The Effect of Polydimethylsiloxane-Ethylcellulose Coating Blends on the Surface Characterization and Drug Release of Ciprofloxacin-Loaded Mesoporous Silica



Abstract
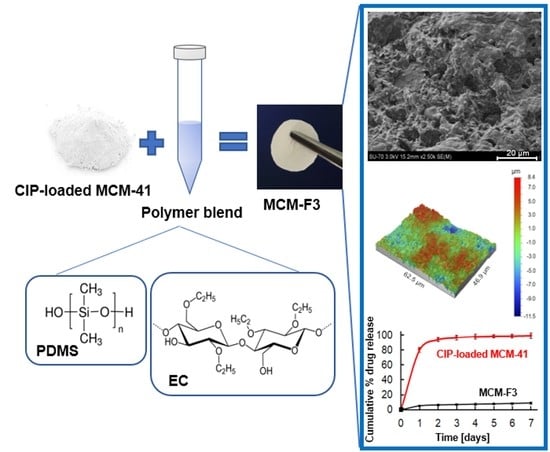
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Mesoporous Silica Materials (MCM-41)
2.3. Ciprofloxacin Adsorption onto MCM-41
2.4. Fabrication of Solid Films Composed of CIP-Loaded MCM-41 and Polymer Blends
2.5. Physicochemical Characterization
2.6. Drug Release Analysis
3. Results and Discussion
3.1. Ciprofloxacin Adsorption onto MCM-41
3.2. Solid Films Formation
3.3. Physicochemical Characterization
3.3.1. FTIR Characterization
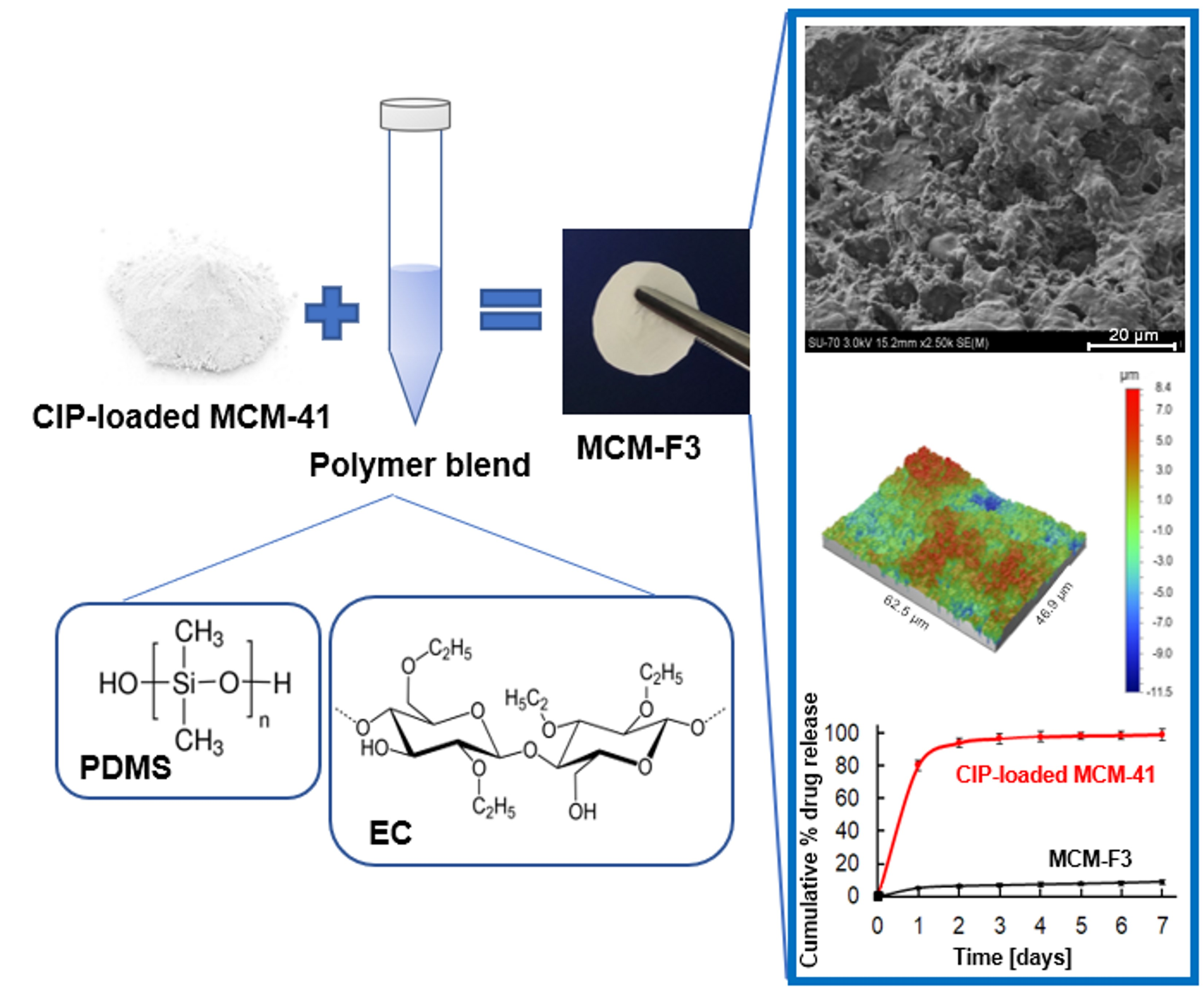
3.3.2. Surface Morphology
3.3.3. Contact Angle and Wetting Properties
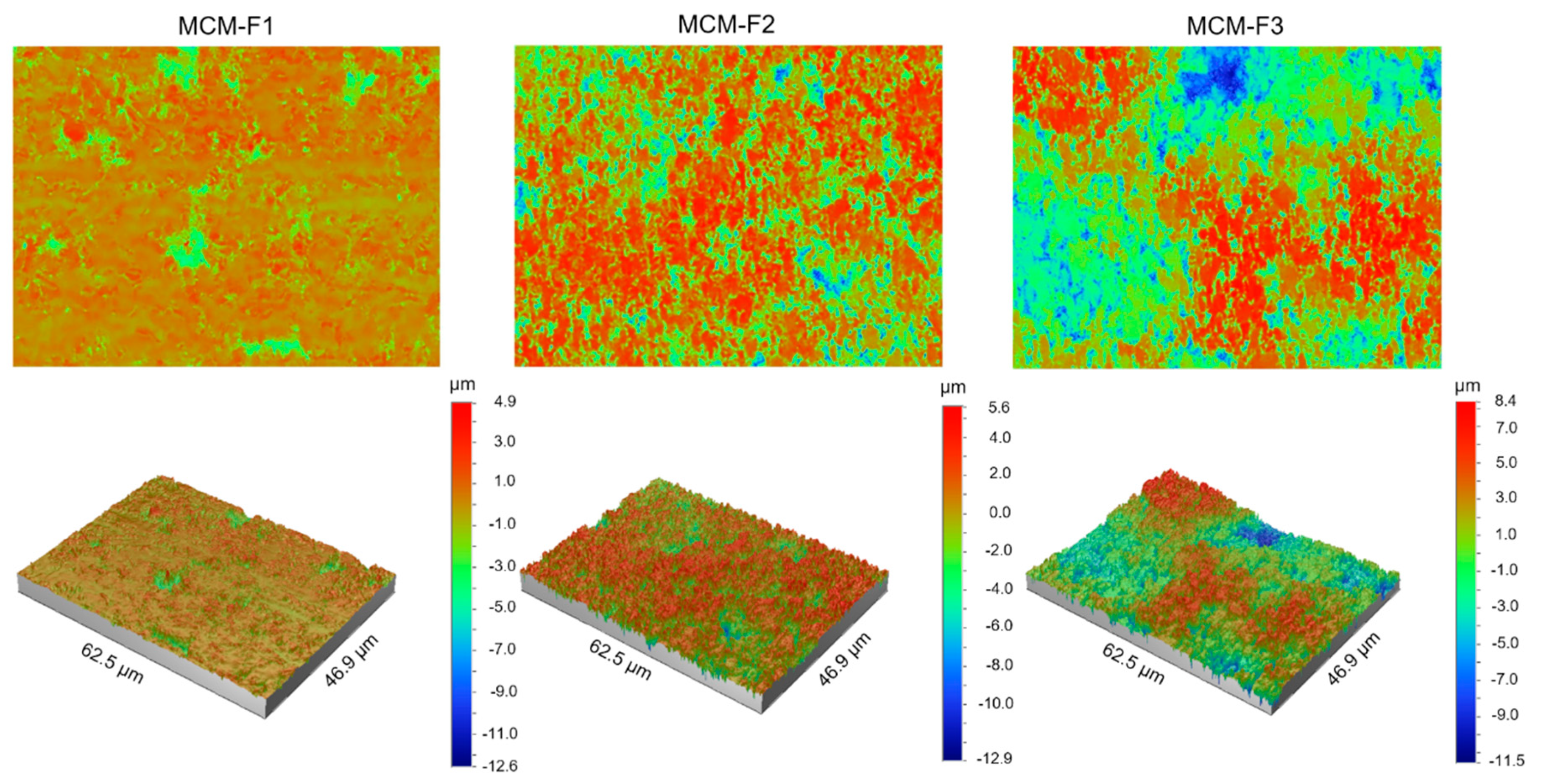
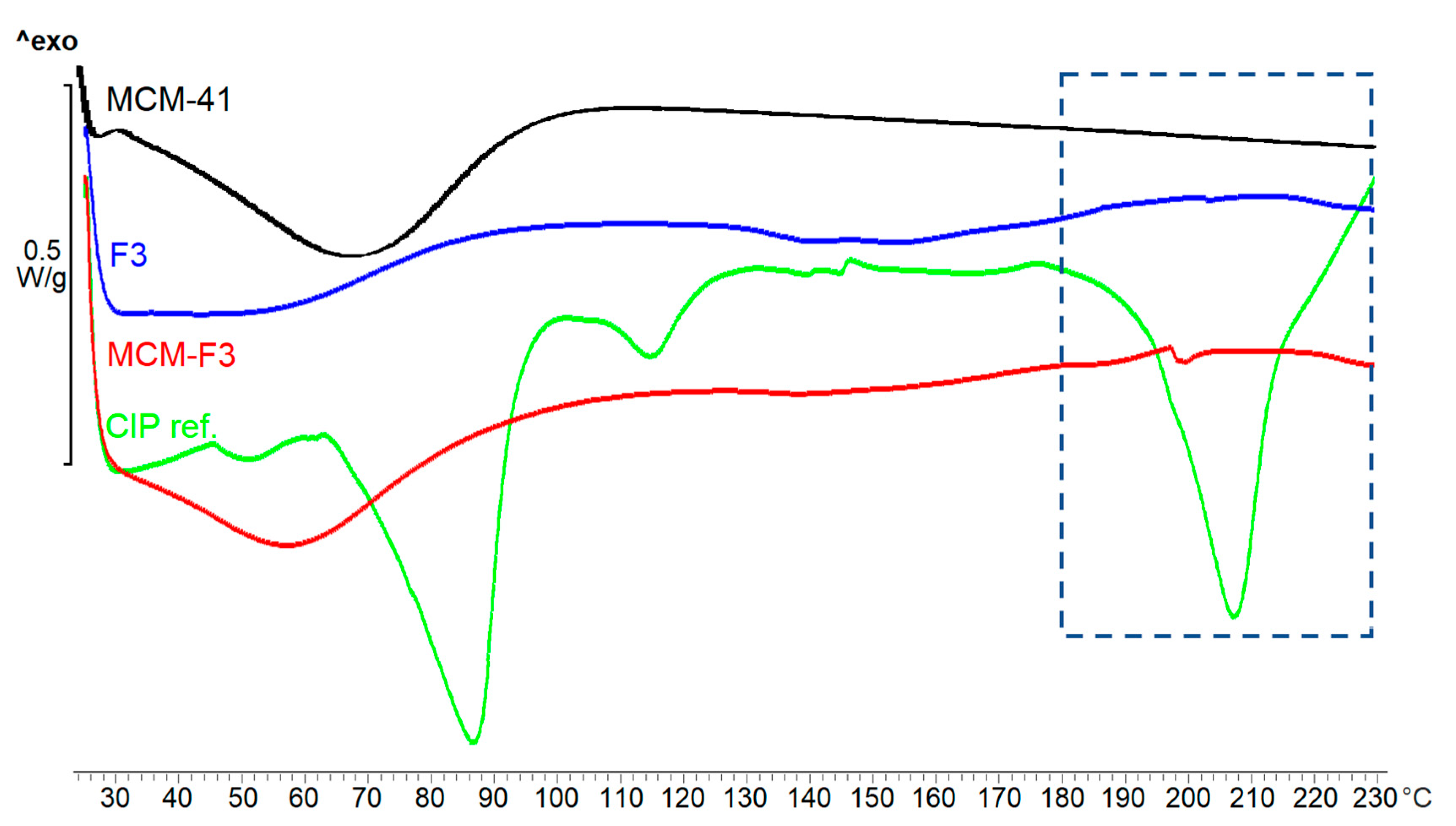
3.3.4. Solid-State Characterization of Ciprofloxacin
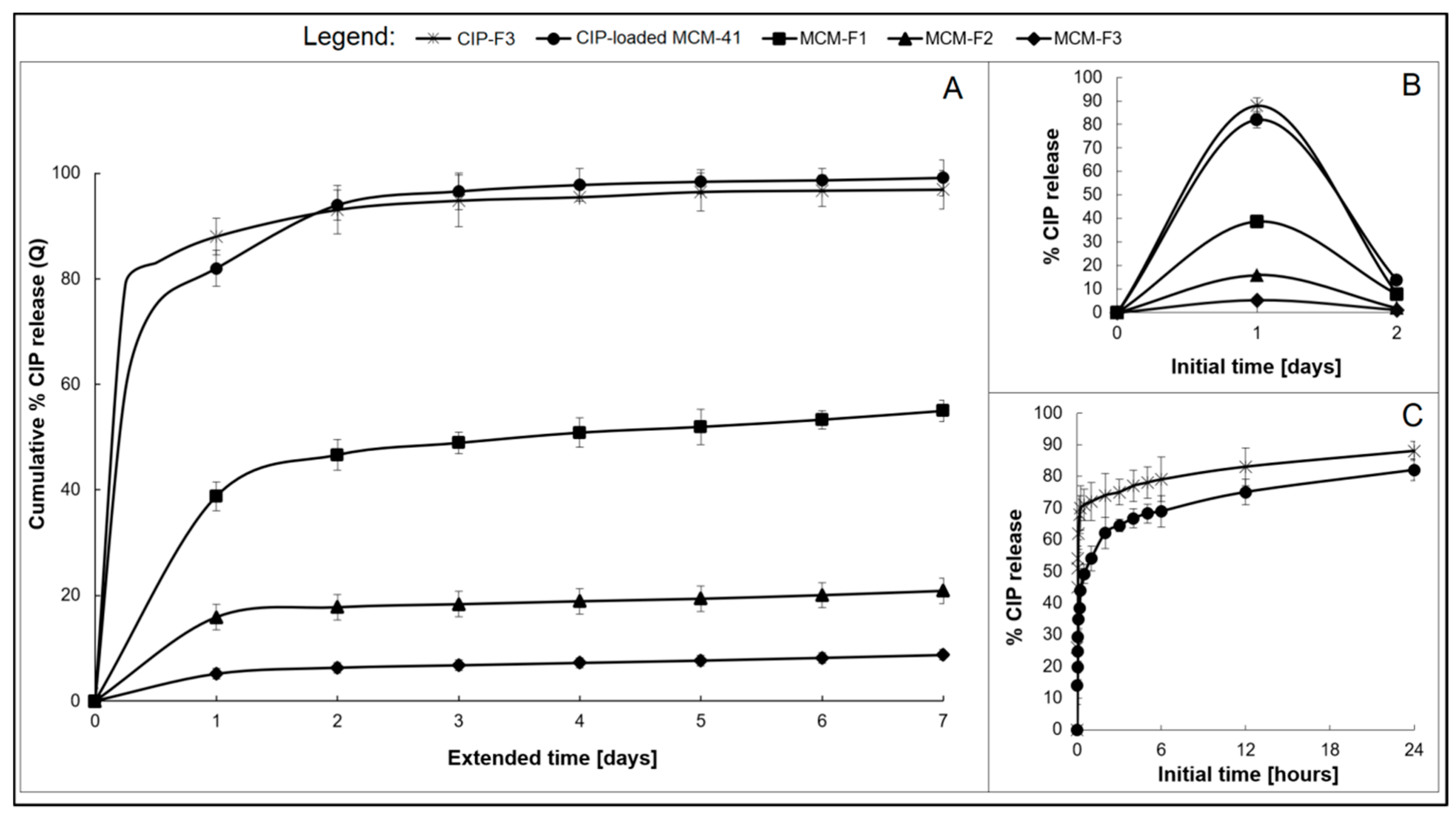
3.4. Ciprofloxacin Release
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nandi, S.K.; Bandyopadhyay, S.; Das, P.; Samanta, I.; Mukherjee, P.; Roy, S.; Kundu, B. Understanding Osteomyelitis and Its Treatment through Local Drug Delivery System. Biotechnol. Adv. 2016, 34, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Mouriño, V.; Boccaccini, A.R. Bone Tissue Engineering Therapeutics: Controlled Drug Delivery in Three-Dimensional Scaffolds. J. R. Soc. Interface 2010, 7, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Controlled Drug Delivery Systems: Past Forward and Future Back. J. Control. Release 2014, 190, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, S. Ordered Mesoporous Materials for Drug Delivery. Microporous Mesoporous Mater. 