Selected Physicochemical and Pharmaceutical Properties of Poly-ε-caprolactone and Poly(d,l-lactide-co-ε-caprolactone) Conjugates of Lamivudine Synthesized via Ring-Opening Polymerization


Abstract
:
1. Introduction
2. Results and Discussion
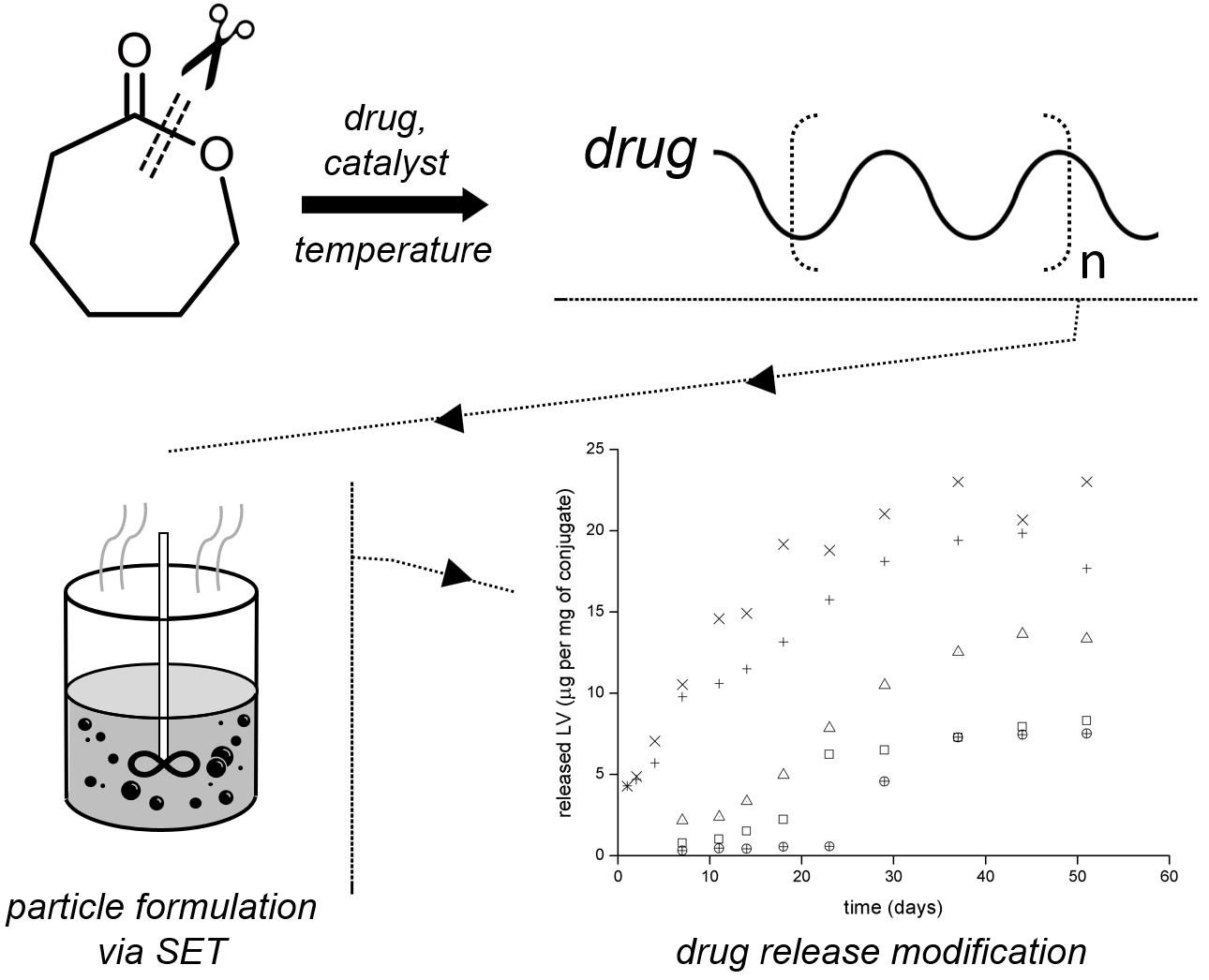
2.1. Lamivudine–Polymer Conjugate Synthesis
2.1.1. Structural Analysis
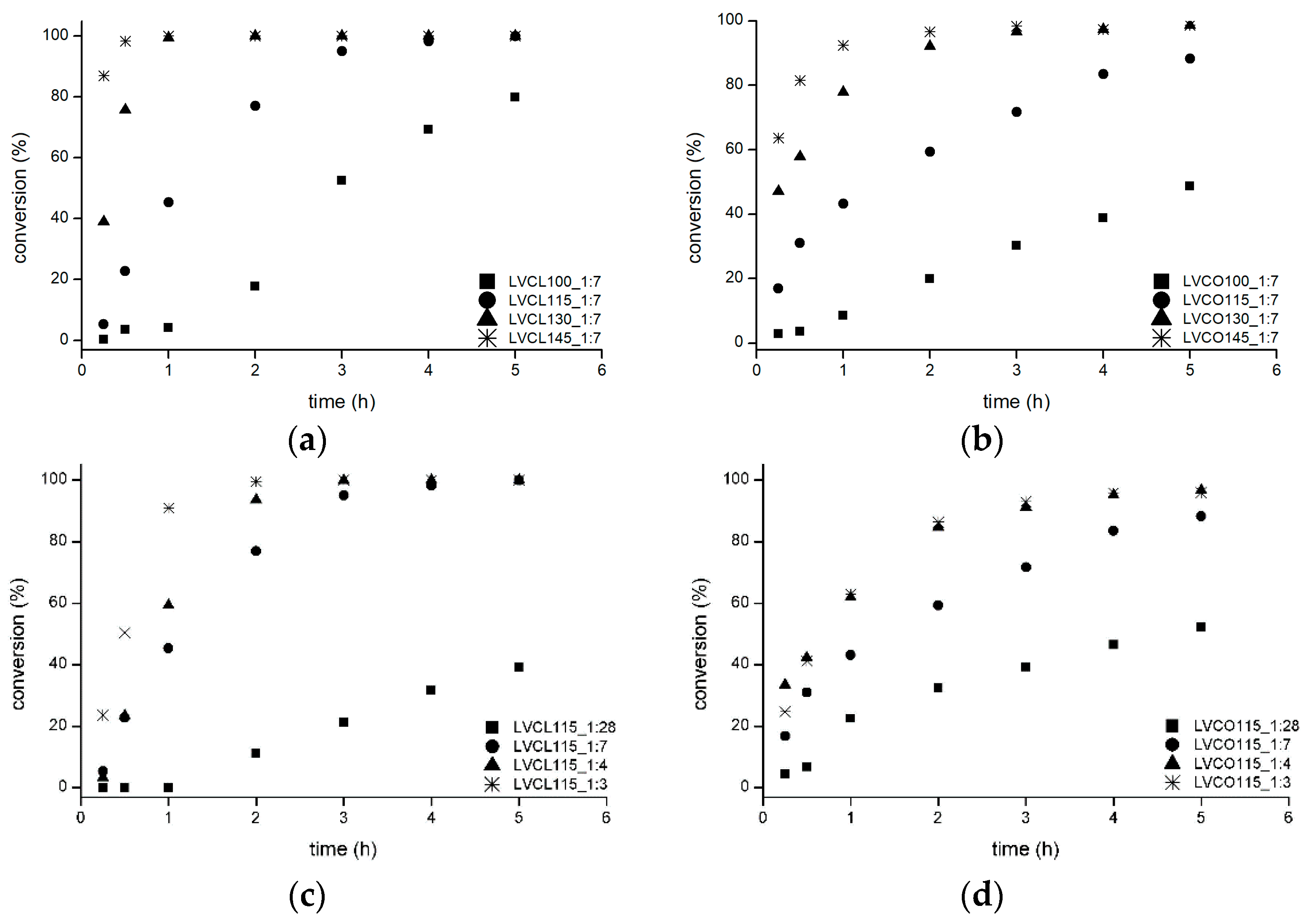
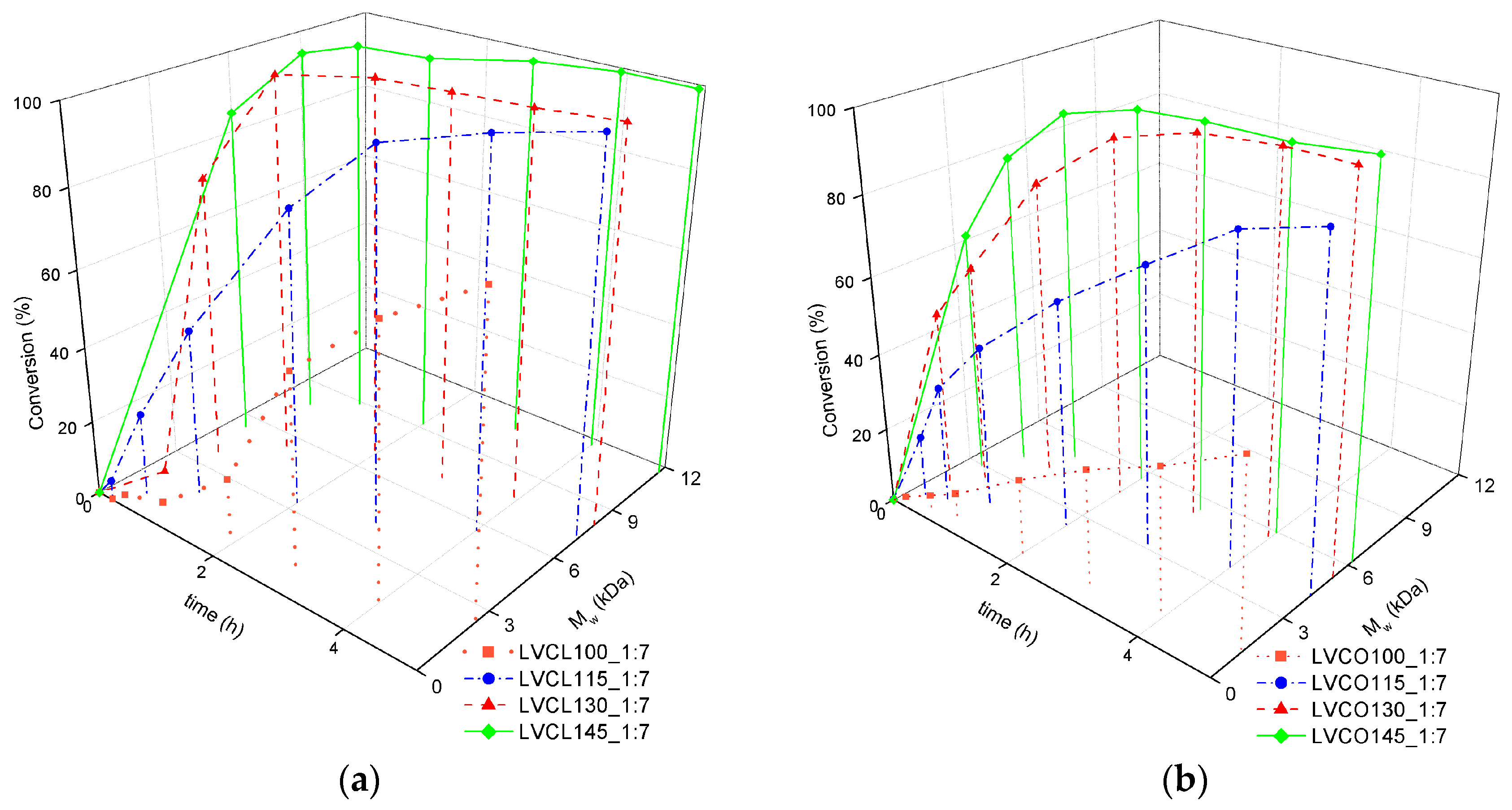
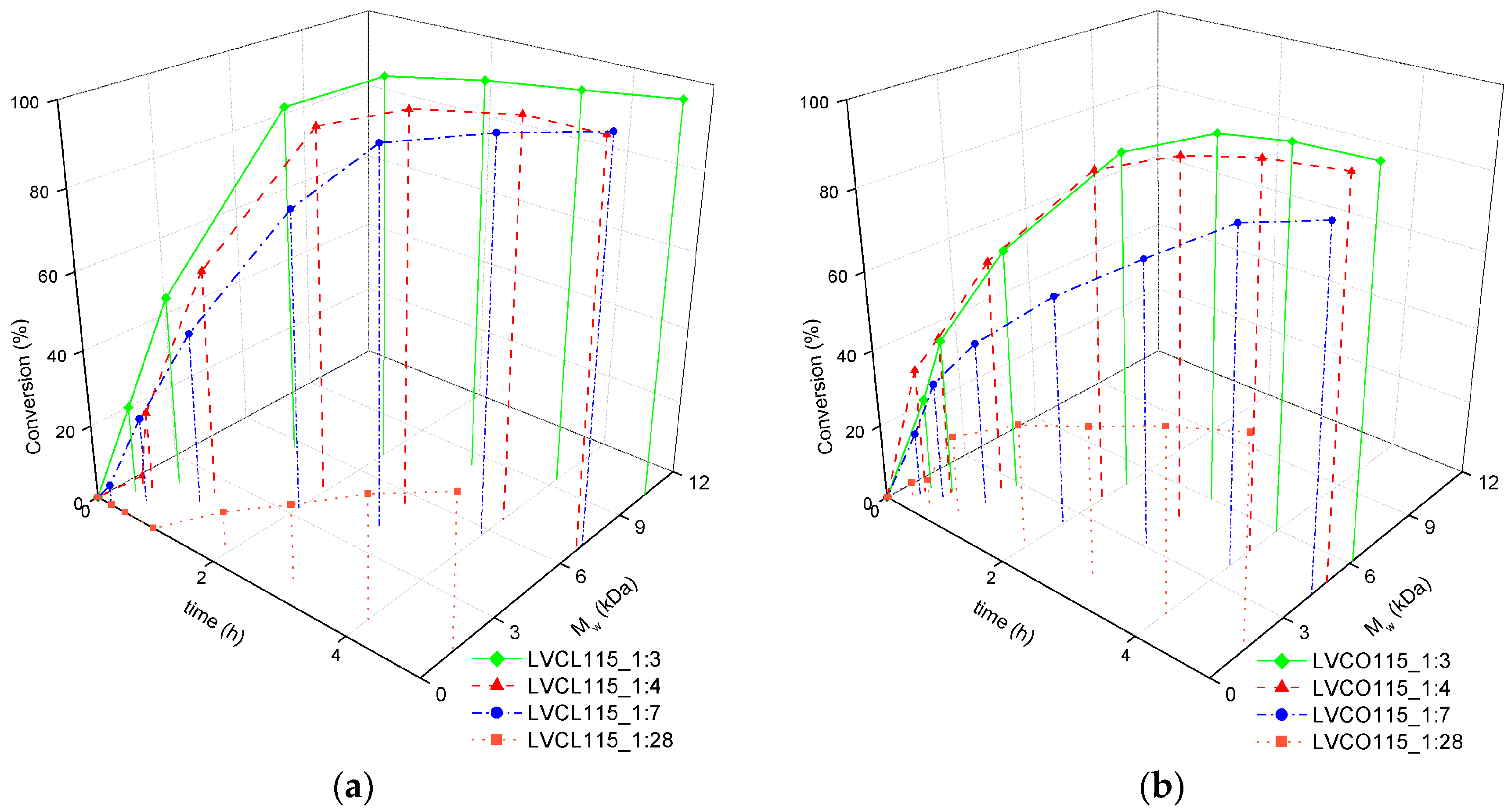
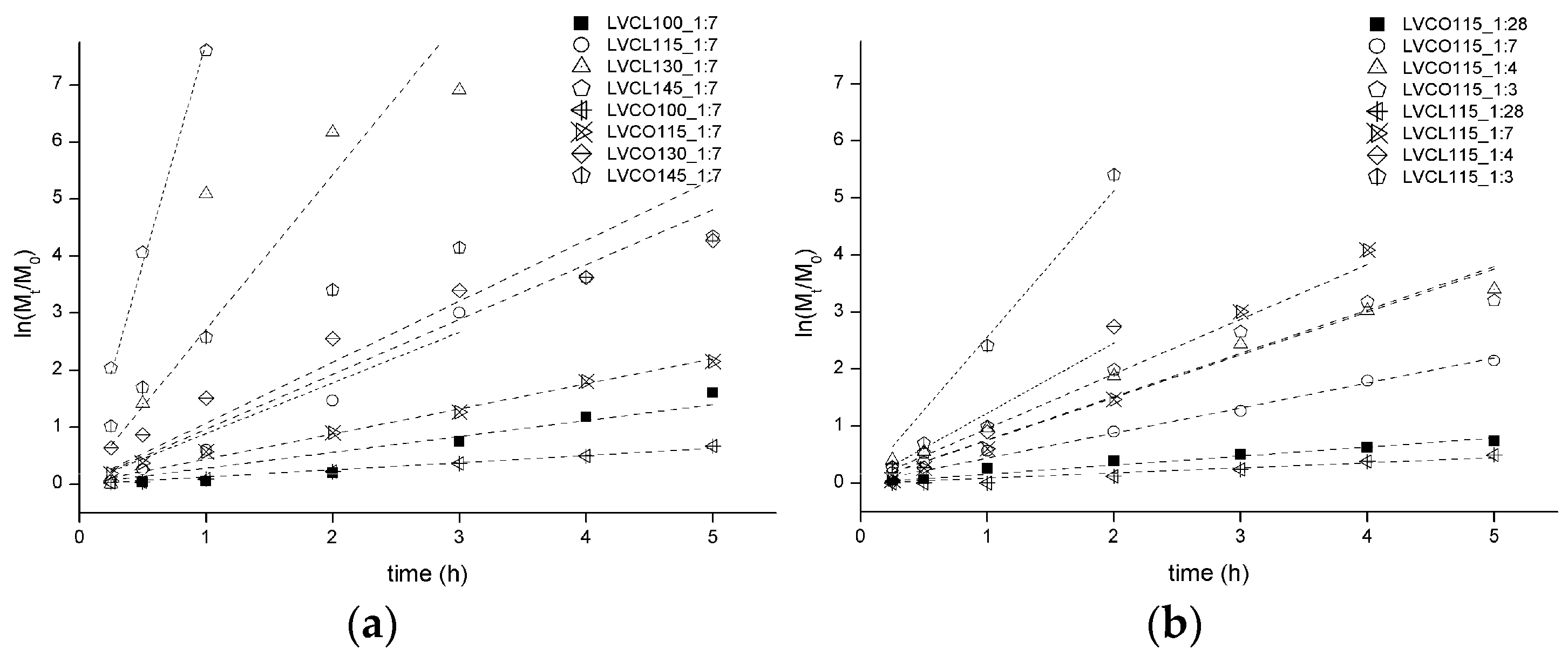
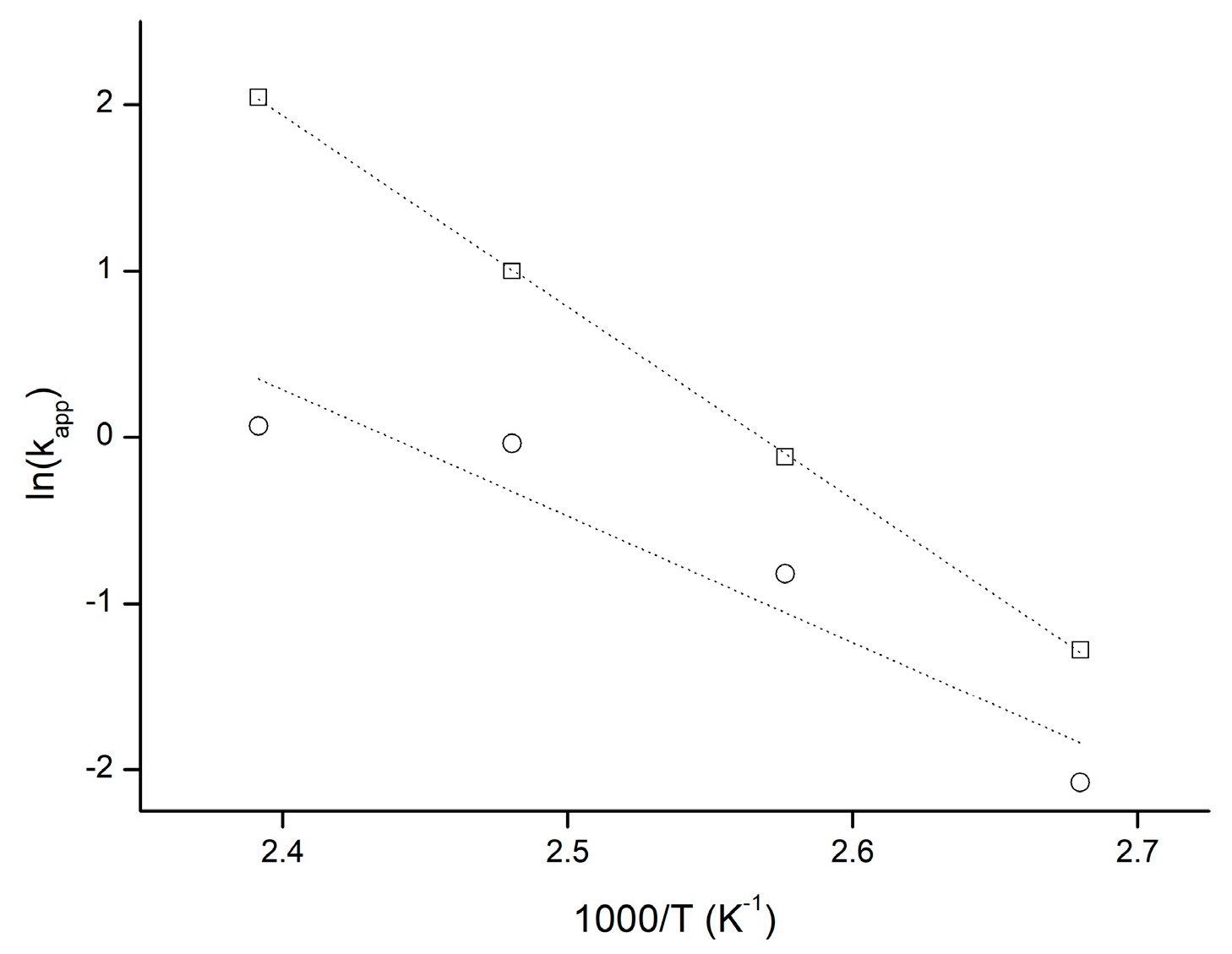
2.1.2. Influence of Synthesis Parameters on Polymerization Reaction Course
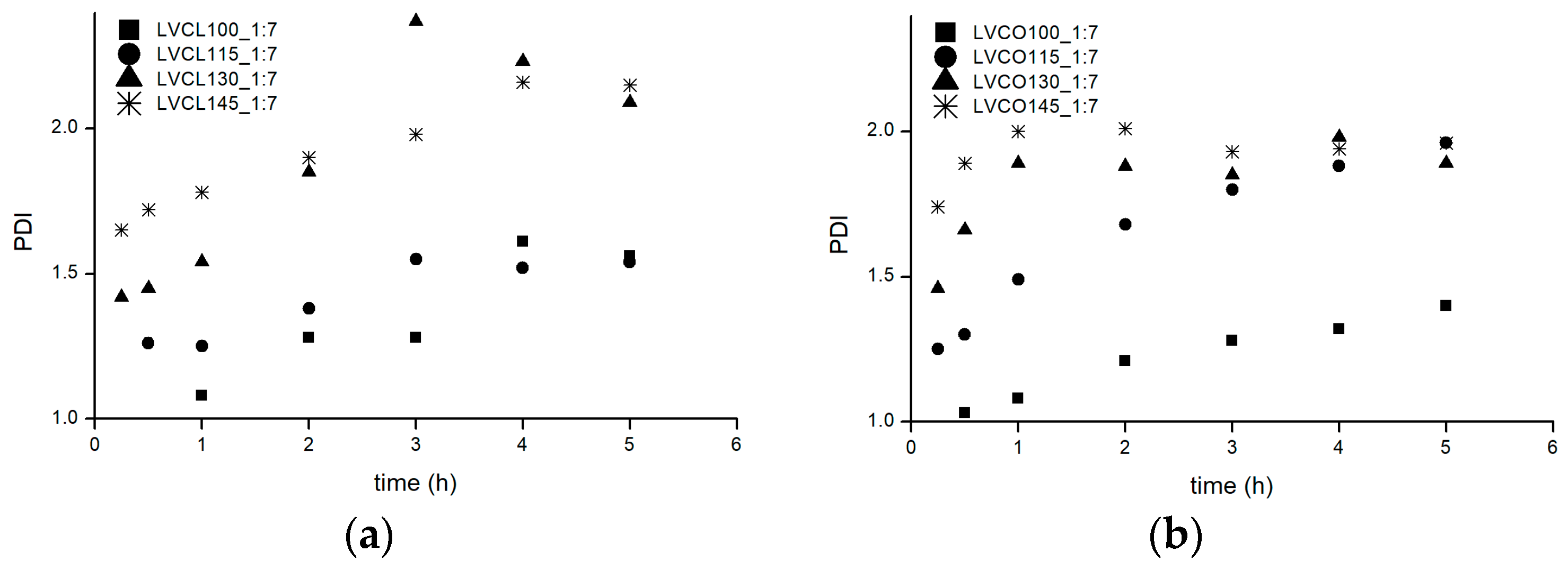
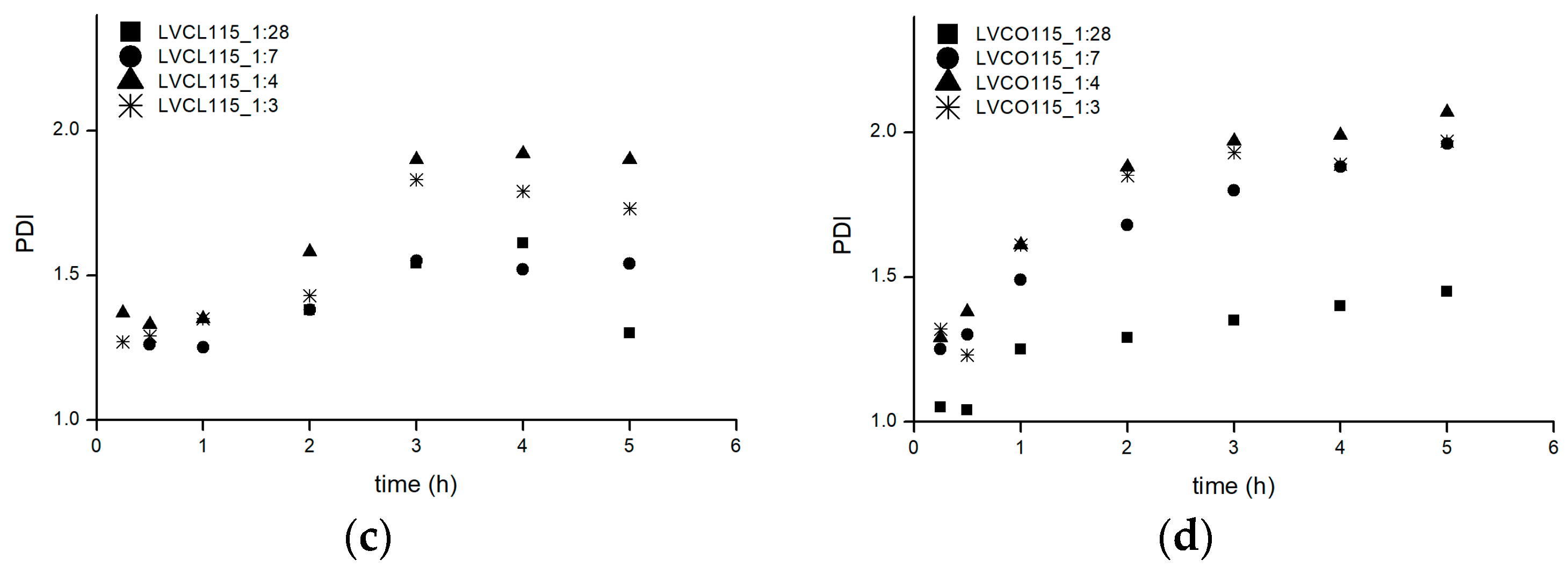
2.1.3. Physicochemical Properties of Obtained Conjugates
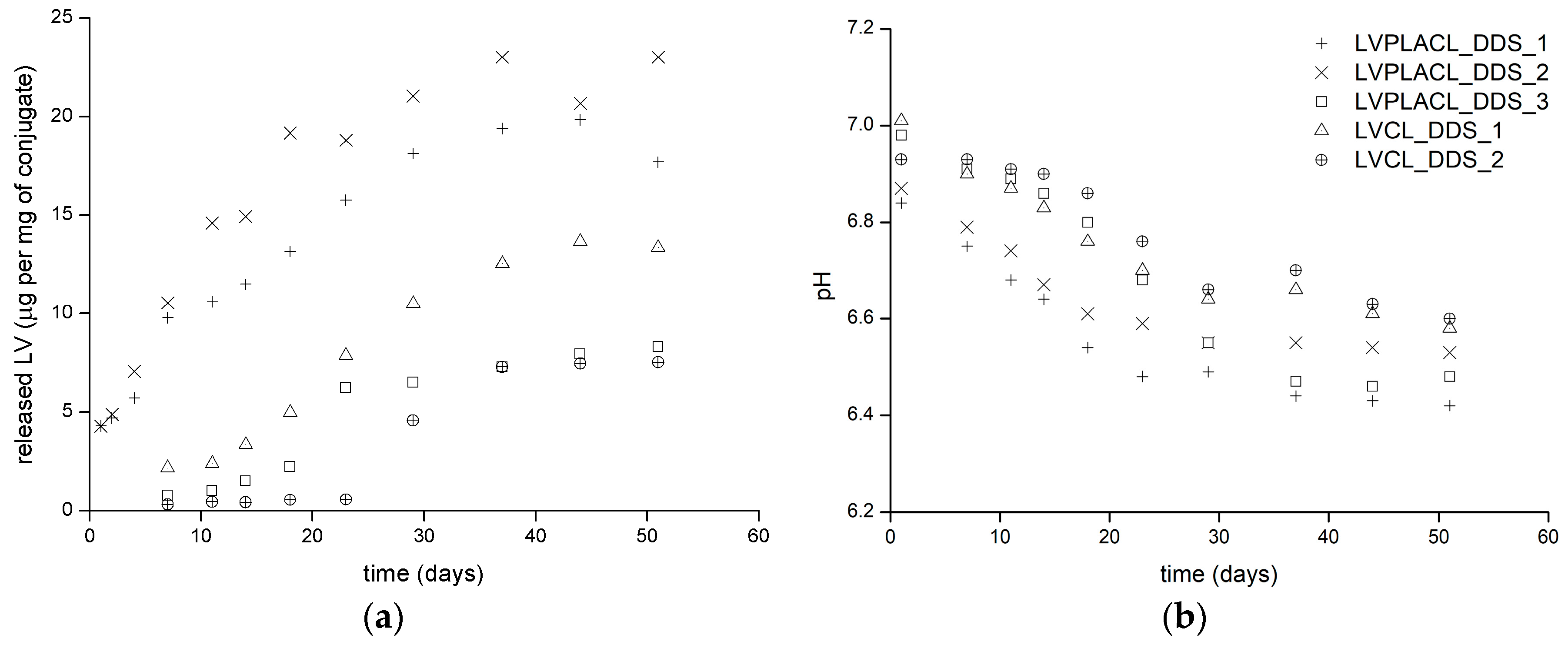
2.2. Drug Release
3. Materials and Methods
3.1. Materials
3.2. Conjugate Synthesis
3.3. Gel Permeation Chromatography
3.4. Proton Nuclear Magnetic Resonance
3.5. Electrospray Ionization Time-of-Flight Mass Spectrometry
3.6. X-ray Powder Diffraction
3.7. Differential Scanning Calorimetry
3.8. Particle Preparation and Drug Release
3.9. Dynamic Light Scattering
3.10. pH Measurements
3.11. High-Performance Liquid Chromatography
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turecek, P.L.; Bossard, M.J.; Schoetens, F.; Ivens, I.A. PEGylation of Biopharmaceuticals: A Review of Chemistry and Nonclinical Safety Information of Approved Drugs. J. Pharm. Sci. 2016, 105, 460–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, N.; Ghandeharia, H. Polymeric conjugates for drug delivery. Chem. Mater. 