
Biomedical PEVA Nanocomposite with Dual Clay Nanofiller: Cytotoxicity, Mechanical Properties, and Biostability


 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
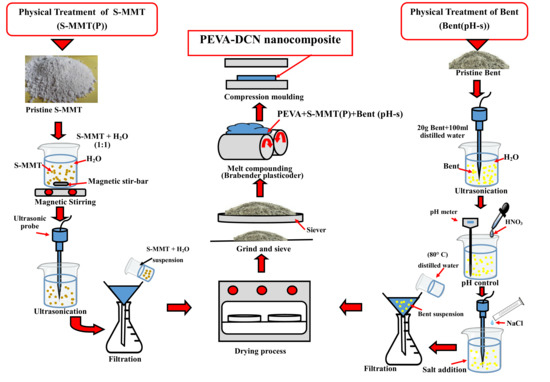
2.2. Preparation of Nanofillers
2.2.1. Physical Treatment by Magnetic Stirring and Ultra-Sonication of Surface Modified Montmorillonite (S-MMT(P))
2.2.2. Physical Treatment by pH Control and Salt Addition of Bentonite(Bent(pH-s))
2.3. Preparation of PEVA Nanocomposites and PEVA-DCN Nanocomposites
2.4. Fourier Transform Infrared Spectroscopy (FTIR)
2.5. X-Ray Diffraction (XRD)
- λ = wavelength of the rays;
- θ = angle between the incident rays and the surface of the crystal;
- d = spacing between the clay layers.
2.6. Fibroblast Cell Cytotoxicity Assay (Biocompatibility Test)
2.7. Tensile Test (Ambient)
2.8. Transmission Electron Microscopy (TEM)
2.9. Scanning Electron Microscope (SEM)
2.10. Biostability Analysis
3. Results and Discussion
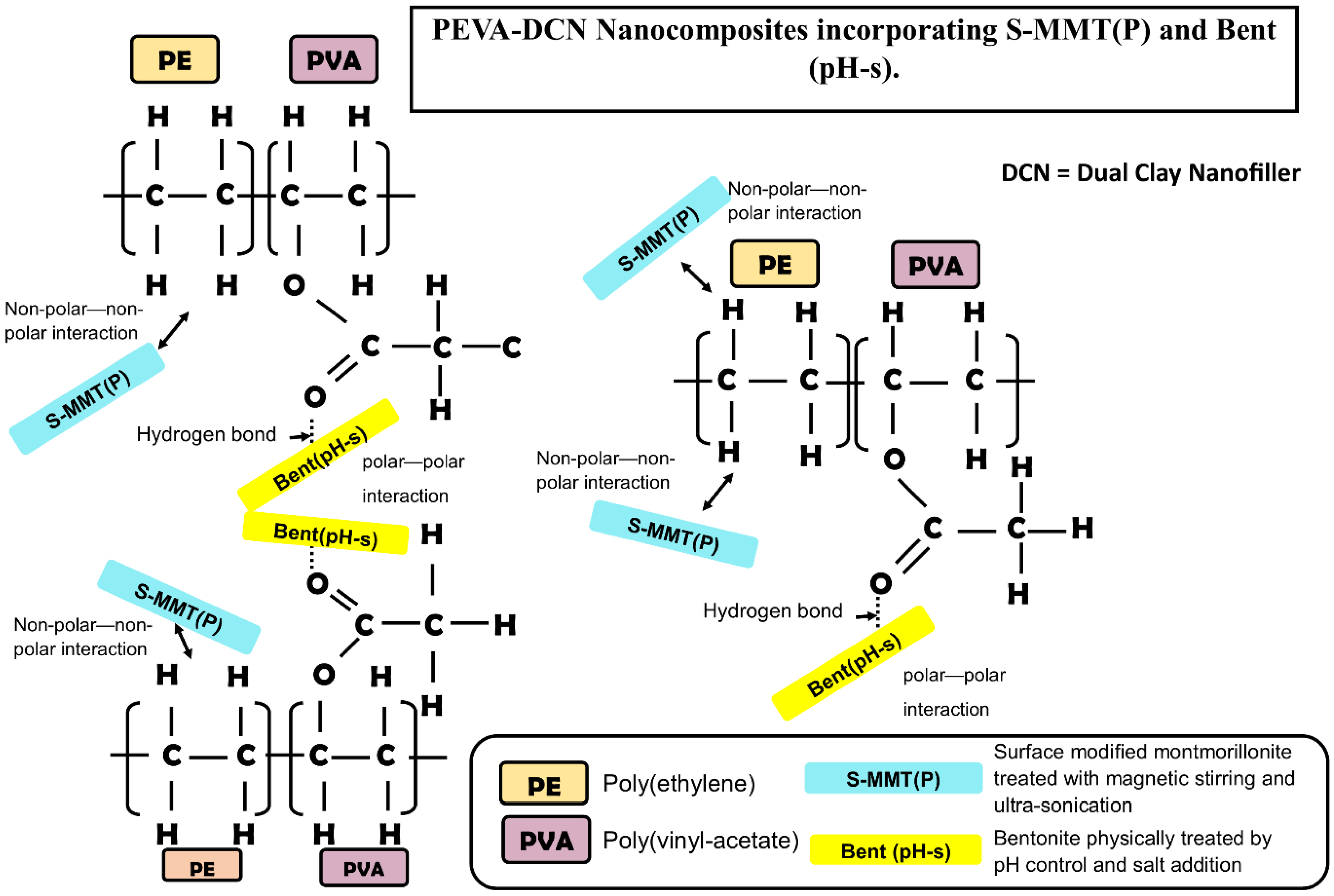
3.1. Characterization and Biocompatibility Analysis of the Physically Treated DCN (S-MMT(P)/Bent(pH-s))
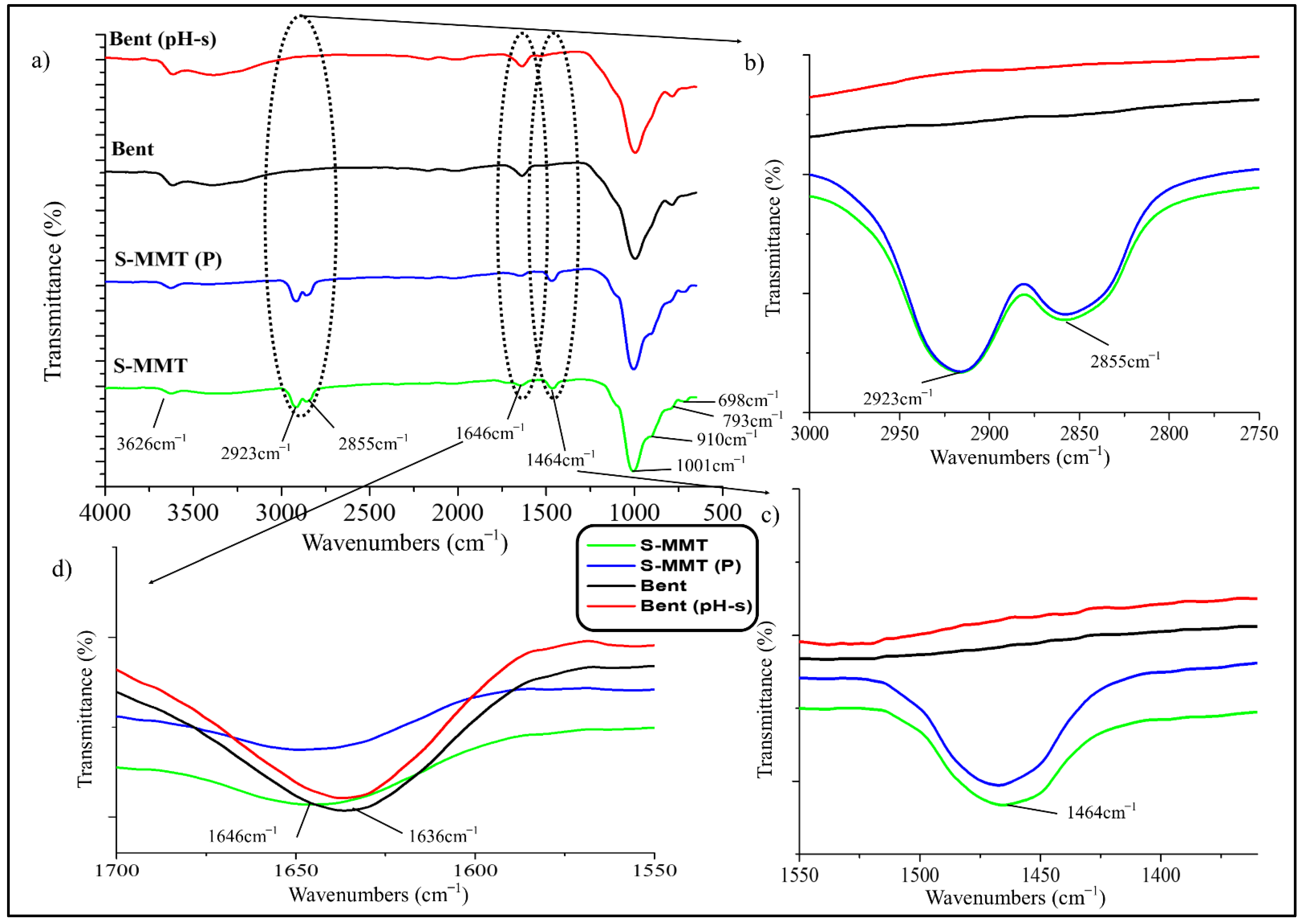
3.1.1. Fourier Transform Infrared Analysis (FTIR)
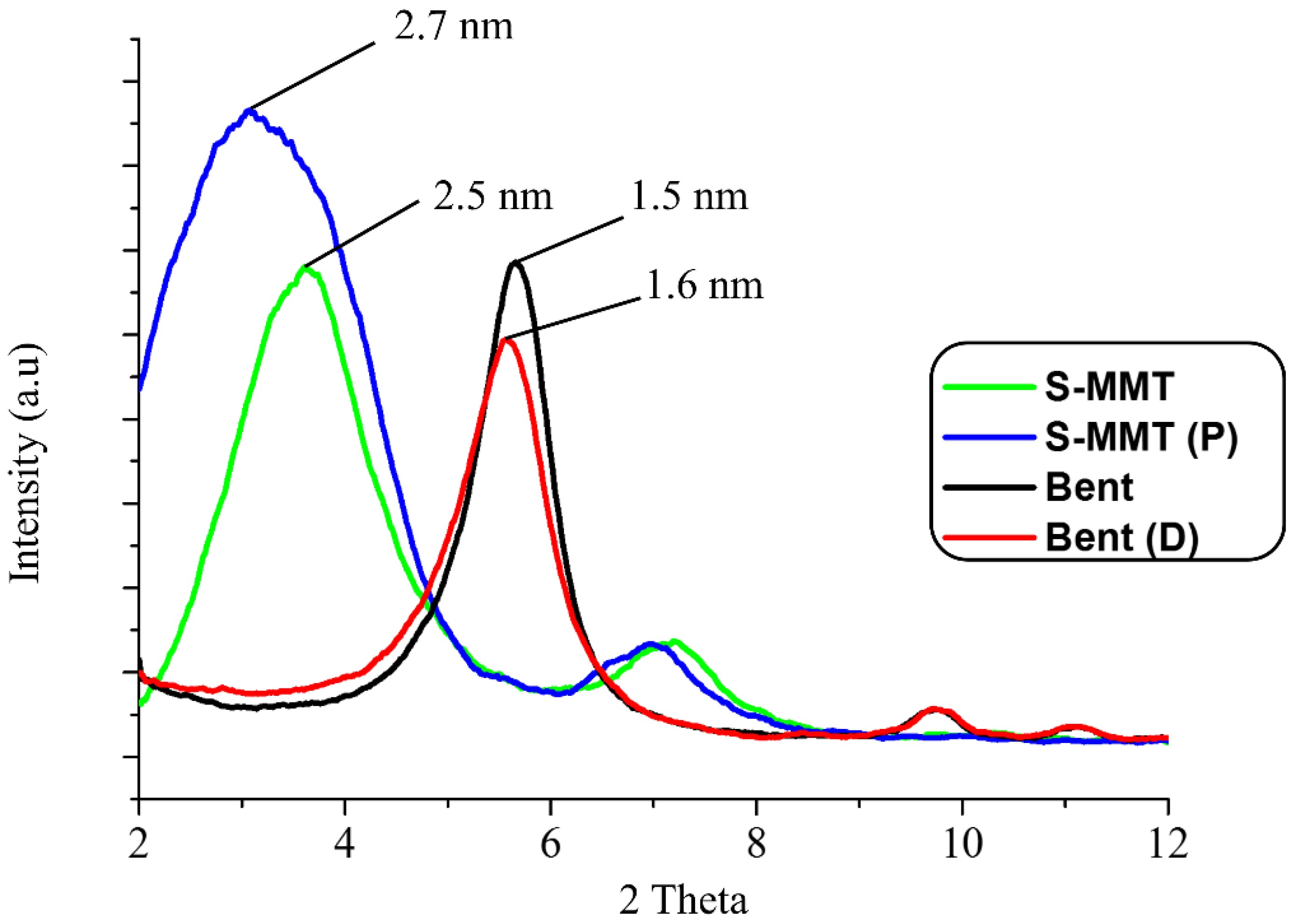
3.1.2. X-ray Diffraction Analysis (XRD)
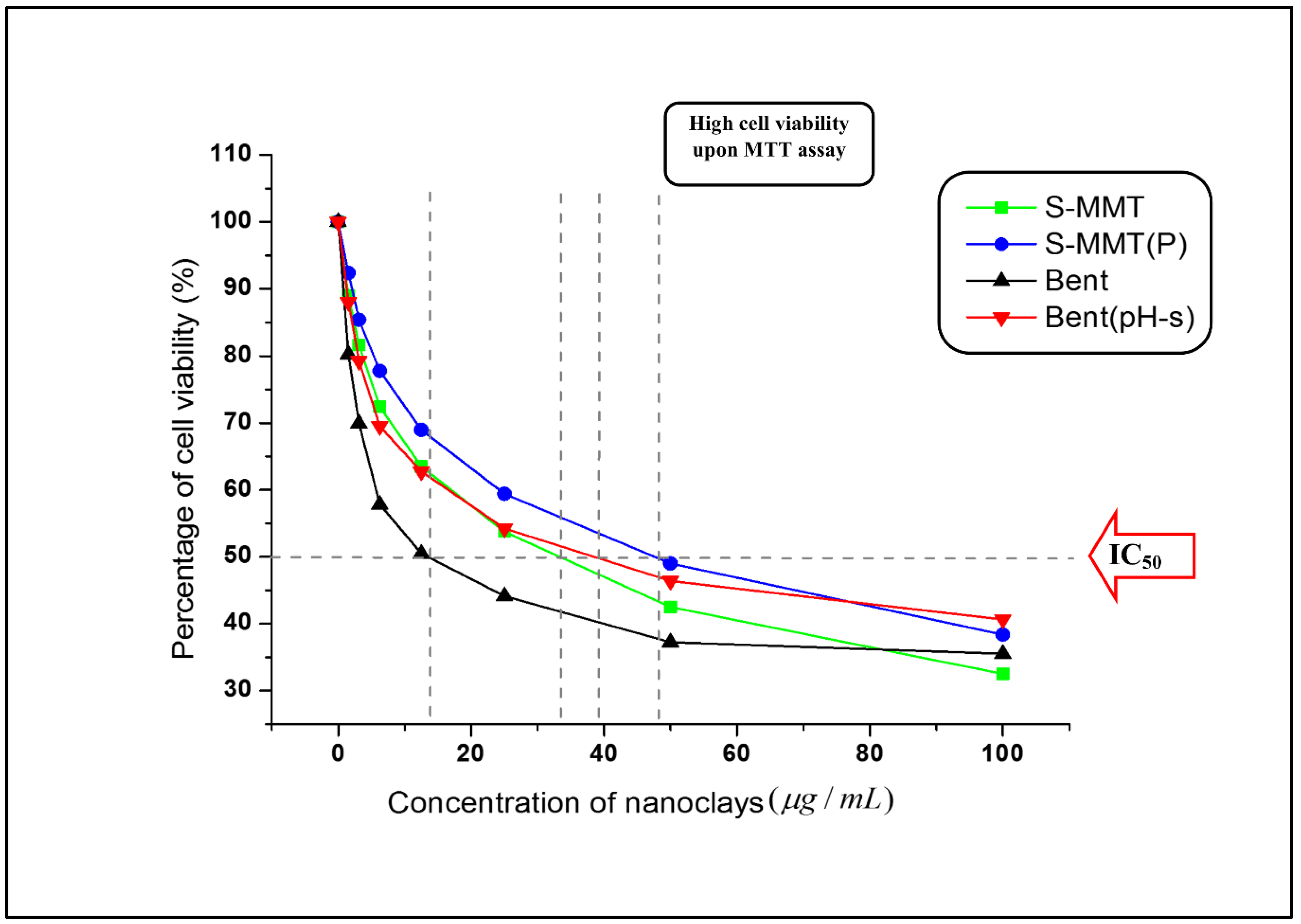
3.1.3. Fibroblast Cell Cytotoxicity Assay
3.2. Mechanical Properties of the Neat PEVA, PEVA Nanocomposites and PEVA-DCN Nanocomposites Incorporating Physically Treated DCN (S-MMT(P)/Bent(pH-s))
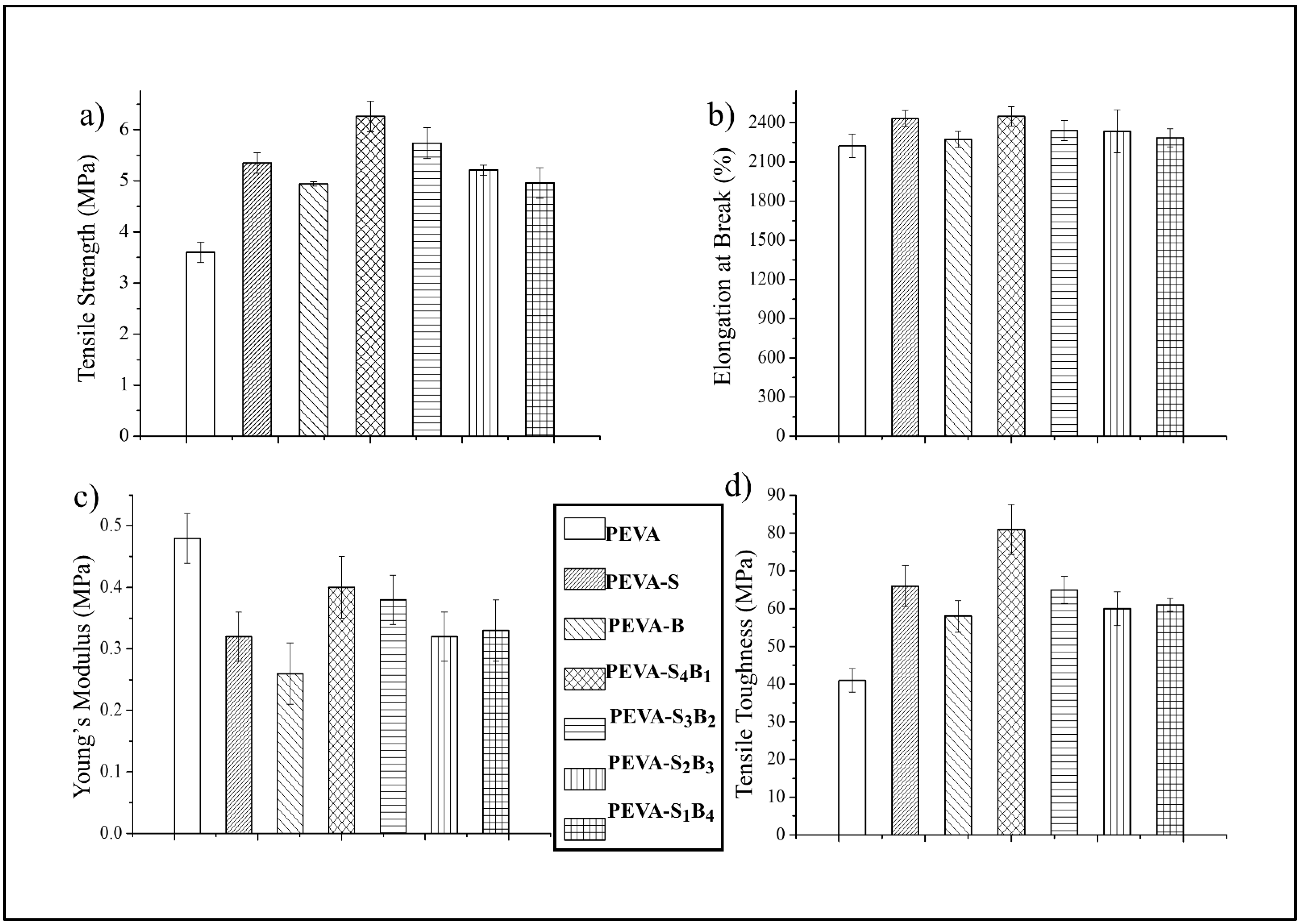
3.2.1. Tensile Properties of Neat PEVA, PEVA Nanocomposites, and PEVA-DCN Nanocomposites
3.2.2. Dispersity Analysis of the PEVA Nanocomposites (PEVA-S and PEVA-B) and Optimum PEVA-DCN Nanocomposite (PEVA-S4B1)
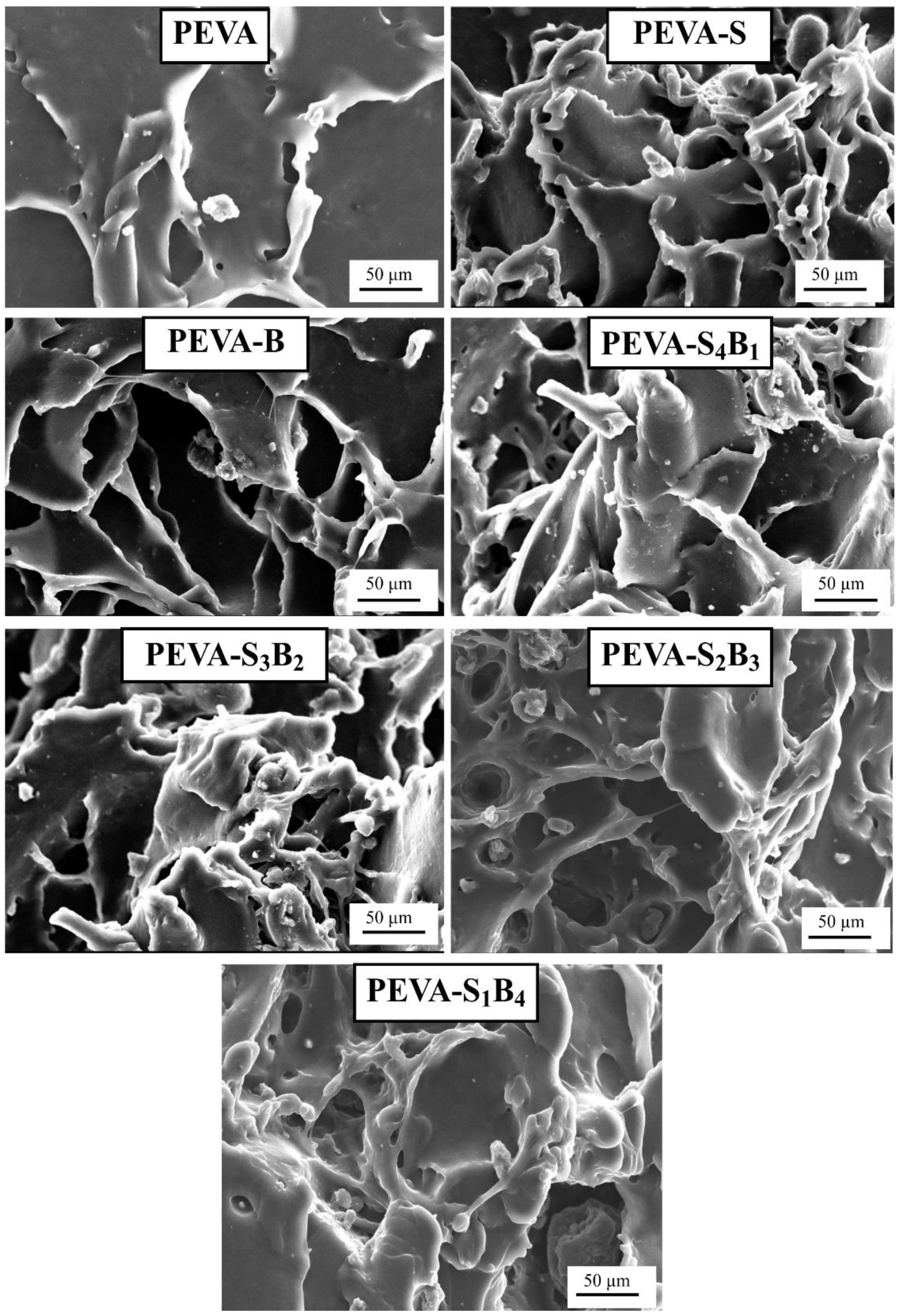
3.2.3. Analyzing Tensile Fractured Surface by SEM
3.3. Biostability Analysis
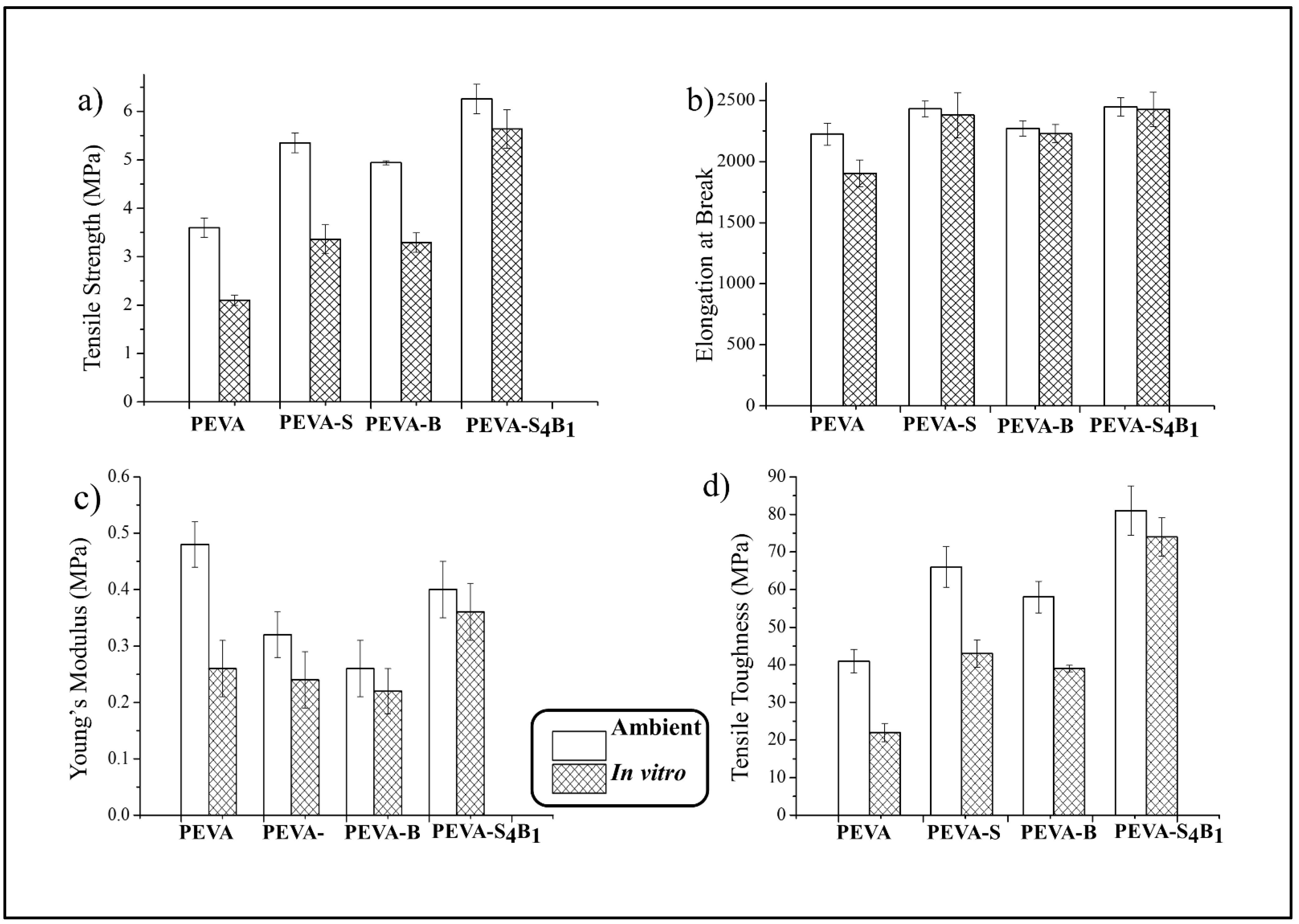
3.3.1. Ambient and In Vitro Mechanical Properties by Tensile Test
