CRISPR/Cas9-Mediated Mutagenesis of RCO in Cardamine hirsuta
by
Claire Lessa Alvim Kamei
1,2, 
Bjorn Pieper
1,
Stefan Laurent
1,
Miltos Tsiantis
1,* and
Peter Huijser
1,* 1
Department of Comparative Development and Genetics, Max Planck Institute for Plant Breeding Research, Carl-von-Linné-Weg 10, 50829 Cologne, Germany
2
Hudson River Biotechnology, Nieuwe Kanaal 7V, 6709 PA Wageningen, The Netherlands
*
Authors to whom correspondence should be addressed.
Plants 2020, 9(2), 268; https://doi.org/10.3390/plants9020268
Submission received: 2 February 2020
/
Revised: 13 February 2020
/
Accepted: 14 February 2020
/
Published: 18 February 2020
(This article belongs to the Special Issue From Genes to Shape and Function: Leaf Morphogenesis at Play)
Abstract
:The small crucifer Cardamine hirsuta bears complex leaves divided into leaflets. This is in contrast to its relative, the reference plant Arabidopsis thaliana, which has simple leaves. Comparative studies between these species provide attractive opportunities to study the diversification of form. Here, we report on the implementation of the CRISPR/Cas9 genome editing methodology in C. hirsuta and with it the generation of novel alleles in the RCO gene, which was previously shown to play a major role in the diversification of form between the two species. Thus, genome editing can now be deployed in C. hirsuta, thereby increasing its versatility as a model system to study gene function and evolution.
1. Introduction
Over the past few years Cardamine hirsuta, a small crucifer related to the reference plant Arabidopsis thaliana, has emerged as a powerful model system for comparative developmental studies in plants. Both species belong to Lineage I of the Brassicaceae, and parallel genetic studies have provided a powerful platform to identify the molecular causes of trait diversity between these two species and for understanding the morphogenetic basis [1,2,3,4,5,6]. Studies of leaf development in particular have led to major advances in identifying both causal genes and regulatory sequences for the diversification of form and the broader logic through which morphological evolution proceeds [2,7,8,9,10,11]. These efforts were underpinned by the development of essential genetic resources, including genetic and cytogenetic maps, a reference genome sequence, recombinant inbred populations, and of a stable gene transformation protocol by floral dipping [12,13,14]. As genome editing has opened considerable new potential for efforts to connect genotype to phenotype through development and evolution [15], it became important to set up a CRISPR/Cas9 genome editing in this system. To develop and evaluate a protocol for applying CRISPR/Cas9-mediated targeted mutagenesis in C. hirsuta, we selected its RCO gene as a first target. A previously identified rco mutant allele causes an easy to observe leaf phenotype with minimal pleiotropic effects elsewhere in the shoot [2].
2. Results
Two sgRNAs were designed to guide the SpCas9 endonuclease to the RCO locus in order to generate additional alleles with the potential to uncover new aspects of RCO function (Figure 1).