2012, 24, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, Y.; Firer, M.; Gellerman, G. Recent innovations in peptide based targeted drug delivery to cancer cells. Biomedicines 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.A.; Saleh, A.M. Applications of nanoparticle systems in drug delivery technology. Saudi Pharm. J. 2018, 26, 64–70. [Google Scholar] [CrossRef]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance Properties of Nano-sized Particles and Molecules as Imagin Agents: Consideration and Caveats. Nanomedicine 2012, 3, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, S.; Untereker, D. Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Mei, L.; Song, C.; Cui, X.; Wang, P. The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [Google Scholar] [CrossRef]
- Woodard, L.N.; Grunlan, M.A. Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. [Google Scholar] [CrossRef] [Green Version]
- Adeosun, S.O.; Lawal, G.I.; Gbenebor, O.P. Characteristics of Biodegradable Implants. J. Miner. Mater. Charact. Eng. 2014, 2, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Patrício, T.; Domingos, M.; Gloria, A.; Bártolo, P. Characterisation of PCL and PCL/PLA scaffolds for tissue engineering. Procedia CIRP 2013, 5, 110–114. [Google Scholar] [CrossRef]
- Lee, B.K.; Yun, Y.; Park, K. PLA micro- and nano-particles. Adv. Drug Deliv. Rev. 2016, 107, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Calzoni, E.; Cesaretti, A.; Polchi, A.; Di Michele, A.; Tancini, B.; Emiliani, C. Biocompatible polymer nanoparticles for drug delivery applications in cancer and neurodegenerative disorder therapies. J. Funct. Biomater. 2019, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuyken, O.; Pask, S. Ring-Opening Polymerization—An Introductory Review. Polymers 2013, 5, 361–403. [Google Scholar] [CrossRef] [Green Version]
