
Plasma Supported Deposition of Amorphous Hydrogenated Carbon (a-C:H) on Polyamide 6: Determining Interlayer Completion and Dehydrogenation Effects during Layer Growth

 ,
,  , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Coating Process
2.2. Surface Morphology
2.3. Infrared Spectroscopy
2.4. X-ray Spectroscopy
3. Results
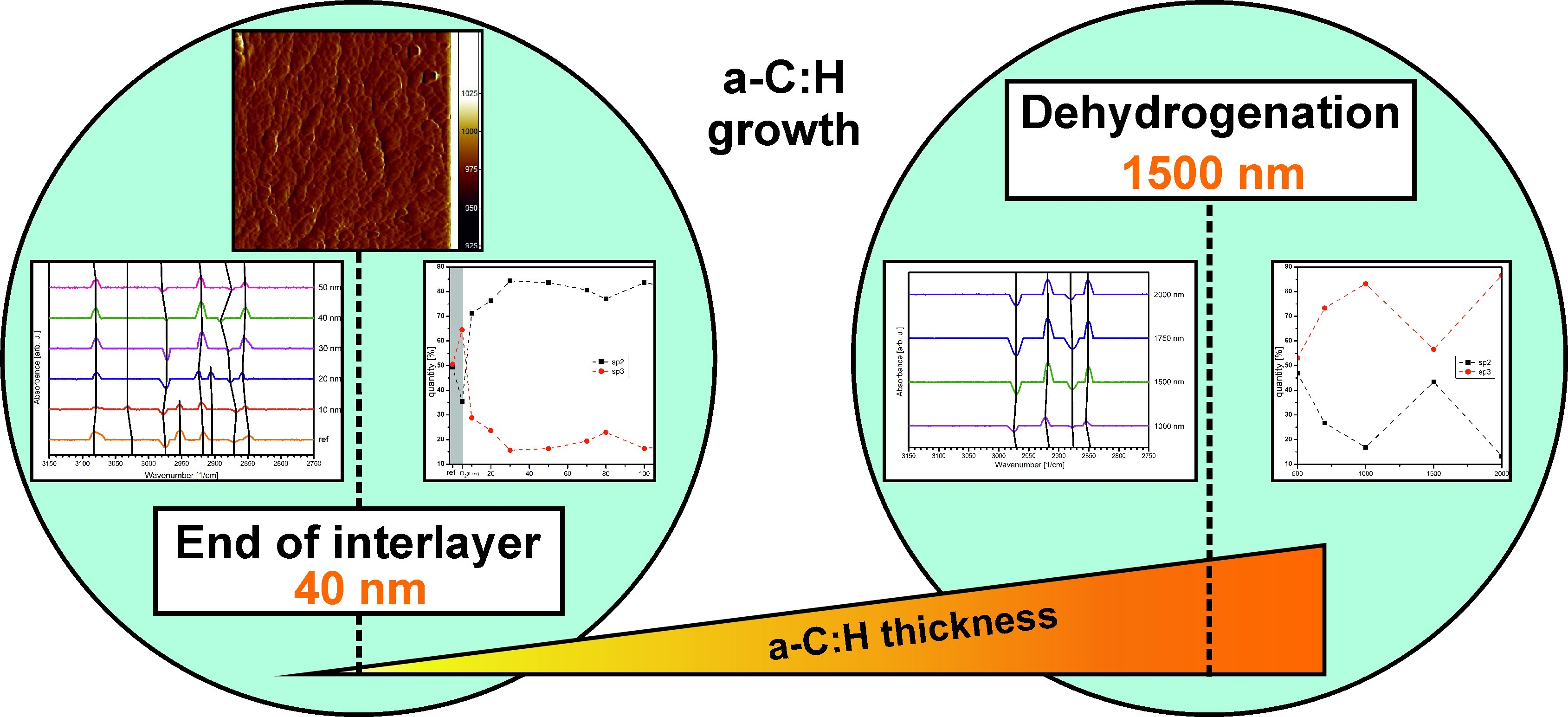
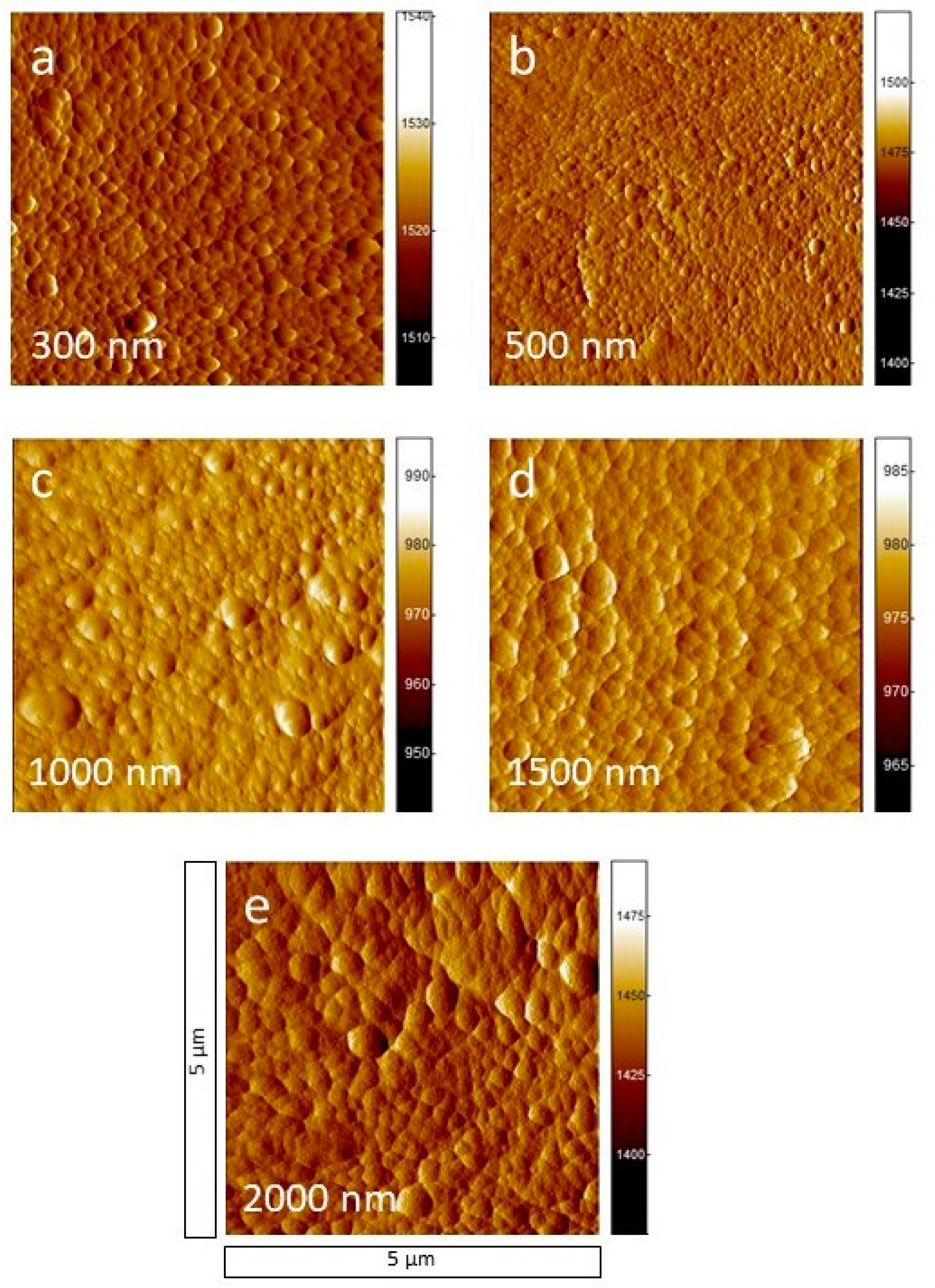
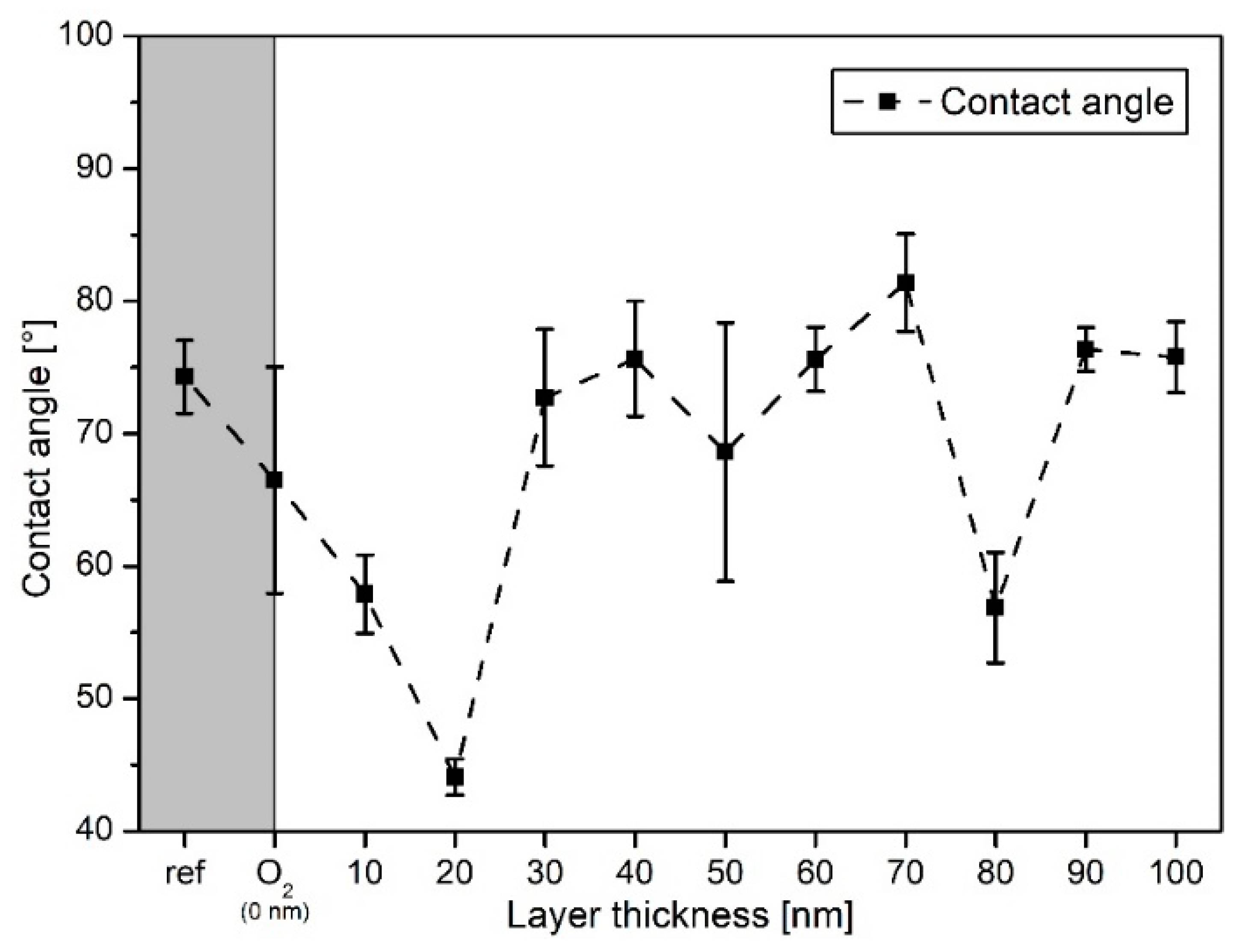
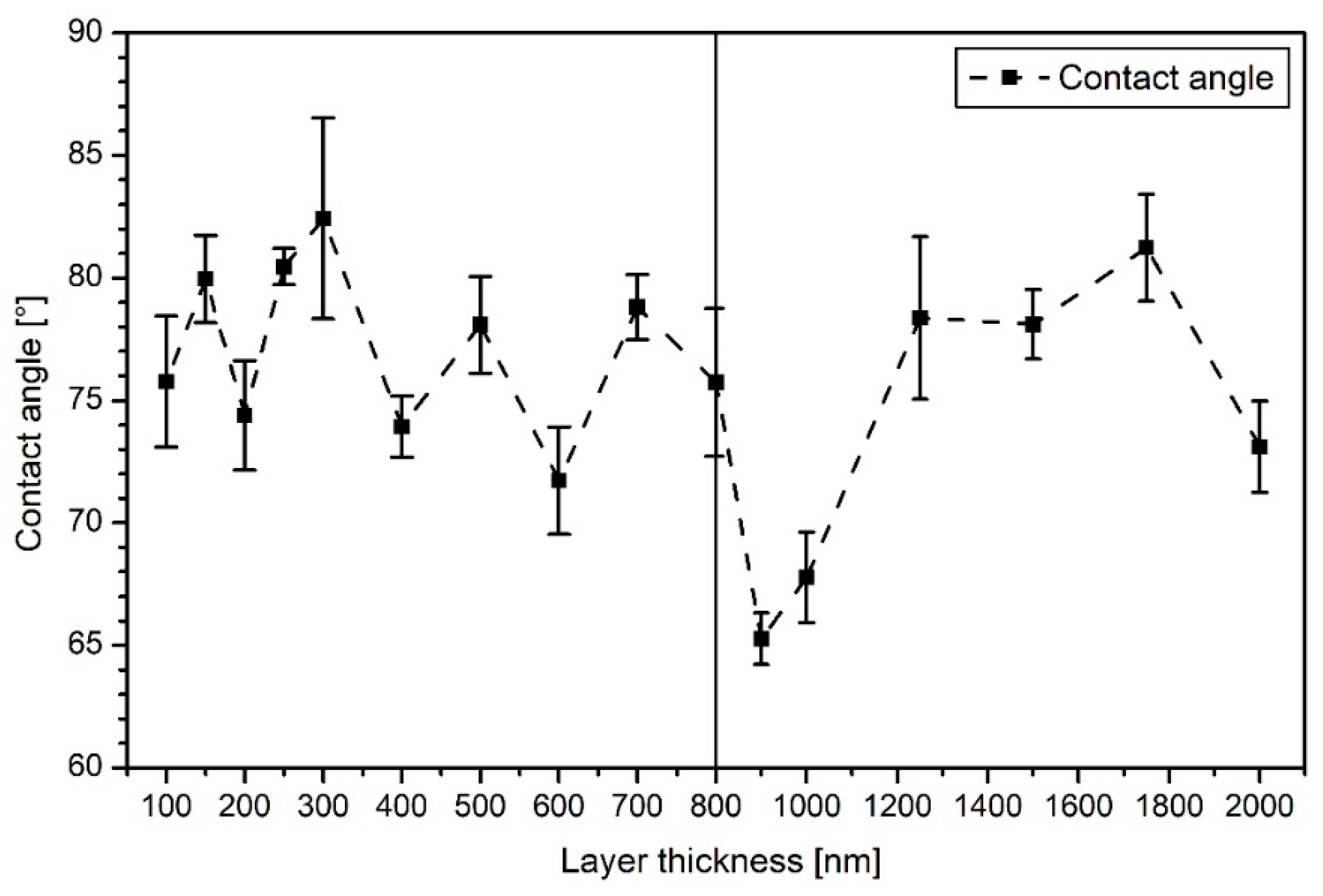
3.1. Surface Morphology
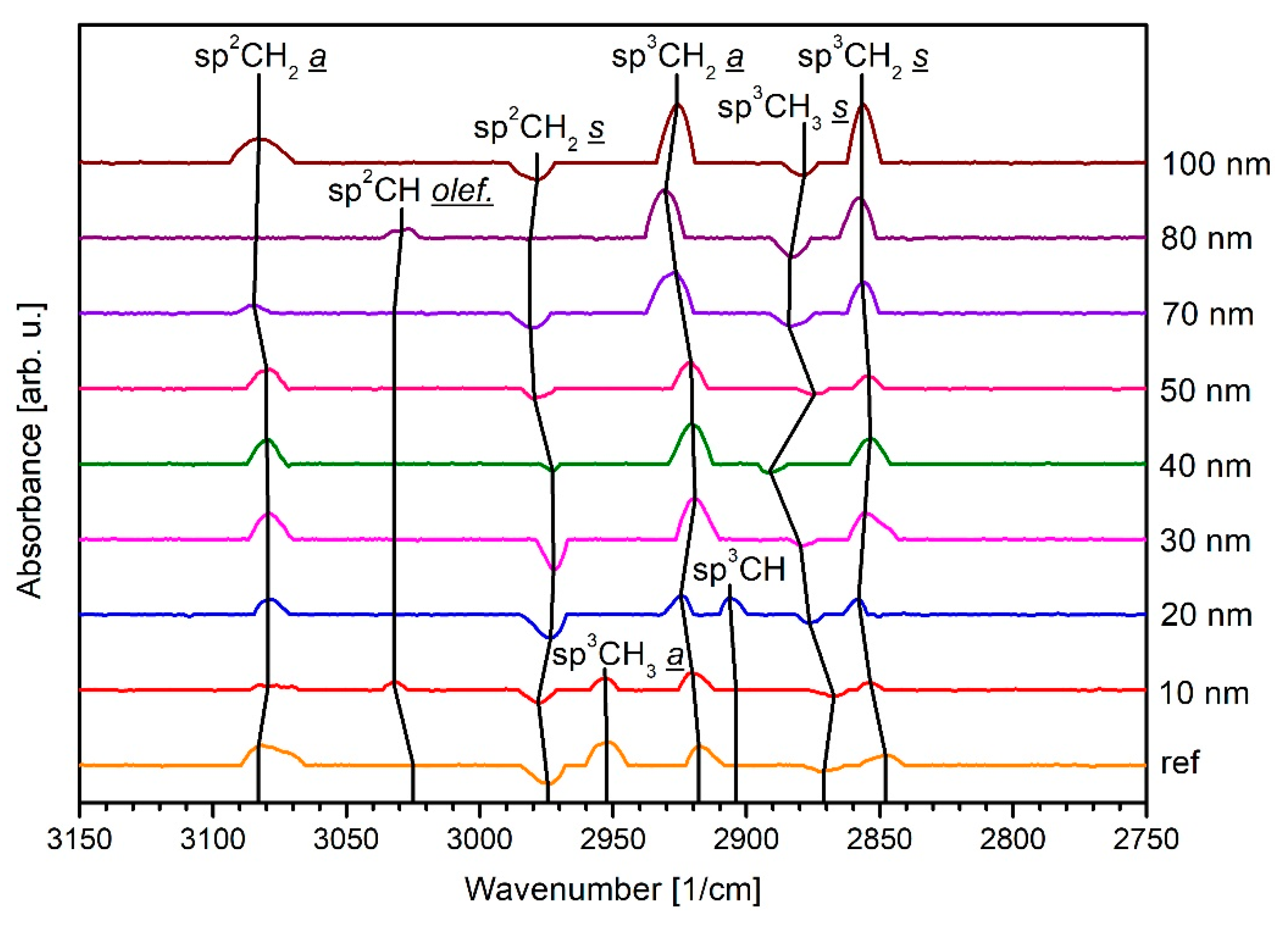
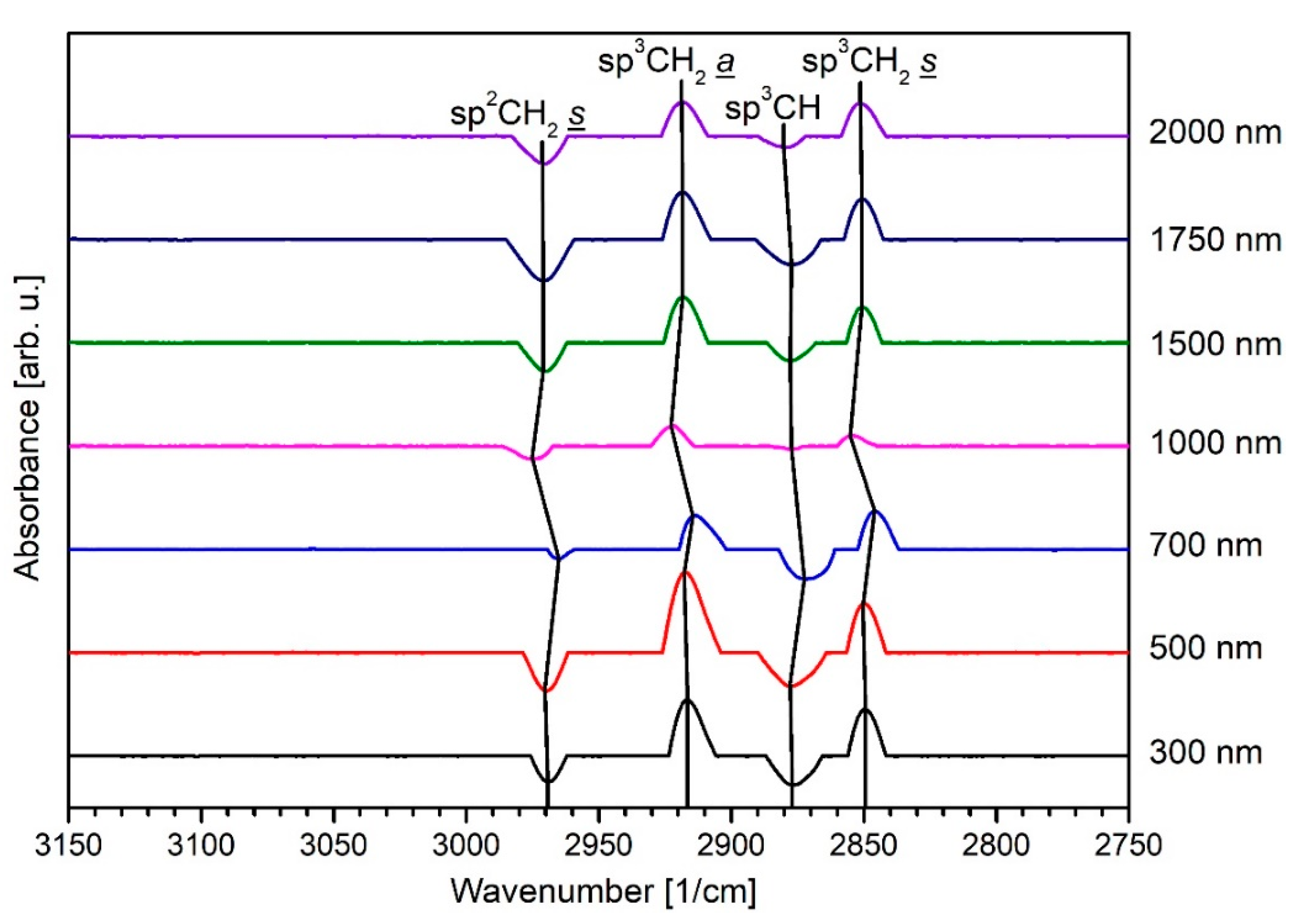
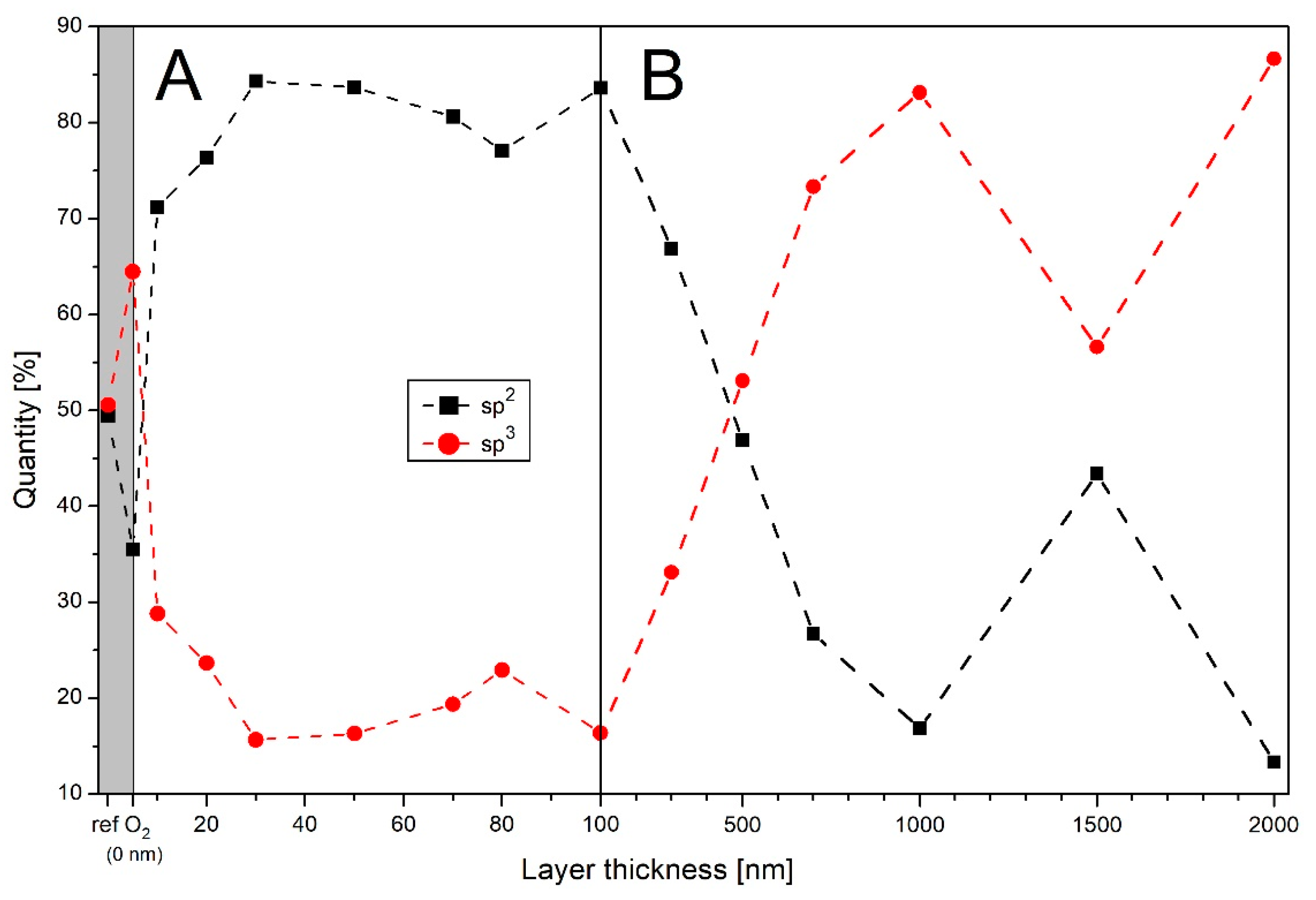
3.2. Infrared Spectroscopy
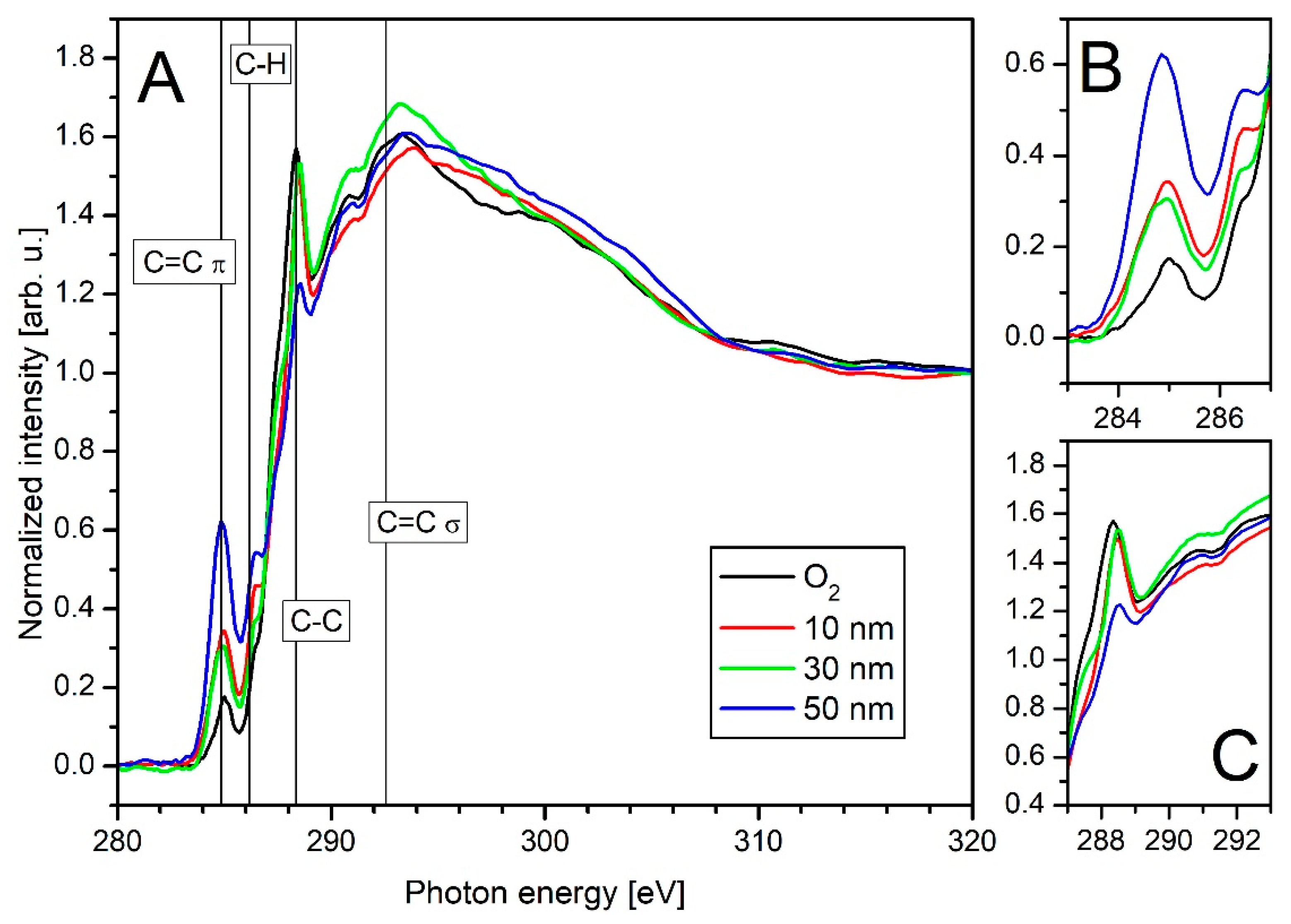
3.3. X-ray Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hellerich, W.; Harsch, G.; Haenle, S. Werkstoff-Führer Kunststoffe: Eigenschaften, Prüfungen, Kennwerte; mit 64 Tabellen, 7. überarb. und erw. Aufl.; Carl Hanser Verlag GmbH: München, Germany, 1996. [Google Scholar]
- Döring, E. Polypropylene and Polyamide Developments in the European Automotive Industry; SAE Technical Paper Series; SAE International: Warrendale, PA, USA, 1983. [Google Scholar]
- Nandhini, R.; Moorthy, M.K.; Muthukumaran, S.; Kumaran, S. Influence of process variables on the characteristics of friction-stir-welded polyamide 6,6 joints. Mater. Werkst. 2019, 50, 1139–1148. [Google Scholar] [CrossRef]
- Félix, J.S.; Monteiro, M.; Manzoli, J.E.; Padula, M.; Pezo, D.; Romero, J.; Nerín, C. Identification and migration of degradation compounds from irradiation of multilayer polyamide 6 films for meat foodstuffs and cheese. Anal. Bioanal. Chem. 2008, 391, 847–857. [Google Scholar] [CrossRef]
- Unal, H.; Mimaroglu, A. Friction and wear performance of polyamide 6 and graphite and wax polyamide 6 composites under dry sliding conditions. Wear 2012, 289, 132–137. [Google Scholar] [CrossRef]