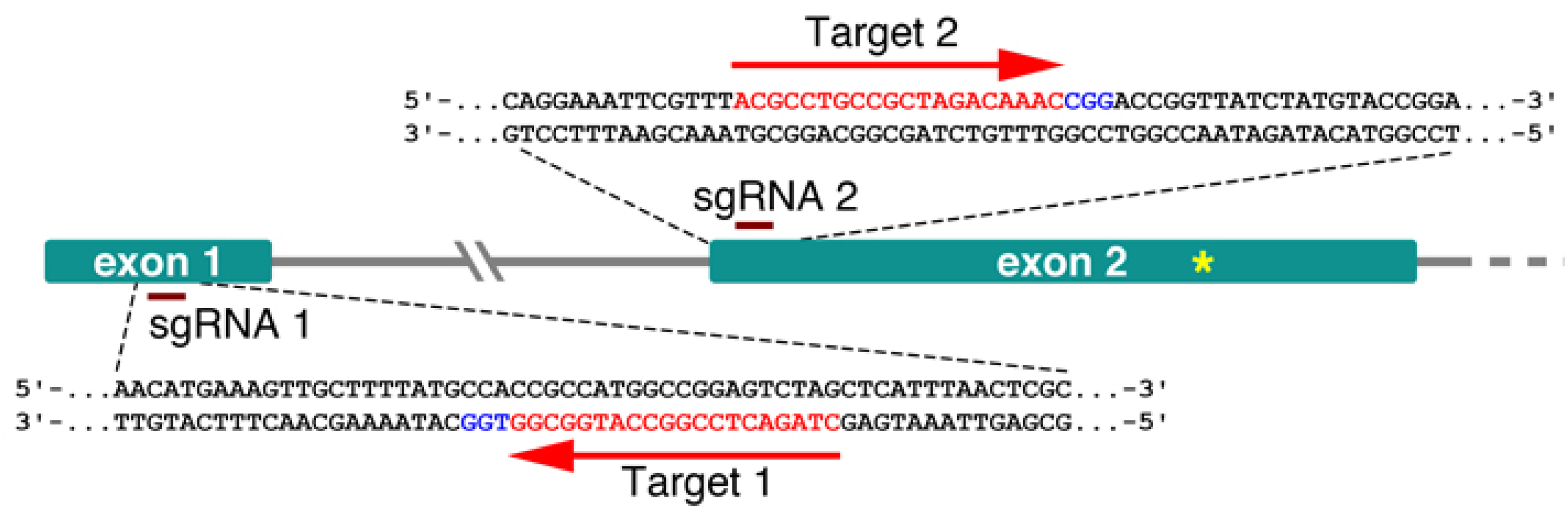
To ensure the high expression of the SpCas9 protein in Cardamine, we focused on two known strong constitutive promoters: CaMV 35S (p35S) and ubiquitin. The first vector construct carrying the 35S promoter driving the expression of a human codon-optimized Cas9 was assembled using the Golden Gate cloning method [16], with minor modifications. In short, level 1 vectors pICSL11017 (pICH47732::NOSp-BAR-NOST, Addgene no. 51145) and pICH47742::2×35S-5′UTR- hCas9(STOP)-NOST (Addgene no. 49771) and a construct carrying the two sgRNAs, synthesized in tandem (GenScript), each with its own A. thaliana U6 RNA polIII promoter, were assembled into the level 2 vector pAGM4723 (Addgene no. 48015). The second construct carrying the ubiquitin promoter has been previously described [17] and takes advantage of the Gateway cloning system. The binary Gateway-compatible destination vector, pDE-Cas9, contains the ubiquitin 4-2 promoter from Petroselinum crispum (parsley; pPcUbi4-2) driving the expression of an Arabidopsis codon-optimized Cas9. With the help of a Gateway-compatible entry vector, the same two sgRNAs, were transferred into pDE-Cas9 through a Gateway LR reaction. Although both promoters are considered constitutive, neither of them shows uniformly high activity in plants. The CaMV 35S promoter in particular, is weakly expressed in pollen and marginally in embryogenic cells [18]. The position of each sgRNA used in this work is illustrated in Figure 1.