3.3.2. SEM Analysis (Surface Degradation after Exposure to the PBS Solution at 37 °C for 3 Months)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, J.; Ghannam, R.; Htet, K.O.; Liu, Y.; Law, M.k.; Roy, V.A.L.; Michel, B.; Imran, M.A.; Heidari, H. Self-Powered Implantable Medical Devices: Photovoltaic Energy Harvesting Review. Adv. Healthc. Mater. 2020, 9, 2000779. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.J.T.; Mishra, A.; Park, I.; Kim, Y.J.; Park, W.T.; Yoon, Y.J. Polymeric Biomaterials for Medical Implants and Devices. ACS Biomater. Sci. Eng. 2016, 2, 454–472. [Google Scholar] [CrossRef]
- Ye, H.; Zhang, K.; Kai, D.; Li, Z.; Loh, X.J. Polyester Elastomers for Soft Tissue Engineering. Chem. Soc. Rev. 2018, 47, 4545–4580. [Google Scholar] [CrossRef]
- Flaig, F.; Bellani, C.F.; Uyumaz, Ö.; Schlatter, G.; Hébraud, A. Elaboration and Mechanical Properties of Elastomeric Fibrous Scaffolds Based on Crosslinked Poly(Glycerol Sebacate) and Cyclodextrin for Soft Tissue Engineering. Mater. Adv. 2021, 2, 1284–1293. [Google Scholar] [CrossRef]
- Reddy, M.S.B.; Ponnamma, D.; Choudhary, R.; Sadasivuni, K.K. A Comparative Review of Natural and Synthetic Biopolymer Composite Scaffolds. Polymers 2021, 13, 1105. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, S.; Scheibel, T. Copolymer/Clay Nanocomposites for Biomedical Applications. Adv. Funct. Mater. 2020, 30, 1908101. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Kitagawa, M.; Hotta, A.; Koizumi, S. Thermo-Responsive Nanocomposite Hydrogels Based on PEG-b-PLGA Diblock Copolymer and Laponite. Polymers 2019, 11, 250. [Google Scholar] [CrossRef] [Green Version]