- Karsli, N.G.; Aytac, A. Tensile and thermomechanical properties of short carbon fiber reinforced polyamide 6 composites. Compos. Part B Eng. 2013, 51, 270–275. [Google Scholar] [CrossRef]
- Deopura, B.L. Polyesters and Polyamides; Woodhead Publishing: Cambridge, UK; Textile Institute: Boca Raton, FL, USA, 2008. [Google Scholar]
- Sabbatini, L. Polymer Surface Characterization; De Gruyter: Boston, MA, USA, 2014. [Google Scholar]
- Andrady, A.L.; Neal, M.A. Applications and societal benefits of plastics. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Couderc, P.; Catherine, Y. Structure and physical properties of plasma-grown amorphous hydrogenated carbon films. Thin Solid Film 1987, 146, 93–107. [Google Scholar] [CrossRef]
- Grill, A. Plasma-deposited diamondlike carbon and related materials. IBM J. Res. Dev. 1999, 43, 147–162. [Google Scholar] [CrossRef]
- Rohrbeck, M.; Fischer, C.; Wehner, S.; Meier, J.; Manz, W. DLC-coated pure bioplastic foil. Vak. Forsch. Prax. 2014, 26, 42–47. [Google Scholar] [CrossRef]
- Rohrbeck, M.; Körsten, S.; Fischer, C.B.; Wehner, S.; Kessler, B. Diamond-like carbon coating of a pure bioplastic foil. Thin Solid Film 2013, 545, 558–563. [Google Scholar] [CrossRef]
- Catena, A.; Agnello, S.; Rösken, L.M.; Bergen, H.; Recktenwald, E.; Bernsmann, F.; Busch, H.; Cannas, M.; Gelardi, F.M.; Hahn, B.; et al. Characteristics of industrially manufactured amorphous hydrogenated carbon (a-C: H) depositions on high-density polyethylene. Carbon 2016, 96, 661–671. [Google Scholar] [CrossRef]