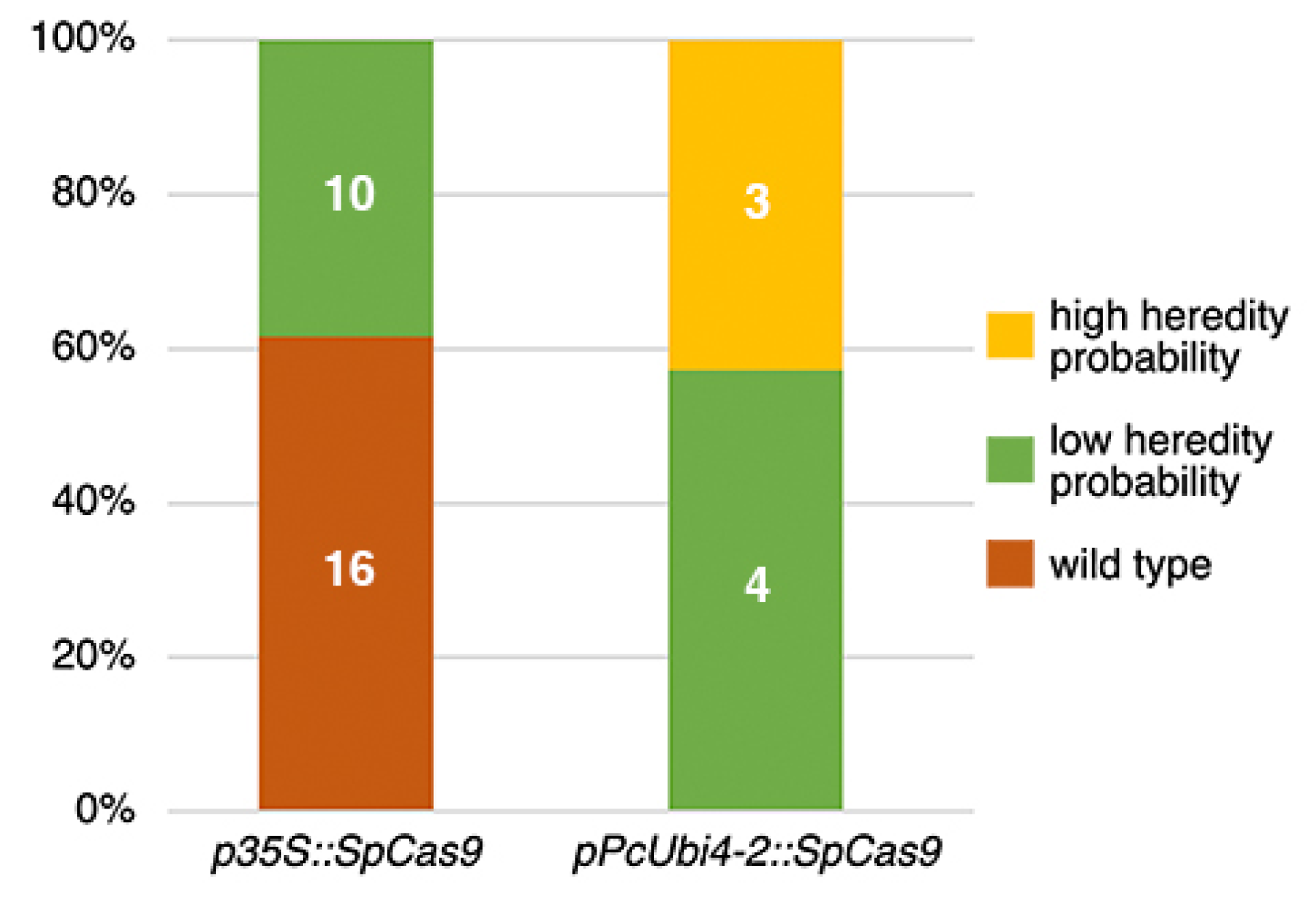
Both vector constructs were transformed into C. hirsuta Ox by means of the floral dip method [14,19] and their editing efficiency was estimated in the T1 generation using the Tracking of Indels by DEcomposition (TIDE) software [20,21]. To get a first impression of how many heritable mutations could be expected, genomic DNA was extracted from the primary inflorescences of randomly picked T1 plants. Primers to isolate the targeted region were designed following the instructions on the website, and amplified PCR products were further sequenced. The TIDE software compares the sequence trace data from a candidate T1 mutant against the wild type and estimates the indel spectrum and editing efficiency. Candidate mutations were found only for the sgRNA 1 target site, ca. 50 nt downstream of the RCO ATG-start codon (Figure 2).
In parallel, the segregation rates of all T1 plants were scored on a selective medium for the presence of the T-DNA-conferred resistance. Only T2 plants with a segregation rate of 3:1 were further analyzed to guarantee the presence of only one copy of Cas9 and thus facilitate the identification of Cas9-free T2 mutants. Of the segregating lines, DNA extracted from the leaves of individual plants was tested by PCR for the absence of Cas9. In the latter case, a second PCR reaction was performed to cover the sgRNA-targeted regions, determine their sequence and analyze using the PolyPeakParser software [22,23] to pinpoint the nature of the generated mutation, e.g., heterozygous, homozygous, or still wild type. A summary of the total number of plants analyzed and their zygosity is presented in Table 1.