- Urbaniak, T.; Musiał, W. Influence of Solvent Evaporation Technique Parameters on Diameter of Submicron Lamivudine-Poly-ε-Caprolactone Conjugate Particles. Nanomaterials 2019, 9, 1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, A.; Libiszowski, J.; Biela, T.; Cypryk, M.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with tin(II) octoate. Polymerization of ε-caprolactone and L,L-Lactide co-initiated with primary amines. Macromolecules 2005, 38, 8170–8176. [Google Scholar] [CrossRef]
- Koppensteiner, H.; Brack-Werner, R.; Schindler, M. Macrophages and their relevance in Human Immunodeficiency Virus Type I infection. Retrovirology 2012, 9, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandru-flaviu, T.; Cornel, C. Macrophages Targeted Drug Delivery as a Key Therapy in Infectious Disease. Biotechnol. Mol. Biol. Nanomed. 2014, 2, 19–21. [Google Scholar]
- Carlone, N.A.; Acocella, G.; Cuffini, A.M.; Forno-Pizzoglio, M. Killing of macrophage-ingested mycobacteria by rifampicin, pyrazinamide, and pyrazinoic acid alone and in combination. Am. Rev. Respir. Dis. 1985, 132, 1274–1277. [Google Scholar]
- Urbaniak, T.; Machová, D.; Janoušková, O.; Musiał, W. Microparticles of Lamivudine—Poly-ε-Caprolactone Conjugate for Drug Delivery via Internalization by Macrophages. Molecules 2019, 24, 723. [Google Scholar] [CrossRef] [Green Version]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484. [Google Scholar] [CrossRef]
- Baimark, Y.; Molloy, R. Synthesis and Characterization of Poly(L-lactide-co-ε-caprolactone) Copolymers: Effects of Stannous Octoate Initiator and Diethylene Glycol Coinitiator Concentrations. ScienceAsia 2004, 30, 327–334. [Google Scholar] [CrossRef]
- Wu, D.; Lv, Y.; Guo, R.; Li, J.; Habadati, A.; Lu, B.; Wang, H.; Wei, Z. Kinetics of Sn(Oct) 2-catalyzed ring opening polymerization of ε-caprolactone. Macromol. Res. 2017, 25, 1070–1075. [Google Scholar] [CrossRef]
