Properties. Membranes 2020, 10, 141. [Google Scholar] [CrossRef]
- Bermeshev, M.V.; Syromolotov, A.V.; Starannikova, L.E.; Gringolts, M.L.; Lakhtin, V.G.; Yampolskii, Y.P.; Finkelshtein, E.S. Glassy Polynorbornenes with Si–O–Si Containing Side Groups. Novel Materials for Hydrocarbon Membrane Separation. Macromolecules 2013, 46, 8973–8979. [Google Scholar] [CrossRef]
- Yampolksii, Y.P.; Banerjee, S. Effects of Bulky Substituents on Transport Properties of Membrane Gas Separation Materials. Pet. Chem. 2018, 57, 1195–1206. [Google Scholar] [CrossRef]
- Masuda, T. Substituted polyacetylenes. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 165–180. [Google Scholar] [CrossRef]
- Pinnau, I.; He, Z.; Morisato, A. Synthesis and gas permeation properties of poly(dialkylacetylenes) containing isopropyl-terminated side-chains. J. Membr. Sci. 2014, 241, 363–369. [Google Scholar] [CrossRef]
- Deshmukh, A.; Boo, C.; Karanikola, V.; Lin, S.; Straub, A.P.; Tong, T.; Warsinger, D.M.; Elimelech, M. Membrane distillation at the water-energy nexus: Limits, opportunities, and challenges. Energy Environ. Sci. 2018, 11, 1177–1196. [Google Scholar] [CrossRef]
- Drioli, E.; Ali, A.; Macedonio, F. Membrane distillation: Recent developments and perspectives. Desalination 2015, 356, 56–84. [Google Scholar] [CrossRef]
- MKhan, M.; Bengtson, G.; Neumann, S.; Rahman, M.M.; Abetz, V.; Filiz, V. Synthesis, characterization and gas permeation properties of anthracene maleimide-based polymers of intrinsic microporosity. RSC Adv. 2014, 4, 32148–32160. [Google Scholar] [CrossRef] [Green Version]
- Halder, K.; Khan, M.M.; Grünauer, J.; Shishatskiy, S.; Abetz, C.; Filiz, V.; Abetz, V. Blend membranes of ionic liquid and polymers of intrinsic microporosity with improved gas separation characteristics. J. Membr. Sci. 2017, 539, 368–382. [Google Scholar] [CrossRef]
- Halder, K.; Georgopanos, P.; Shishatskiy, S.; Filiz, V.; Abetz, V. Investigation of gas transport and other physical properties in relation to the bromination degree of polymers of intrinsic microporosity. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 2752–2761. [Google Scholar] [CrossRef]
- Du, N.; Song, J.; Robertson, G.P.; Pinnau, I.; Guiver, M.D. Linear High Molecular Weight Ladder Polymer via Fast Polycondensation of 5,5′,6,6′-Tetrahydroxy-3,3,3′,3′-tetramethylspirobisindane with 1,4-Dicyanotetrafluorobenzene. Macromol. Rapid Commun. 2008, 29, 783–788. [Google Scholar] [CrossRef] [Green Version]
- Nagai, K.; Freeman, B.D.; Hill, A.J. Effect of physical aging of poly(1-trimethylsilyl-1-propyne) films synthesized with TaCl5 and NbCl5 on gas permeability, fractional free volume, and positron annihilation lifetime spectroscopy parameters. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 1222–1239. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Krevelen, D.W.V.; Nijenhuis, K.T. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009; p. 1032. [Google Scholar]
- Lillepärg, J.; Georgopanos, P.; Emmler, T.; Shishatskiy, S. Effect of the reactive amino and glycidyl ether terminated polyethylene oxide additives on the gas transport properties of Pebax® bulk and thin film composite membranes. RSC Adv. 2016, 6, 11763–11772. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, D.; Bengtson, G.; Carta, M.; McKeown, N.B. Synthesis and Gas Permeation Properties of Spirobischromane-Based Polymers of Intrinsic Microporosity. Macromol. Chem. Phys. 2011, 212, 1137–1146. [Google Scholar] [CrossRef]