2009, 117, 1–9. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Czarnobaj, K.; Szewczyk, A.; Sawicki, W. Preparation and in Vitro Characterisation of Bioactive Mesoporous Silica Microparticles for Drug Delivery Applications. Mater. Sci. Eng. C 2016, 60, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Beck, G.R.; Ha, S.W.; Camalier, C.E.; Yamaguchi, M.; Li, Y.; Lee, J.K.; Weitzmann, M.N. Bioactive Silica-Based Nanoparticles Stimulate Bone-forming Osteoblasts, Suppress Bone-Resorbing Osteoclasts, and Enhance Bone Mineral Density in Vivo. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 793–803. [Google Scholar] [CrossRef]
- Braun, K.; Pochert, A.; Beck, M.; Fiedler, R.; Gruber, J.; Lindén, M. Dissolution Kinetics of Mesoporous Silica Nanoparticles in Different Simulated Body Fluids. J. Sol-Gel Sci. Technol. 2016, 79, 319–327. [Google Scholar] [CrossRef]
- Götz, W.; Tobiasch, E.; Witzleben, S.; Schulze, M. Effects of Silicon Compounds on Biomineralization, Osteogenesis, and Hard Tissue Formation. Pharmaceutics 2019, 11, 117. [Google Scholar] [CrossRef]
- Pérez-Esteve, É.; Ruiz-Rico, M.; De La Torre, C.; Villaescusa, L.A.; Sancenón, F.; Marcos, M.D.; Amorós, P.; Martínez-Máñez, R.; Barat, J.M. Encapsulation of Folic Acid in Different Silica Porous Supports: A Comparative Study. Food Chem. 2016, 196, 66–75. [Google Scholar] [CrossRef]
- Lee, J.H.; Yeo, Y. Controlled Drug Release from Pharmaceutical Nanocarriers. Chem. Eng. Sci. 2015, 125, 75–84. [Google Scholar] [CrossRef]