- Catena, A.; Guo, Q.; Kunze, M.R.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Morphological and Chemical Evolution of Gradually Deposited Diamond-Like Carbon Films on Polyethylene Terephthalate: From Subplantation Processes to Structural Reorganization by Intrinsic Stress Release Phenomena. ACS Appl. Mater. Interfaces 2016, 8, 10636–10646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catena, A.; Kunze, M.R.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Amorphous hydrogenated carbon (a-C: H) depositions on polyoxymethylene: Substrate influence on the characteristics of the developing coatings. Surf. Coat. Technol. 2016, 307, 658–665. [Google Scholar] [CrossRef]
- Fruth, W.; Meerkamm, H.; Krumpiegl, T.; Schaufler, C.; Erkens, G.; Ruttor, M. Tribological behaviour of PVD-coated PA plastic material sliding against metal counterparts. Surf. Coat. Technol. 1999, 120–121, 470–475. [Google Scholar] [CrossRef]
- Paul, R.; Das, S.N.; Dalui, S.; Gayen, R.N.; Roy, R.K.; Bhar, R.; Pal, A.K.; Das, S. Synthesis of DLC films with different sp2/sp3 ratios and their hydrophobic behaviour. J. Phys. D Appl. Phys. 2008, 41, 1–7. [Google Scholar] [CrossRef]
- Mohagheghpour, E.; Rajabi, M.; Gholamipour, R.; Larijani, M.M.; Sheibani, S. Ion beam energy dependence of surface and structural properties of amorphous carbon films deposited by IBSD method on Ni–Cu alloy. J. Mater. Res. 2017, 32, 1258–1266. [Google Scholar] [CrossRef]
- Robertson, J. Plasma Deposition of Diamond-Like Carbon. Jpn. J. Appl. Phys. 2011, 50, 1–8. [Google Scholar] [CrossRef]
- Robertson, J. Diamond-like amorphous carbon. Mater. Sci. Eng. R Rep. 2002, 37, 129–281. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Tai, F.C.; Wei, C.H. Correlation between sp2/sp3 ratio or hydrogen content and water contact angle in hydrogenated DLC film. Mater. Trans. 2007, 48, 2534–2538. [Google Scholar] [CrossRef] [Green Version]