- Yin, H.; Yang, B.; Chua, Y.Z.; Szymoniak, P.; Carta, M.; Malpass-Evans, R.; McKeown, N.B.; Harrison, W.J.; Budd, P.M.; Schick, C.; et al. Effect of Backbone Rigidity on the Glass Transition of Polymers of Intrinsic Microporosity Probed by Fast Scanning Calorimetry. ACS Macro Lett. 2019, 8, 1022–1028. [Google Scholar] [CrossRef]
- Harms, S.; Rätzke, K.; Faupel, F.; Chaukura, N.; Budd, P.M.; Egger, W.; Ravelli, L. Aging and Free Volume in a Polymer of Intrinsic Microporosity (PIM-1). J. Adhes. 2017, 88, 608–619. [Google Scholar] [CrossRef]
- Guiver, M.D.; Yahia, M.; Dal-Cin, M.M.; Robertson, G.P.; Saeedi Garakani, S.; Du, N.; Tavajohi, N. Gas Transport in a Polymer of Intrinsic Microporosity (PIM-1) Substituted with Pseudo-Ionic Liquid Tetrazole-Type Structures. Macromolecules 2020, 53, 8951–8959. [Google Scholar] [CrossRef]
- Song, J.; Du, N.; Dai, Y.; Robertson, G.P.; Guiver, M.D.; Thomas, S.; Pinnau, I. Linear High Molecular Weight Ladder Polymers by Optimized Polycondensation of Tetrahydroxytetramethylspirobisindane and 1,4-Dicyanotetrafluorobenzene. Macromolecules 2008, 41, 7411–7417. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, C.; Zhang, P.; Sun, B.; Li, J. Pervaporation Properties of Polyimide Membranes for Separation of Ethanol + Water Mixtures. J. Chem. Eng. Data 2006, 51, 1841–1845. [Google Scholar] [CrossRef]
- Huang, J.; Cranford, R.J.; Matsuura, T.; Roy, C. Water vapor permeation properties of aromatic polyimides. J. Membr. Sci. 2003, 215, 129–140. [Google Scholar] [CrossRef]
- Jiang, L.Y.; Wang, Y.; Chung, T.-S.; Qiao, X.Y.; Lai, J.-Y. Polyimides membranes for pervaporation and biofuels separation. Prog. Polym. Sci. 2009, 34, 1135–1160. [Google Scholar] [CrossRef]
- Petrov, A.V.; Smirnov, M.A.; Sokolova, M.P.; Toikka, A.M. Influence of Water Concentration on Its Mobility in Matrimid®. Coatings 2019, 9, 466. [Google Scholar] [CrossRef] [Green Version]
- Maya, E.M.; Benavente, J.; de Abajo, J. Effect of the carboxylic acid groups on water sorption, thermal stability and dielectric properties of polyimide films. Mater. Chem. Phys. 2012, 131, 581–588. [Google Scholar] [CrossRef]
- Kim, J.W.; Chang, J.-H. Syntheses of Colorless and Transparent Polyimide Membranes for Microfiltration. Polymers 2020, 12, 1610. [Google Scholar] [CrossRef]
- Stern, S.A.; Shah, V.M.; Hardy, B.J. Structure-permeability relationships in silicone polymers. J. Polym. Sci. Part B Polym. Phys. 1987, 25, 1263–1298. [Google Scholar] [CrossRef]
- Borisov, I.L.; Ushakov, N.V.; Volkov, V.V.; Finkel’shtein, E.S. Polydimethylsilalkylene-dimethylsiloxanes as advanced membrane materials for thermopervaporative recovery of oxygenates from aqueous reaction media. Pet. Chem. 2016, 56, 798–804. [Google Scholar] [CrossRef]
- Chen, G.Q.; Scholes, C.A.; Qiao, G.G.; Kentish, S.E. Water vapor permeation in polyimide membranes. J. Membr. Sci. 2011, 379, 479–487. [Google Scholar] [CrossRef]
- Barrie, J.A.; Platt, B. The diffusion and clustering of water vapour in polymers. Polymer 1963, 4, 303–313. [Google Scholar] [CrossRef]
- Modesti, M.; Dall’Acqua, C.; Lorenzetti, A.; Florian, E. Mathematical model and experimental validation of water cluster influence upon vapour permeation through hydrophilic dense membrane. J. Membr. Sci. 2004, 229, 211–223. [Google Scholar] [CrossRef]
- Kelkar, A.J.; Paul, D.R. Water vapor transport in a series of polyarylates. J. Membr. Sci. 2001, 181, 199–212. [Google Scholar] [CrossRef]
- Arce, A.; Fornasiero, F.; Rodríguez, O.; Radke, C.J.; Prausnitz, J.M. Sorption and transport of water vapor in thin polymer films at 35 °C. Phys. Chem. Chem. Phys. 2004, 6, 103–108. [Google Scholar] [CrossRef]