- Siepmann, F.; Siepmann, J.; Walther, M.; MacRae, R.J.; Bodmeier, R. Polymer Blends for Controlled Release Coatings. J. Control. Release 2008, 125, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Duo, Y.; Li, Y.; Chen, C.; Liu, B.; Wang, X.; Zeng, X.; Chen, H. DOX-Loaded pH-Sensitive Mesoporous Silica Nanoparticles Coated with PDA and PEG Induce Pro-Death Autophagy in Breast Cancer. RSC Adv. 2017, 7, 39641–39650. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Wang, H.; Ducheyne, P. Polymer-Coated Mesoporous Silica Nanoparticles for the Controlled Release of Macromolecules. Acta Biomater. 2012, 8, 3429–3435. [Google Scholar] [CrossRef] [PubMed]
- Trendafilova, I.; Szegedi, Á.; Yoncheva, K.; Shestakova, P.; Mihály, J.; Ristić, A.; Konstantinov, S.; Popova, M. A pH Dependent Delivery of Mesalazine from Polymer Coated and Drug-Loaded SBA-16 Systems. Eur. J. Pharm. Sci. 2016, 81, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Lamprou, D.A. Polymer Coatings for Biomedical Applications: A Review. Trans. IMF 2014, 92, 9–19. [Google Scholar] [CrossRef]
- Lecomte, F.; Siepmann, J.; Walther, M.; MacRae, R.J.; Bodmeier, R. Polymer Blends used for the Coating of Multiparticulates: Comparison of Aqueous and Organic Coating Techniques. Pharm. Res. 2004, 21, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Ponche, A.; Bigerelle, M.; Anselme, K. Relative Influence of Surface Topography and Surface Chemistry on Cell Response to Bone Implant Materials. Part 1: Physico-Chemical Effects. Proc. Inst. Mech. Eng. Part H J. Eng. Med. 2010, 224, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Hokmabad, V.R.; Davaran, S.; Aghazadeh, M.; Rahbarghazi, R.; Salehi, R.; Ramazani, A. Fabrication and characterization of Novel ethyl Cellulose-Grafted-Poly (ɛ-caprolactone)/alginate Nanofibrous/Macroporous Scaffolds Incorporated with Nano-Hydroxyapatite for Bone Tissue Engineering. J. Biomater. Appl. 2019, 33, 1128–1144. [Google Scholar] [CrossRef]