- Rapacz-Kmita, A.; Szaraniec, B.; MikoŁajczyk, M.; Stodolak-Zych, E.; Dzierzkowska, E.; Gajek, M.; Dudek, P. Multifunctional biodegradable polymer/clay nanocomposites with antibacterial properties in drug delivery systems. Acta Bioeng. Biomech. 2020, 22, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Andriani, Y.; Morrow, I.C.; Taran, E.; Edwards, G.A.; Schiller, T.L.; Osman, A.F.; Martin, D.J. In vitro biostability of poly(dimethyl siloxane/hexamethylene oxide)-based polyurethane/layered silicate nanocomposites. Acta Biomater. 2013, 9, 8308–8317. [Google Scholar] [CrossRef]
- Willerth, S. Engineering Neural Tissue from Stem Cells; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 127–158. [Google Scholar] [CrossRef]
- Meng, N.; Zhou, N.L.; Zhang, S.Q.; Shen, J. Synthesis and Antimicrobial Activities of Polymer/Montmorillonite-Chlorhexidine Acetate Nanocomposite Films. Appl. Clay Sci. 2009, 42, 667–670. [Google Scholar] [CrossRef]
- Song, K. Micro- and Nano-Fillers Used in the Rubber Industry. In Progress in Rubber Nanocomposites; Thomas, S., Maria, H.J., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2017; pp. 41–80. [Google Scholar] [CrossRef]
- Wang, K.; Deng, Q. The Thermal and Mechanical Properties of Microinhomogeneous Materials. Polymers 2019, 11, 1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarte, N.H.; Cui, L.; Woo, S. Polyolefins/Layered Silicate Nanocomposites Prepared by In Situ Polymerization. In Advances in Polymer Science (Polyolefins: 50 Years after Ziegler and Natta II- Polyolefins by Metallocenes and Other Single-Site Catalysts); Kaminsky, W., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; Volume 258, pp. 311–340. [Google Scholar] [CrossRef]
- Schneider, C.; Langer, R.; Loveday, D.; Hair, D. Applications of Ethylene Vinyl Acetate Copolymers (EVA) in Drug Delivery Systems. J. Control. Release 2017, 262, 284–295. [Google Scholar] [CrossRef]
- Yousaf, S.S.; Houacine, C.; Khan, I.; Ahmed, W.; Jackson, M.J. Importance of Biomaterials in Biomedical Engineering. In Advances in Medical and Surgical Engineering, 1st ed.; Ahmed, W., Phoenix, D.A., Jackson, M.J., Charalambous, C.P., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; pp. 151–177. [Google Scholar] [CrossRef]