- Schlebrowski, T.; Rouabeh, W.; Wehner, S.; Fischer, C.B. Specifying the interlayer turning point and dehydrogenation in a-C:H layers plasma deposited on high-density polyethylene with X-ray synchrotron techniques. Thin Solid Film 2019, 691, 1–6. [Google Scholar] [CrossRef]
- Schlebrowski, T.; Beucher, L.; Bazzi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. Prediction of a-C: H layer failure on industrial relevant biopolymer polylactide acide (PLA) foils based on the sp2/sp3 ratio. Surf. Coat. Technol. 2019, 368, 79–87. [Google Scholar] [CrossRef]
- Schlebrowski, T.; Beucher, L.; Bazzi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. Changing Contents of Carbon Hybridizations in Amorphous Hydrogenated Carbon Layers (a-C: H) on Sustainable Polyhydroxybutyrate (PHB) Exhibit a Significant Deterioration in Stability, Depending on Thickness. C 2019, 5, 52. [Google Scholar] [CrossRef] [Green Version]
- Schlebrowski, T.; Acharchi, H.; Hahn, B.; Wehner, S.; Fischer, C.B. 202 Refinement of Sustainable Polybutylene Adipate Terephthalate (PBAT) with Amorphous Hydrogenated Carbon Films (a-C:H) Revealing Film Instabilities Influenced by a Thickness-Dependent Change of sp2/sp3 Ratio. Materials 2020, 13, 1077. [Google Scholar] [CrossRef] [Green Version]
- Koidl, P.; Wild, C.; Dischler, B.; Wagner, J.; Ramsteiner, M. Plasma Deposition, Properties and Structure of Amorphous Hydrogenated Carbon Films. Mater. Sci. Forum 1990, 52, 41–70. [Google Scholar] [CrossRef]
- Fischer, C.B.; Rohrbeck, M.; Wehner, S.; Richter, M.; Schmeißer, D. Interlayer formation of diamond-like carbon coatings on industrial polyethylene: Thickness dependent surface characterization by SEM, AFM and NEXAFS. Appl. Surf. Sci. 2013, 271, 381–389. [Google Scholar] [CrossRef]