- Le Guéhennec, L.; Soueidan, A.; Layrolle, P.; Amouriq, Y. Surface Treatments of Titanium Dental Implants for Rapid Osseointegration. Dent. Mater. 2007, 23, 844–854. [Google Scholar] [CrossRef]
- Krishna Alla, R.; Ginjupalli, K.; Upadhya, N.; Shammas, M.; Krishna Ravi, R.; Sekhar, R. Surface Roughness of Implants: A review. Trends Biomater. Artif. Organs 2011, 25, 112–118. [Google Scholar]
- Linez-Bataillon, P.; Monchau, F.; Bigerelle, M.; Hildebrand, H.F. In Vitro MC3T3 Osteoblast Adhesion with Respect to Surface Roughness of Ti6Al4V Substrates. Biomol. Eng. 2002, 19, 133–141. [Google Scholar] [CrossRef]
- Wirth, J.; Tahriri, M.; Khoshroo, K.; Rasoulianboroujeni, M.; Dentino, A.R.; Tayebi, L. Surface Modification of Dental Implants. In Biomaterials for Oral and Dental Tissue Engineering; Woodhead Publishing: Cambridge, MA, USA, 2017; pp. 85–96. [Google Scholar]
- Garg, H.; Bedi, G.; Garg, A. Implant Surface Modifications: A review. J. Clin. Diagn. Res. 2012, 6, 319–324. [Google Scholar]
- Bose, S.; Bogner, R.H. Solventless Pharmaceutical Coating Processes: A review. Pharm. Dev. Technol. 2007, 12, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.J.T.; Mishra, A.; Park, I.; Kim, Y.J.; Park, W.T.; Yoon, Y.J. Polymeric Biomaterials for Medical Implants and Devices. ACS Biomater. Sci. Eng. 2016, 2, 454–472. [Google Scholar] [CrossRef]
- Blanco, I. Polysiloxanes in Theranostics and Drug Delivery: A Review. Polymers (Basel) 2018, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G. Ethylcellulose Microparticles: A review. Acta Pol. Pharm.-Drug Res. 2012, 69, 11–22. [Google Scholar]