- Ehsani, M.; Khodabakhshi, K.; Asgari, M. Lactide synthesis optimization: Investigation of the temperature, catalyst and pressure effects. E-Polymers 2014, 14, 353–361. [Google Scholar] [CrossRef]
- Puaux, J.P.; Banu, I.; Nagy, I.; Bozga, G. A study of L-lactide ring-opening polymerization kinetics. Macromol. Symp. 2007, 259, 318–326. [Google Scholar] [CrossRef]
- Mezzasalma, L.; Dove, A.P.; Coulembier, O. Organocatalytic ring-opening polymerization of L-lactide in bulk: A long standing challenge. Eur. Polym. J. 2017, 95, 628–634. [Google Scholar] [CrossRef]
- Storey, R.F.; Sherman, J.W. Kinetics and mechanism of the stannous octoate-catalyzed bulk polymerization of ε-caprolactone. Macromolecules 2002, 35, 1504–1512. [Google Scholar] [CrossRef]
- Nguyen, N.T.; Greenhalgh, E.; Kamaruddin, M.J.; El Harfi, J.; Carmichael, K.; Dimitrakis, G.; Kingman, S.W.; Robinson, J.P.; Irvine, D.J. Understanding the acceleration in the ring-opening of lactones delivered by microwave heating. Tetrahedron 2014, 70, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Giachi, G.; Frediani, M.; Rosi, L.; Frediani, P. Synthesis and Processing of Biodegradable and Bio-Based Polymers by Microwave Irradiation; IntechOpen Limited: London, UK, 2011; ISBN 978-953-307-573-0. [Google Scholar]
- De SB Monteiro, M.S.; Tavares, M.I.B. The Development and Characterization of Polycaprolactone and Titanium Dioxide Hybrids. Adv. Nanopart. 2018, 7, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Guang-Mei, C.; Tie-Mei, Z.; Lei, C.; Yi-Ping, H. Crystallization properties of polycaprolactone induced by different hydroxyapatite nano-particles. Asian J. Chem. 2010, 22, 5902–5912. [Google Scholar]