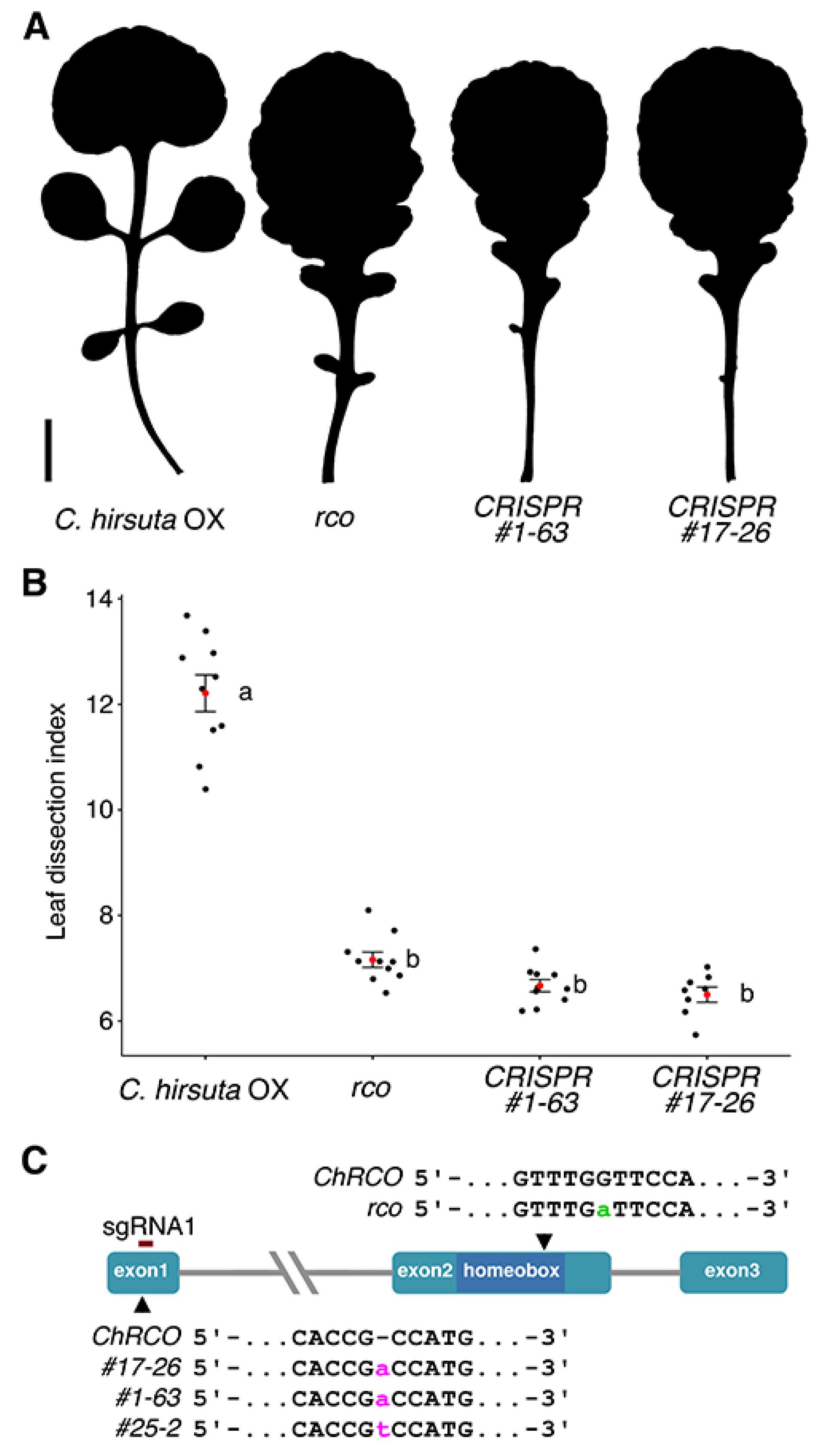
Phenotypic and genotypic analysis of the Cas9-free homozygous mutant progeny from two independent pPcUbi4-2::Cas9 lines, resp. #1-63 and #17-26, and one p35S::SpCas9 derived line, #25-2, is presented in Figure 3. The leaf shape of all three lines (only #1-63 and #17-26 shown) clearly differs from the wild type (Figure 3A), e.g., much less dissected (Figure 3B), but is virtually indistinguishable from those of the previously isolated rco mutant. Consistent with this, the new CRISPR mutants were found to be allelic to the original rco mutant allele in an allelism test.