- Tian, B.; Tang, S.; Li, Y.; Long, T.; Qu, X.H.; Yu, D.G.; Guo, Y.J.; Guo, Y.P.; Zhu, Z.A. Fabrication, Characterization, and Biocompatibility of Ethyl Cellulose/Carbonated Hydroxyapatite Composite coatings on Ti6Al4V. J. Mater. Sci. Mater. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.Y.; Missaghi, S.; Tiwari, S.B.; Rajabi-Siahboomi, A.R. Application of Ethylcellulose Coating to Hydrophilic Matrices: A Strategy to Modulate Drug Release Profile and Reduce Drug Release Variability. AAPS Pharm. Sci. Tech. 2014, 15, 1049–1059. [Google Scholar] [CrossRef] [Green Version]
- Mata, A.; Fleischman, A.J.; Roy, S. Characterization of Polydimethylsiloxane (PDMS) Properties for Biomedical Micro/Nanosystems. Biomed. Microdevices 2005, 7, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Tsourvakas, S. Local Antibiotic Therapy in the Treatment of Bone and Soft Tissue Infections. Sel. Top. Plast. Reconstr. Surg. 2012, 17–46. [Google Scholar]
- Vallet-Regi, M.; Rámila, A.; Del Real, R.P.; Pérez-Pariente, J. A new property of MCM-41: Drug delivery system. Chem. Mater. 2001, 13, 308–311. [Google Scholar] [CrossRef]
- Hanoosh, W.S.; Abdelrazaq, E.M. Polydimethyl Siloxane Toughened Epoxy Resins: Tensile Strength and Dynamic Mechanical Analysis. Malaysian Polym. J. 2009, 4, 52–61. [Google Scholar]
- Mesallati, H.; Umerska, A.; Paluch, K.J.; Tajber, L. Amorphous Polymeric Drug Salts as Ionic Solid Dispersion Forms of Ciprofloxacin. Mol. Pharm. 2017, 14, 2209–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, M.K.; Branton, A.; Trivedi, D.; Nayak, G.; Mishra, R.K.; Jana, S. Characterization of Physicochemical and Thermal Properties of Biofield Treated Ethyl Cellulose and Methyl Cellulose. Int. J. Biomed. Mater. Res. 2015, 3, 83–91. [Google Scholar]