- Singh, C.; Purohit, S.; Pandey, B.L.; Singh, M. Solvent Evaporation Technique of Microencapsulation: A Systemic Review. Int. J. Adv. Pharm. Anal. 2014, 4, 96–104. [Google Scholar]
- Zhang, X.; Peng, X.; Zhang, S.W. Synthetic Biodegradable Medical Polymers: Polymer Blends; Elsevier Ltd.: Amsterdam, The Netherlands, 2016; ISBN 9780081003930. [Google Scholar]
- Lassalle, V.; Ferreira, M.L. PLA nano- and microparticles for drug delivery: An overview of the methods of preparation. Macromol. Biosci. 2007, 7, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Dash, T.K.; Konkimalla, V.B. Poly-є-caprolactone based formulations for drug delivery and tissue engineering: A review. J. Control. Release 2012, 158, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Tshweu, L.; Katata, L.; Kalombo, L.; Swai, H. Nanoencapsulation of water soluble drug, lamivudine using a double emulsion spray drying technique for improving HIV treatment Lesego Tshweu. J. Nanopart. Res. 2013, 15, 2040. [Google Scholar] [CrossRef]
- Wang, B.; Chen, G.Q.; Mao, Z.W.; Zhang, Y.Y.; Yu, D.H.; Gao, C.Y. Preparation and cellular uptake of PLGA particles loaded with lamivudine. Chin. Sci. Bull. 2012, 57, 3985–3993. [Google Scholar] [CrossRef] [Green Version]
- Tamizhrasi, S.; Shukla, A.; Shivkumar, T.; Rathi, V.; Rathi, J.C. Formulation and evaluation of lamivudine loaded polymethacrylic acid nanoparticles. Int. J. PharmTech Res. 2009, 1, 411–415. [Google Scholar]
- Sansare, V.; Patel, N.; Patankar, N. Design, Optimization and Characterization of Lamivudine Loaded Solid Lipid Nanoparticles for Targeted Delivery to Brain. Int. Res. J. Pharm. 2019, 10, 143–148. [Google Scholar] [CrossRef]
- Save, M.; Soum, A. Controlled ring-opening polymerization of lactones and lactide initiated by lanthanum isopropoxide, 2a: Mechanistic studies. Macromol. Chem. Phys. 2002, 203, 2591–2603. [Google Scholar] [CrossRef]