To check for the presence of possible off-target mutations, whole genomes of the three respective mutants were sequenced in depth and compared against the C. hirsuta Ox reference. For both guide RNAs, no mutations (Indels or SNPs) were found in four off-target intervals, including ChLMI1-LIKE3 (CARHR209480) predicted by Cas-OFFinder9 (v2.4) [24,25], with an edit distance of up to 4 bp. Within the intended location in the RCO gene (CARHR209490) an insertion of an A was found in samples #1-63 and #17-26 and an insertion of a T in #25-2 as depicted in Figure 3C. As expected, the respective genotypes lacked the T-DNA insertion containing Cas9, were homozygous for the mutant alleles and the insertions located within the sgRNA 1 target site, 4 nt upstream of the PAM site (Figure 1).
3. Discussion
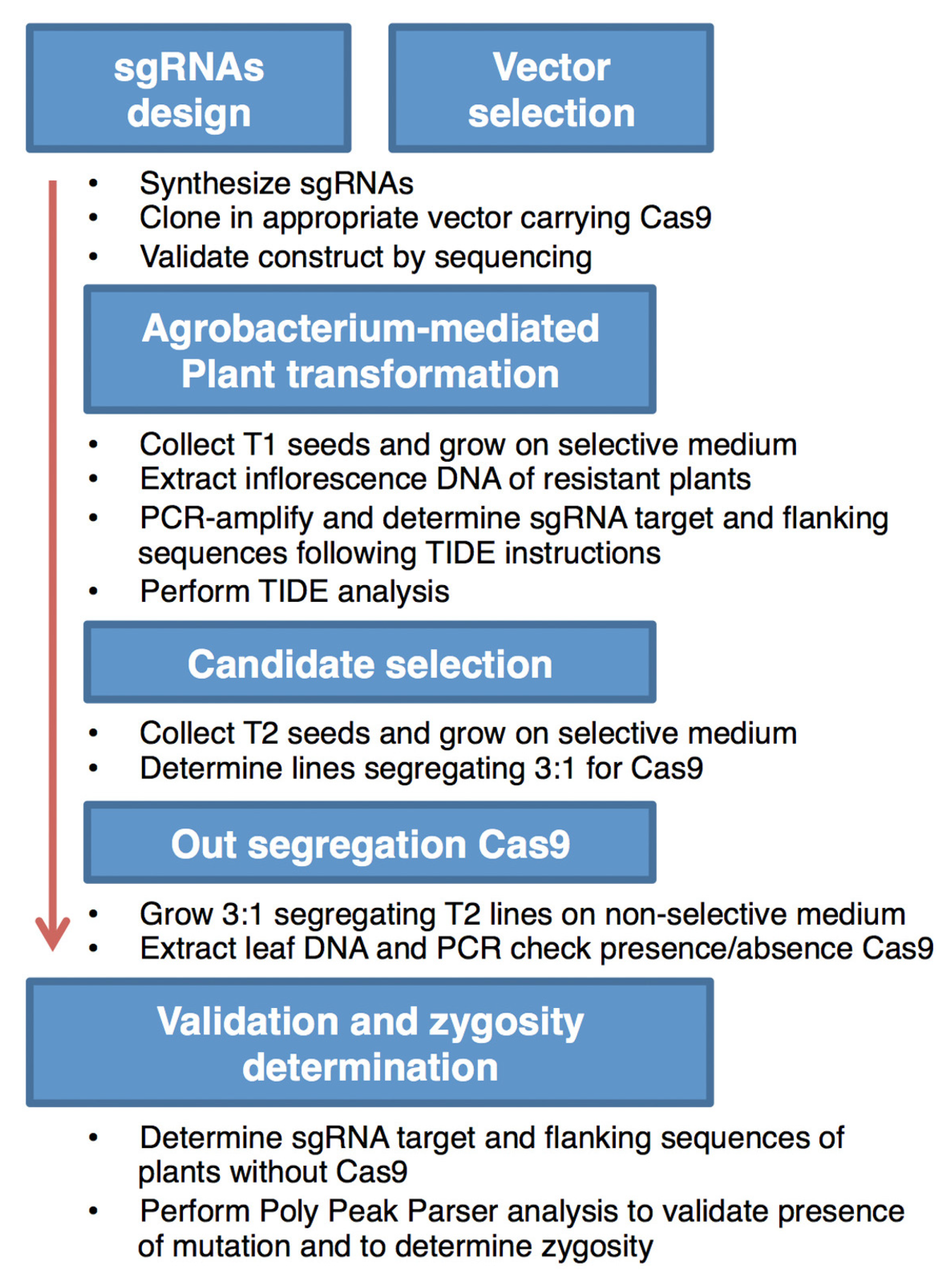
Using two available and previously described CRISPR/Cas9 expression constructs and following the protocol, as depicted in Figure 4, we successfully generated targeted mutations in the form of 1 bp insertions in the RCO locus of C. hirsuta.
For the construct carrying p35S::SpCas9, such mutations could be detected only in ca. 40% of the T1 plants which, moreover, all had low probability of heredity, i.e., present with low frequency in reproductive tissue. In this respect, the Arabidopsis codon-optimized Cas9 driven by the parsley PcUbi4-2 promoter clearly performed better. For the latter, all tested T1 plants carried targeted mutations with ca. 60% of these with low and ca. 40% with a high probability of heredity. The 0.2% versus the 3.8% homozygous and Cas9 free mutant plants found in the T2 generation supported this difference in efficiency and heredity.