- Catena, A.; McJunkin, T.; Agnello, S.; Gelardi, F.M.; Wehner, S.; Fischer, C.B. Surface morphology and grain analysis of successively industrially grown amorphous hydrogenated carbon films (a-C:H) on silicon. Appl. Surf. Sci. 2015, 347, 657–667. [Google Scholar] [CrossRef]
- Armaroli, T.; Bécue, T.; Gautier, S. Diffuse Reflection Infrared Spectroscopy (Drifts). Oil Gas Sci. Technol. 2004, 59, 215–237. [Google Scholar] [CrossRef]
- D’Souza, L.; Devi, P.; Kamat, T.; Naik, C.G. Diffuse reflectance infrared fourier transform spectroscopic (DRIFTS) investigation of E.coli, Staphylococcus aureus and Candida albicans. Indian J. Mar. Sci. 2009, 38, 45–51. [Google Scholar]
- Günzler, H.; Gremlich, H.U. IR Spectroscopy: An. Introduction; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Fang, D.; He, F.; Xie, J.; Xue, L. Calibration of Binding Energy Positions with C1s for XPS Results. J. Wuhan Univ. Technol. Mat. Sci. Ed. 2020, 35, 711–718. [Google Scholar] [CrossRef]
- Nefedov, A.; Wöll, C. Advanced Applications of NEXAFS Spectroscopy for Functionalized Surfaces. In Book Surface Science Techniques; Bracco, G., Holst, B., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Watts, B.; Thomsen, L.; Dastoor, P.C. Methods in carbon K-edge NEXAFS: Experiment and Analysis. J. Chem. Phys. 2014, 140, 105–120. [Google Scholar] [CrossRef]
- Banerjee, D.; Mukherjee, S.; Chattopadhyay, K. Controlling the surface topology and hence the hydrophobicity of amorphous carbon thin films. Carbon 2010, 48, 1025–1031. [Google Scholar] [CrossRef]
- Barthlott, W.; Neinhuis, C. Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 1997, 202, 1–8. [Google Scholar] [CrossRef]
- Feng, L.; Li, S.; Li, Y.; Li, H.; Zhang, L.; Zhai, J.; Song, Y.; Liu, B.; Jiang, L.; Zhu, D. Super-Hydrophobic Surfaces. Adv. Mater. 2002, 14, 1857–1860. [Google Scholar] [CrossRef]