- United States Pharmacopeial Convention. United States Pharmacopeia and National Formulary (USP41–NF36); United States Pharmacopeial Convention: Rockville, MD, USA, 2016. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis | Structure Matched to Simulation * | Adduct/s |
|---|---|---|
| LVCO115_1:28 | Conjugated poly(d,l-lactide-co-ε-caprolactone)chain | H+ |
| Conjugated PLA chain | H+ | |
| Conjugated PCL chain | H+ | |
| PLA chain | K+ | |
| LVCO115_1:7 | Conjugated poly(d,l-lactide-co-ε-caprolactone)chain | H+ |
| Conjugated PLA chain | H+ | |
| Conjugated PCL chain | H+ | |
| LVCO115_1:4 | Conjugated poly(d,l-lactide-co-ε-caprolactone) chain | H+ |
| PLA chains | Na+ | |
| Conjugated PLA chain | H+ | |
| LVCO115_1:3 | Conjugated poly(d,l-lactide-co-ε-caprolactone) chain | H+ |
| Conjugated PLA chain | H+ | |
| LVCL115_1:28 | Conjugated PCL chain | H+ |
| LVCL115_1:7 | Conjugated PCL chain | Na+, H+ |
| LVCL115_1:4 | Conjugated PCL chain | H+ |
| LVCL115_1:3 | Conjugated PCL chain | H+ |
| LVCO100_1:7 | Conjugated poly(d,l-lactide-co-ε-caprolactone) chain | H+ |
| Conjugated PLA chain | H+ | |
| LVCO130_1:7 | Conjugated poly(d,l-lactide-co-ε-caprolactone) chain | H+ |
| Conjugated PCL chain | H+ | |
| LVCO145_1:7 | Conjugated poly(d,l-lactide-co-ε-caprolactone) chain | H+ |
| Conjugated PLA chain | H+ | |
| LVCL 100_1:7 | Conjugated PCL chain | H+ |
| PCL chain | Na+ | |
| LVCL130_1:7 | Conjugated PCL chain | H+ |
| PCL chain | Na+ | |
| LVCL145_1:7 | Conjugated PCL chain | H+ |
| PCL chain | Na+ |
| Synthesis | Apparent Rate Constant k (h−1) | Standard Error | R-Square |
|---|---|---|---|
| LVCO115_1:28 | 0.159 | 0.007 | 0.98 |
| LVCO115_1:7 | 0.440 | 0.013 | 0.99 |
| LVCO115_1:4 | 0.750 | 0.036 | 0.98 |
| LVCO115_1:3 | 0.759 | 0.050 | 0.97 |