- Mohammed, M.T. Nanocomposites in Total Hip Joint Replacements. In Applications of Nanocomposite Materials in Orthopedics; Inamuddin Asiri, A.M., Mohammad, A., Eds.; Woodhead Publishing: Sawston, UK, 2018; pp. 221–252. [Google Scholar] [CrossRef]
- Kashi, A.; Saha, S. Failure Mechanisms of Medical Implants and Their Effects on Outcomes. In Biointegration of Medical Implant Materials; Sharma, C.P., Ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2019; pp. 407–432. [Google Scholar] [CrossRef]
- Fauzi, A.A.A.; Osman, A.F.; Abdullah, M.A.A.; Mandal, S.; Ananthakrishan, R. Ethylene Vinyl Acetate Nanocomposites with Hybrid Silicate Nanofillers of Destabilized Natural and Commercial Bentonites and Organomontmorillonites. J. Vinyl Addit. Technol. 2019, 25, 396–411. [Google Scholar] [CrossRef]
- Tombácz, E.; Szekeres, M. Colloidal behavior of aqueous montmorillonite suspensions: The specific role of pH in the presence of indifferent electrolytes. Appl. Clay Sci. 2004, 27, 75–94. [Google Scholar] [CrossRef]
- Kelessidis, V.C.; Tsamantaki, C.; Dalamarinis, P. Effect of pH and electrolyte on the rheology of aqueous Wyoming bentonite-dispersions. Appl. Clay Sci. 2007, 38, 86–96. [Google Scholar] [CrossRef]
- Hamid, A.R.A.; Osman, A.F. Effects of Ultrasonication Time on Thermal Stability and Swelling Behaviour of the Commercial Organo-Montmorillonite (O-MMT). Int. J. Eng. Technol. 2018, 7, 288–291. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, A.D.; Beatrice, C.A.G. Polymer Nanocomposites with Different Types of Nanofiller. In Nanocomposites—Recent Evolutions; Sivasankaran, S., Ed.; IntechOpen: London, UK, 2019; pp. 104–127. [Google Scholar] [CrossRef] [Green Version]
- Agubra, V.A.; Owuor, P.S.; Hosur, M.V. Influence of Nanoclay Dispersion Methods on the Mechanical Behavior of E-Glass/Epoxy Nanocomposites. Nanomaterials 2013, 3, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.L.; Liao, Y.C.; Pan, G.T.; Chong, S. Hybrid Nanocomposite Film with Enhanced Moisture Barrier Properties. J. Taiwan Inst. Chem. Eng. 2018, 83, 168–173. [Google Scholar] [CrossRef]