- Werder, T.; Walther, J.H.; Jaffe, R.L.; Halicioglu, T.; Koumoutsakos, P. On the Water-Carbon Interaction for Use in Molecular Dynamics Simulations of Graphite and Carbon Nanotubes. J. Phys. Chem. B 2003, 107, 1345–1352. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, B.; Song, X.; Li, E.; Li, G.; Zhao, S.; Yan, H. Control over the wettability of amorphous carbon films in a large range from hydrophilicity to super-hydrophobicity. Appl. Surf. Sci. 2006, 253, 2690–2694. [Google Scholar] [CrossRef]
- Bismarck, A.; Brostow, W.; Chiu, R.; Lobland, H.E.H.; Ho, K.K. Effects of surface plasma treatment on tribology of thermoplastic polymers. Polym. Eng. Sci. 2008, 48, 1971–1976. [Google Scholar] [CrossRef]
- Ostrovskaya, L.; Perevertailo, V.; Ralchenko, V.; Dementjev, A.; Loginova, O. Wettability and surface energy of oxidized and hydrogen plasma-treated diamond films. Diam. Relat. Mater. 2002, 11, 845–850. [Google Scholar] [CrossRef]
- Ostrovskaya, L. Studies of diamond and diamond-like film surfaces using XAES, AFM and wetting. Vacuum 2002, 68, 219–238. [Google Scholar] [CrossRef]
- Piazza, F.; Morell, G. Wettability of hydrogenated tetrahedral amorphous carbon. Diam. Relat. Mater. 2009, 18, 43–50. [Google Scholar] [CrossRef]
- Tay, B.K.; Sheeja, D.; Lau, S.P.; Guo, J. Study of surface energy of tetrahedral amorphous carbon films modified in various gas plasma. Diam. Relat. Mater. 2003, 12, 2072–2076. [Google Scholar] [CrossRef]
- Extrand, C.W. Water contact angles and hysteresis of polyamide surfaces. J. Colloid Interface Sci. 2002, 248, 136–142. [Google Scholar] [CrossRef]
- Tomasella, E.; Thomas, L.; Dubois, M.; Meunier, C. Structural and mechanical properties of a-C. Diam. Relat. Mater. 2004, 13, 1618–1624. [Google Scholar] [CrossRef]