- Johnson, L.M.; Gao, L.; Shields, C.W.; Smith, M.; Efimenko, K.; Cushing, K.; Genzer, J.; López, G.P. Elastomeric microparticles for acoustic mediated bioseparations. J. Nanobiotechnology 2013, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Machigashira, M.; Miyamoto, M.; Takeuchi, H.; Noguchi, K.; Izumi, Y.; Ban, S. Effect of surface roughness on initial responses of osteoblast-like cells on two types of zirconia. Dent. Mater. J. 2009, 28, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, B.; Quan, G.; Li, F.; Wu, Q.; Dian, L.; Dong, Y.; Li, G.; Wu, C. Increasing the oral bioavailability of poorly water-soluble carbamazepine using immediate-release pellets supported on SBA-15 mesoporous silica. Int. J. Nanomedicine 2012, 7, 5807–5818. [Google Scholar] [PubMed] [Green Version]
- Prokopowicz, M. Correlation between physicochemical properties of doxorubicin-loaded silica/polydimethylsiloxane xerogel and in vitro release of drug. Acta Biomater. 2009, 5, 193–207. [Google Scholar] [CrossRef]
- Nandi, S.K.; Mukherjee, P.; Roy, S.; Kundu, B.; De, D.K.; Basu, D. Local antibiotic delivery systems for the treatment of osteomyelitis - A review. Mater. Sci. Eng. C 2009, 29, 2478–2485. [Google Scholar] [CrossRef]
- Costa, P.; Sousa Lobo, J.M. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Huynh, C.T.; Lee, D.S. Controlled Release. Encycl. Polym. Nanomater. 2015, 439–449. [Google Scholar] [CrossRef]