- Osman, A.F.; Alakrach, A.M.; Kalo, H.; Azmi, W.N.W.; Hashim, F. In vitro biostability and biocompatibility of ethyl vinyl acetate (EVA) nanocomposites for biomedical applications. RSC Adv. 2015, 40, 31485–31495. [Google Scholar] [CrossRef]
- Lewkowitz-Shpuntoff, H.M.; Wen, M.C.; Singh, A.; Brenner, N.; Gambino, R.; Pernodet, N.; Isseroff, R.; Rafailovich, M.; Sokolov, J. The Effect of Organo Clay and Adsorbed FeO3 Nanoparticles on Cells Cultured on Ethylene Vinyl Acetate Substrates and Fibers. Biomaterials 2009, 30, 8–18. [Google Scholar] [CrossRef]
- Peña-Morán, O.A.; Villarreal, M.L.; Álvarez-Berber, L.; Meneses-Acosta, A.; Rodríguez-López, V. Cytotoxicity, Post-Treatment Recovery, and Selectivity Analysis of Naturally Occurring Podophyllotoxins from Bursera Fagaroides Var. Fagaroides on Breast Cancer Cell Lines. Molecules 2016, 21, 1013. [Google Scholar] [CrossRef] [PubMed]
- Nordin, M.L.; Abdul Kadir, A.; Zakaria, Z.A.; Abdullah, R.; Abdullah, M.N.H. In Vitro Investigation of Cytotoxic and Antioxidative Activities of Ardisia Crispa against Breast Cancer Cell Lines, MCF-7 and MDA-MB-231. BMC Complement. Altern. Med. 2018, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Tong, D.; Yu, W. 7-Smectite Nanomaterials: Preparation, Properties, and Functional Applications. In Nanomaterials from Clay Minerals: A New Approach to Green Functional Materials; Wang, A., Wang, W., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 335–364. [Google Scholar] [CrossRef]
- Uddin, F. Montmorillonite: An Introduction to Properties and Utilization. In Current Topics in the Utilization of Clay in Industrial and Medical Applications; Zoveidavianpoor, M., Ed.; IntechOpen: London, UK, 2018; pp. 3–24. [Google Scholar] [CrossRef] [Green Version]