| LVCL115_1:28 | 0.090 | 0.007 | 0.96 |
| LVCL115_1:7 | 0.958 | 0.057 | 0.98 |
| LVCL115_1:4 | 1.229 | 0.155 | 0.94 |
| LVCL115_1:3 | 2.566 | 0.190 | 0.98 |
| LVCO100_1:7 | 0.125 | 0.004 | 0.99 |
| LVCO130_1:7 | 0.962 | 0.069 | 0.96 |
| LVCO145_1:7 | 1.069 | 0.156 | 0.87 |
| LVCL 100_1:7 | 0.279 | 0.028 | 0.93 |
| LVCL130_1:7 | 2.714 | 0.377 | 0.91 |
| LVCL145_1:7 | 7.724 | 0.156 | 0.99 |
| Synthesis | Mn (kDa) | Mw (kDa) | PDI | Cryst. (%) | Tm (˚C) | LA:ε-CL Ratio | Conv. (%) |
|---|---|---|---|---|---|---|---|
| LVCL100_1:7 | 1.54 | 2.4 | 1.56 | 66 | 55 | - | 80% |
| LVCL115_1:7 | 4.59 | 7.08 | 1.54 | 60 | 60.8 | - | 100% |
| LVCL130_1:7 | 3.84 | 8.04 | 2.09 | 61 | 60.1 | - | 100% |
| LVCL145_1:7 | 5.47 | 11.76 | 2.15 | 59 | 62.8 | - | 100% |
| LVCL115_1:28 | 0.99 | 1.28 | 1.3 | 62 | 54.7 | - | 39% |
| LVCL115_1:4 | 3.87 | 6.78 | 1.9 | 56 | 61.5 | - | 100% |
| LVCL115_1:3 | 6.01 | 10.4 | 1.73 | 60 | 62.8 | - | 100% |
| LVCO100_1:7 | 0.88 | 1.23 | 1.4 | 0 | - | 1.3166 | 51% |
| LVCO115_1:7 | 2.16 | 4.23 | 1.96 | 3 | - | 0.891 | 88% |
| LVCO130_1:7 | 2.78 | 5.27 | 1.89 | 0 | - | 0.8328 | 99% |
| LVCO145_1:7 | 3.16 | 6.21 | 1.96 | 0 | - | 0.9156 | 99% |
| LVCO115_1:28 | 0.92 | 1.33 | 1.45 | 0 | - | 3.8382 | 52% |
| LVCO115_1:4 | 2.38 | 4.92 | 1.97 | 0 | - | 0.662 | 97% |
| LVCO115_1:3 | 3.12 | 6.15 | 2.07 | 7 | - | 0.7278 | 96% |
| Preparation | Conjugate | Hydrodynamic Diameter (nm) | PDIHd |
|---|---|---|---|
| LVPLACL_DDS_1 | LVCO100_1:7 | 389.6 ± 5.4 | 0.46 ± 0.02 |
| LVPLACL_DDS_2 | LVCO115_1:28 | 380.9 ± 18.6 | 0.51 ± 0.06 |
| LVPLACL_DDS_3 | LVCO145_1:7 | 440.8 ± 11.1 | 0.39 ± 0.02 |
| LVCL_DDS_1 | LVCL115_1:28 | 353.4 ± 4.6 | 0.45 ± 0.05 |
| LVCL_DDS_2 | LVCL115_1:4 | 506.9 ± 6.964 | 0.26 ± 0.02 |
| Synthesis | SO: Initiator: LA: ε-CL Molar Ratio | Reaction Temperature (°C) | Initiator | Monomer |