- Kanehashi, S.; Tomita, Y.; Obokata, H.; Kidesaki, T.; Sato, S.; Miyakoshi, T.; Nagai, K. Effect of substituted groups on characterization and water vapor sorption property of polyhedral oligomeric silsesquioxane (POSS)-containing methacryl polymer membranes. Polymer 2013, 54, 2315–2323. [Google Scholar] [CrossRef]
- Du, A.; Koo, D.; Theryo, G.; Hillmyer, M.A.; Cairncross, R.A. Water transport and clustering behavior in homopolymer and graft copolymer polylactide. J. Membr. Sci. 2012, 396, 50–56. [Google Scholar] [CrossRef]
- Davis, E.M.; Minelli, M.; Baschetti, M.G.; Sarti, G.C.; Elabd, Y.A. Nonequilibrium Sorption of Water in Polylactide. Macromolecules 2012, 45, 7486–7494. [Google Scholar] [CrossRef]
- Jansen, J.C.; Friess, K.; Drioli, E. Organic vapour transport in glassy perfluoropolymer membranes: A simple semi-quantitative approach to analyze clustering phenomena by time lag measurements. J. Membr. Sci. 2011, 367, 141–151. [Google Scholar] [CrossRef]
- Friess, K.; Jansen, J.C.; Poživil, J.; Hanta, V.; Hynek, V.; Vopička, O.; Zgažar, M.; Bernardo, P.; Izák, P.; Drioli, E. Anomalous Phenomena Occurring during Permeation and Sorption of C1–C6 Alcohol Vapors in Teflon AF 2400. Ind. Eng. Chem. Res. 2013, 52, 10406–10417. [Google Scholar] [CrossRef]
- Levitt, M.; Perutz, M.F. Aromatic rings act as hydrogen bond acceptors. J. Mol. Biol. 1988, 201, 751–754. [Google Scholar] [CrossRef]
- Vojislavljević, D.Z.; Janjić, G.V.; Ninković, D.B.; Kapor, A.; Zarić, S.D. The influence of water molecule coordination onto the water–aromatic interaction. Strong interactions of water coordinating to a metal ion. CrystEngComm 2013, 15, 2099–2105. [Google Scholar] [CrossRef]
- Jain, A.; Ramanathan, V.; Sankararamakrishnan, R. Lone pair … pi interactions between water oxygens and aromatic residues: Quantum chemical studies based on high-resolution protein structures and model compounds. Protein Sci. 2009, 18, 595–605. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PIM-1 | Methyl-100 | Ethyl-100 | Propyl-100 | i-Propyl-100 | t-Butyl-100 | Cyclohexyl-100 | Phenyl-100 | |
|---|---|---|---|---|---|---|---|---|
| Water permeability (Barrer *) | 79,300 | 110,000 | 87,800 | 56,300 | 64,100 | 72,500 | 46,800 | 48,600 |
| Methyl-50 | Ethyl-50 | Propyl-50 | i-Propyl-50 | t-Butyl-50 | Cyclohexyl-50 | Phenyl-50 | ||
| Water permeability (Barrer *) | 114,500 | 62,700 | 60,100 | 54,800 | 88,500 | 60,600 | 56,400 |
| Polymer | Time [Time-Lags] |
|---|---|
| PIM-1 | 4.44 |
| Methyl-100 | 4.49 |
| t-Butyl-100 | 4.90 |
| Cyclohexyl-100 | 4.17 |
| Phenyl-100 | 7.16 |
| Phenyl-50 | 3.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliskan, E.; Shishatskiy, S.; Neumann, S.; Abetz, V.; Filiz, V. Investigation of the Side Chain Effect on Gas and Water Vapor Transport Properties of Anthracene-Maleimide Based Polymers of Intrinsic Microporosity. Polymers 2022, 14, 119. https://doi.org/10.3390/polym14010119
Caliskan E, Shishatskiy S, Neumann S, Abetz V, Filiz V. Investigation of the Side Chain Effect on Gas and Water Vapor Transport Properties of Anthracene-Maleimide Based Polymers of Intrinsic Microporosity. Polymers. 2022; 14(1):119. https://doi.org/10.3390/polym14010119
Chicago/Turabian StyleCaliskan, Esra, Sergey Shishatskiy, Silvio Neumann, Volker Abetz, and Volkan Filiz. 2022. "Investigation of the Side Chain Effect on Gas and Water Vapor Transport Properties of Anthracene-Maleimide Based Polymers of Intrinsic Microporosity" Polymers 14, no. 1: 119. https://doi.org/10.3390/polym14010119