- Jones, D.S.; Medlicott, N.J. Casting solvent controlled release of chlorhexidine from ethylcellulose films prepared by solvent evaporation. Int. J. Pharm. 1995, 114, 257–261. [Google Scholar] [CrossRef]
- Prokopowicz, M. Atomic force microscopy technique for the surface characterization of sol–gel derived multi-component silica nanocomposites. Colloids Surfaces A Physicochem. Eng. Asp. 2016, 504, 350–357. [Google Scholar] [CrossRef]
- Kim, H.J.; Matsuda, H.; Zhou, H.; Honma, I. Ultrasound-triggered smart drug release from a poly(dimethylsiloxane)- mesoporous silica composite. Adv. Mater. 2006, 18, 3083–3088. [Google Scholar] [CrossRef]
- Popova, M.; Trendafilova, I.; Zgureva, D.; Kalvachev, Y.; Boycheva, S.; Novak Tušar, N.; Szegedi, A. Polymer-coated mesoporous silica nanoparticles for controlled release of the prodrug sulfasalazine. J. Drug Deliv. Sci. Technol. 2018, 44, 415–420. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before solvent evaporation | |||
| Formulation | PDMS:EC ratio | PDMS content [µL] | EC ethanolic solution content [µL] |
| F1 | 0:100 | - | 250.0 |
| F2 | 1:99 | 2.5 | 247.5 |
| F3 | 2:98 | 5.0 | 245.0 |
| After solvent evaporation | |||
| Formulation | PDMS:EC ratio | PDMS content [mg] | EC content [mg] |
| F1 | 0:1 | - | 10.0 |
| F2 | 1:4 | 2.4 | 9.9 |
| F3 | 1:2 | 4.8 | 9.8 |
| Formulation | Ra ± SD [µm] | Rq ± SD [µm] | Rt ± SD [µm] |
|---|---|---|---|
| MCM-F1 | 0.57 ± 0.10 | 0.86 ± 0.17 | 11.22 ± 2.49 |
| MCM-F2 | 1.86 ± 0.40 | 2.30 ± 0.45 | 19.12 ± 2.85 |
| MCM-F3 | 2.49 ± 0.54 | 3.14 ± 0.70 | 23.67 ± 6.06 |
| Sample | Average Contact Angle [θ, °] | Surface Free Energy [mJ/m2] | ||||
|---|---|---|---|---|---|---|
| Measuring Liquid | ||||||