- Mirila, D.C.; Pîrvan, M.-Ș.; Platon, N.; Georgescu, A.-M.; Zichil, V.; Nistor, I.D. Total Mineralization of Malachite Green Dye by Advanced Oxidation Processes. Acta Chem. Iasi 2018, 26, 263–280. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, M.; Rajulapati, S.B.; Sonawane, S.; Girdhar, A. Synthesis and Implication of Novel Poly(Acrylic Acid)/Nanosorbent Embedded Hydrogel Composite for Lead Ion Removal. Sci. Rep. 2017, 7, 16413. [Google Scholar] [CrossRef]
- Tang, N.; Yang, J.; Cen, W.; Pan, W.; Wu, L.; Xu, C. Preparation of Organic Montmorillonite Promoter for Improving the Adhesion between Bitumen and Acidic Aggregate. Constr. Build. Mater. 2021, 274, 121833. [Google Scholar] [CrossRef]
- Tyagi, B.; Chudasama, C.D.; Jasra, R.V. Determination of Structural Modification in Acid Activated Montmorillonite Clay by FT-IR Spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2006, 64, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Barani, H. Modification of Bentonite with Different Surfactants and Substitute as a Mordant in Wool Natural Dyeing. Chiang Mai J. Sci. 2018, 45, 492–504. [Google Scholar]
- Terpiłowska, S.; Siwicka-Gieroba, D.; Siwicki, A.K. Cell Viability in Normal Fibroblasts and Liver Cancer Cells after Treatment with Iron (III), Nickel (II), and Their Mixture. J. Vet. Res. 2018, 62, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Khazaei, S.; Esa, N.M.; Ramachandran, V.; Hamid, R.A.; Pandurangan, A.K.; Etemad, A.; Ismail, P. In Vitro Antiproliferative and Apoptosis Inducing Effect of Allium Atroviolaceum Bulb Extract on Breast, Cervical, and Liver Cancer Cells. Front. Pharmacol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Latif, M.A.; Ibrahim, F.W.; Arshad, S.A.; Hui, C.K.; Jufri, N.F.; Hamid, A. Cytotoxicity, Proliferation and Migration Rate Assessments of Human Dermal Fibroblast Adult Cells Using Zingiber Zerumbet Extract. Sains Malaysiana 2019, 48, 121–127. [Google Scholar] [CrossRef]