- Veres, M.; Koos, M.; Pócsik, I. IR study of the formation process of polymeric hydrogenated amorphous carbon film. Diam. Relat. Mater. 2002, 11, 1110–1114. [Google Scholar] [CrossRef]
- Dietrich, P.M.; Horlacher, T.; Girard-Lauriault, P.L.; Gross, T.; Lippitz, A.; Min, H.; Wirth, T.; Castelli, R.; Seeberger, P.H.; Unger, W.E.S. Adlayers of Dimannoside Thiols on Gold: Surface Chemical Analysis. Langmuir 2011, 27, 4808–4815. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.; Madix, R.; Stöhr, J. Orientation and absolute coverage of benzene, aniline, and phenol on Ag (110) determined by NEXAFS and XPS. Surf. Sci. 1991, 255, 12–30. [Google Scholar] [CrossRef]
- Tai, F.C.; Lee, S.C.; Wei, C.H.; Tyan, S.L. Correlation between ID/IG Ratio from Visible Raman Spectra and sp2/sp3 Ratio from XPS Spectra of Annealed Hydrogenated DLC Film. Mater. Trans. 2006, 47, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Dhez, O.; Ade, H.; Urquhart, S.; Urquhart, S. Calibrated NEXAFS spectra of some common polymers. J. Electron. Spectrosc. Relat. Phenom. 2003, 128, 85–96. [Google Scholar] [CrossRef]
- Diaz, J.; Monteiro, O.R.; Hussain, Z. Structure of amorphous carbon from near-edge and extended x-ray absorption spectroscopy. Phys. Rev. B 2007, 76, 094201. [Google Scholar] [CrossRef]
- Díaz, J.; Anders, S.; Zhou, X.; Moler, E.J.; Kellar, S.A.; Hussain, Z. Analysis of the π* and σ* bands of the x-ray absorption spectrum of amorphous carbon. Phys. Rev. B 2001, 64, 125204. [Google Scholar] [CrossRef]
- Stöhr, J. NEXAFS Spectroscopy; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar]
- Brüster, B.; Amozoqueño, C.; Grysan, P.; Peral, I.; Watts, B.; Raquez, J.-M.; Dubois, P.; Addiego, F. Resolving Inclusion Structure and Deformation Mechanisms in Polylactide Plasticized by Reactive Extrusion. Macromol. Mater. Eng. 2017, 302, 1700326. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ra [nm] | Rq [nm] |