|---|---|---|---|---|
| ACCL | 1:31:0:1948 | 165 | AC | ε-CL |
| CLARCL | 1:12:0:1948 | 115 | CLAR | ε-CL |
| RIFCL | 1:12:0:1948 | 115 | RIF | ε-CL |
| LVCL100_1:7 | 1:7:0:438 | 100 | LV | ε-CL |
| LVCL115_1:7 | 1:7:0:438 | 115 | LV | ε-CL |
| LVCL130_1:7 | 1:7:0:438 | 130 | LV | ε-CL |
| LVCL145_1:7 | 1:7:0:438 | 145 | LV | ε-CL |
| LVCL115_1:28 | 1:28:0:1753 | 115 | LV | ε-CL |
| LVCL115_1:4 | 1:4:0:250 | 115 | LV | ε-CL |
| LVCL115_1:3 | 1:3:0:175 | 115 | LV | ε-CL |
| LVCO100_1:7 | 1:7:77:204 | 100 | LV | ε-CL, LA |
| LVCO115_1:7 | 1:7:77:204 | 115 | LV | ε-CL, LA |
| LVCO130_1:7 | 1:7:77:204 | 130 | LV | ε-CL, LA |
| LVCO145_1:7 | 1:7:77:204 | 145 | LV | ε-CL, LA |
| LVCO115_1:28 | 1:28:310:818 | 115 | LV | ε-CL, LA |
| LVCO115_1:4 | 1:4:44:116 | 115 | LV | ε-CL, LA |
| LVCO115_1:3 | 1:3:31:81 | 115 | LV | ε-CL, LA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbaniak, T.; Musiał, W. Selected Physicochemical and Pharmaceutical Properties of Poly-ε-caprolactone and Poly(d,l-lactide-co-ε-caprolactone) Conjugates of Lamivudine Synthesized via Ring-Opening Polymerization. Polymers 2019, 11, 2124. https://doi.org/10.3390/polym11122124
Urbaniak T, Musiał W. Selected Physicochemical and Pharmaceutical Properties of Poly-ε-caprolactone and Poly(d,l-lactide-co-ε-caprolactone) Conjugates of Lamivudine Synthesized via Ring-Opening Polymerization. Polymers. 2019; 11(12):2124. https://doi.org/10.3390/polym11122124
Chicago/Turabian StyleUrbaniak, Tomasz, and Witold Musiał. 2019. "Selected Physicochemical and Pharmaceutical Properties of Poly-ε-caprolactone and Poly(d,l-lactide-co-ε-caprolactone) Conjugates of Lamivudine Synthesized via Ring-Opening Polymerization" Polymers 11, no. 12: 2124. https://doi.org/10.3390/polym11122124