- Campoccia, D.; Ravaioli, S.; Santi, S.; Mariani, V.; Santarcangelo, C.; De Filippis, A.; Montanaro, L.; Arciola, C.R.; Daglia, M. Exploring the Anticancer Effects of Standardized Extracts of Poplar-Type Propolis: In Vitro Cytotoxicity toward Cancer and Normal Cell Lines. Biomed. Pharmacother. 2021, 141, 111895. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Lordan, S.; Kennedy, J.E.; Higginbotham, C.L. Cytotoxic Effects Induced by Unmodified and Organically Modified Nanoclays in the Human Hepatic HepG2 Cell Line. J. Appl. Toxicol. 2011, 31, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Jeevanandam, J.; Barhoum, A.; Chan, Y.S.; Dufresne, A.; Danquah, M.K. Review on Nanoparticles and Nanostructured Materials: History, Sources, Toxicity and Regulations. Beilstein J. Nanotechnol. 2018, 9, 1050–1074. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Li, Z.; Huang, J.; Wu, Y.; Katayama, S. Microstructure and Softening of Laser-Welded 960 MPa Grade High Strength Steel Joints. J. Mater. Eng. Perform. 2014, 23, 538–544. [Google Scholar] [CrossRef]
- Huang, N.J.; Zang, J.; Zhang, G.D.; Guan, L.Z.; Li, S.N.; Zhao, L.; Tang, L.C. Efficient Interfacial Interaction for Improving Mechanical Properties of Polydimethylsiloxane Nanocomposites Filled with Low Content of Graphene Oxide Nanoribbons. RSC Adv. 2017, 7, 22045–22053. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Yang, Y.; Xu, Z.; Shi, Q. Effect of Martensitic Transformation on Elastic Modulus Anisotropy of Ti-6Al-4V Alloy. Mater. Res. 2018, 21, 1–8. [Google Scholar] [CrossRef]
- Lyu, S.; Untereker, D. Degradability of Polymers for Implantable Biomedical Devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [Green Version]
- Stewart, S.A.; Domínguez-Robles, J.; Donnelly, R.F.; Larrañeta, E. Implantable Polymeric Drug Delivery Devices: Classification, Manufacture, Materials, and Clinical Applications. Polymers 2018, 10, 1379. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Samples | Acronym | Weight (%) | ||
|---|---|---|---|---|