|---|---|---|
| Reference | 4.74 ± 0.37 | 6.21 ± 0.68 |
| O2 | 5.25 ± 0.76 | 6.81 ± 1.10 |
| 10 nm | 6.25 ± 1.05 | 8.22 ± 1.31 |
| 20 nm | 6.52 ± 0.25 | 8.25 ± 0.40 |
| 30 nm | 5.98 ± 0.04 | 7.61 ± 0.27 |
| 40 nm | 6.86 ± 0.49 | 9.04 ± 0.92 |
| 50 nm | 6.53 ± 0.44 | 8.73 ± 0.58 |
| 100 nm | 6.73 ± 0.02 | 9.20 ± 0.11 |
| 300 nm | 5.15 ± 0.11 | 6.74 ± 0.27 |
| 500 nm | 4.81 ± 0.62 | 6.14 ± 0.91 |
| 1000 nm | 4.86 ± 0.12 | 6.28 ± 0.18 |
| 1500 nm | 5.09 ± 1.90 | 6.63 ± 2.12 |
| 2000 nm | 3.98 ± 0.29 | 5.12 ± 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlebrowski, T.; Lüber, H.; Beucher, L.; Fritz, M.; Benjillali, Y.; Bentaouit, M.; Hahn, B.; Wehner, S.; Fischer, C.B. Plasma Supported Deposition of Amorphous Hydrogenated Carbon (a-C:H) on Polyamide 6: Determining Interlayer Completion and Dehydrogenation Effects during Layer Growth. Polymers 2021, 13, 1886. https://doi.org/10.3390/polym13111886
Schlebrowski T, Lüber H, Beucher L, Fritz M, Benjillali Y, Bentaouit M, Hahn B, Wehner S, Fischer CB. Plasma Supported Deposition of Amorphous Hydrogenated Carbon (a-C:H) on Polyamide 6: Determining Interlayer Completion and Dehydrogenation Effects during Layer Growth. Polymers. 2021; 13(11):1886. https://doi.org/10.3390/polym13111886
Chicago/Turabian StyleSchlebrowski, Torben, Henriette Lüber, Lucas Beucher, Melanie Fritz, Youssef Benjillali, Mohammed Bentaouit, Barbara Hahn, Stefan Wehner, and Christian B. Fischer. 2021. "Plasma Supported Deposition of Amorphous Hydrogenated Carbon (a-C:H) on Polyamide 6: Determining Interlayer Completion and Dehydrogenation Effects during Layer Growth" Polymers 13, no. 11: 1886. https://doi.org/10.3390/polym13111886