| Water | Diiodomethane | Total (γs) | Dispersive (γsd) | Polar (γsp) | ||
| A | F1 | 82.2 | 63.0 | 29.27 | 21.35 | 7.92 |
| F2 | 86.3 | 54.2 | 32.03 | 27.81 | 4.22 | |
| F3 | 83.5 | 54.4 | 32.50 | 26.99 | 5.51 | |
| B | MCM-F1 | 112.9 | 81.9 | 16.40 | 16.07 | 0.33 |
| MCM-F2 | 125.1 | 67.5 | 29.28 | 27.97 | 1.31 | |
| MCM-F3 | 124.5 | 62.9 | 32.86 | 31.21 | 1.65 | |
| Formulation | Korsmeyer-Peppas Model | Higuchi Model | Zero Order Kinetics * | |||
|---|---|---|---|---|---|---|
| n | R2 | kH | R2 | k0 | R2 | |
| CIP-F3 | 0.51 | 0.95 | 27.01 | 0.94 | 0.03 | 0.89 |
| CIP-loaded MCM-41 | 0.34 | 0.92 | 4.93 | 0.85 | 0.02 | 0.82 |
| MCM-F1 | 0.17 | 0.97 | 8.92 | 0.93 | 1.60 | 0.99 |
| MCM-F2 | 0.33 | 0.87 | 6.80 | 0.98 | 0.61 | 0.99 |
| MCM-F3 | 0.55 | 0.93 | 6.49 | 0.98 | 0.48 | 0.99 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skwira, A.; Szewczyk, A.; Prokopowicz, M. The Effect of Polydimethylsiloxane-Ethylcellulose Coating Blends on the Surface Characterization and Drug Release of Ciprofloxacin-Loaded Mesoporous Silica. Polymers 2019, 11, 1450. https://doi.org/10.3390/polym11091450
Skwira A, Szewczyk A, Prokopowicz M. The Effect of Polydimethylsiloxane-Ethylcellulose Coating Blends on the Surface Characterization and Drug Release of Ciprofloxacin-Loaded Mesoporous Silica. Polymers. 2019; 11(9):1450. https://doi.org/10.3390/polym11091450
Chicago/Turabian StyleSkwira, Adrianna, Adrian Szewczyk, and Magdalena Prokopowicz. 2019. "The Effect of Polydimethylsiloxane-Ethylcellulose Coating Blends on the Surface Characterization and Drug Release of Ciprofloxacin-Loaded Mesoporous Silica" Polymers 11, no. 9: 1450. https://doi.org/10.3390/polym11091450