| PEVA | S-MMT (P) | Bent (pH-s) | ||
| Neat PEVA | PEVA | 100 | - | - |
| PEVA + S-MMT(P) | PEVA-S | 96 | 4 | - |
| PEVA + Bent(pH-s) | PEVA-B | 96 | - | 4 |
| PEVA + 3.2 wt% S-MMT(P) + 0.8 wt% Bent(pH-s) | PEVA-S4B1 | 96 | 3.2 | 0.8 |
| PEVA + 2.4 wt% S-MMT(P) + 1.6 wt% Bent(pH-s) | PEVA-S3B2 | 96 | 2.4 | 1.6 |
| PEVA + 1.6 wt% S-MMT(P) + 2.4 wt% Bent(pH-s) | PEVA-S2B3 | 96 | 1.6 | 2.4 |
| PEVA + 0.8 wt% S-MMT(P) + 3.2 wt% Bent(pH-s) | PEVA-S1B4 | 96 | 0.8 | 3.2 |
| Type of Sample | Tensile Strength (TS) (MPa) | Elongation at Break (EB) (%) | Young’s Modulus (YM) (MPa) | Tensile Toughness (TS) (MPa) |
|---|---|---|---|---|
| PEVA | 3.60 ± 0.2 | 2224 ± 89 | 0.48 ± 0.04 | 41 ± 3.1 |
| PEVA-S | 5.35 ± 0.2 | 2431 ± 64 | 0.32 ± 0.04 | 66 ± 5.4 |
| PEVA-B | 4.94 ± 0.04 | 2272 ± 63 | 0.26 ± 0.05 | 58 ± 4.2 |
| PEVA-S4B1 | 6.26 ± 0.3 | 2448 ± 75 | 0.40 ± 0.05 | 81 ± 6.6 |
| PEVA-S3B2 | 5.74 ± 0.3 | 2341 ± 76 | 0.38 ± 0.04 | 65 ± 3.6 |
| PEVA-S2B3 | 5.21 ± 0.1 | 2335 ± 164 | 0.32 ± 0.04 | 60 ± 4.5 |
| PEVA-S1B4 | 4.96 ± 0.3 | 2284 ± 71 | 0.33 ± 0.05 | 61 ± 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed Fitri, T.F.; Osman, A.F.; Alosime, E.M.; Othman, R.; Hashim, F.; Abdullah, M.A.A. Biomedical PEVA Nanocomposite with Dual Clay Nanofiller: Cytotoxicity, Mechanical Properties, and Biostability. Polymers 2021, 13, 4345. https://doi.org/10.3390/polym13244345
Mohammed Fitri TF, Osman AF, Alosime EM, Othman R, Hashim F, Abdullah MAA. Biomedical PEVA Nanocomposite with Dual Clay Nanofiller: Cytotoxicity, Mechanical Properties, and Biostability. Polymers. 2021; 13(24):4345. https://doi.org/10.3390/polym13244345
Chicago/Turabian StyleMohammed Fitri, Tuty Fareyhynn, Azlin Fazlina Osman, Eid M. Alosime, Rahimah Othman, Fatimah Hashim, and Mohd Aidil Adhha Abdullah. 2021. "Biomedical PEVA Nanocomposite with Dual Clay Nanofiller: Cytotoxicity, Mechanical Properties, and Biostability" Polymers 13, no. 24: 4345. https://doi.org/10.3390/polym13244345