Despite the relatively low number of 33 T1 plants tested for both constructs in total, it is noteworthy that targeted mutations could be detected for sgRNA 1 only. The reason for this difference in sgRNA efficiency is unclear, but it endorses the general advice to design and test multiple sgRNAs to maximize the chance of mutating a target gene. In conclusion, we provided a workflow for genome editing that will increase the versatility of C. hirsuta as a model for comparative studies as well as additional alleles of rco which will help further dissection of genetic pathways determining leaf shape.
4. Materials and Methods
4.1. Plant Material, Vector Constructs, and Cardamine hirsuta Transformation
C. hirsuta plants, Oxford (Ox) accession, were grown in long day conditions (16 h light) in the greenhouse. The origin of the plasmid vectors used, the design and synthesis of the sgRNAs, and their assembly using either the Golden Gate or Gateway cloning method has been described with the appropriate references in Section 3 of the main text. All vectors were transformed into C. hirsuta Ox by Agrobacterium tumefaciens (strain GV3101)-mediated floral dip, as previously described [14,19].
4.2. Primers Used for PCR Analysis
Primer pairs (forward/reverse) used to PCR-amplify sgRNA target regions:
ChRCO_sgRNA1 TCGTATGGTCCAACAAAACC/GTGAAATTTAACGGCCACTTAC
ChRCO_sgRNA2 GTGATTCTTCCGCTTTCTCTTC/GAACCAAACCGCTACCTGAC
For TIDE analysis of T1 plants:
TIDE_sgRNA1 CACCTCCAAAAGACAGTATAGAAGA/TAACTGCTGCCTAGGAATGTTT
TIDE_sgRNA2 GTTTTTCTTGGGAACCAGCTC/CTCTCAGCTTATTCACCTGCATTAT
To check presence/absence of Cas9:
T2Gateway GTATGTATATATGTAGATCTGG/GAAGTTAGACTTGAAGTTAGG
T2GoldenGate CTAGATCGACGCTTGCTGAA/TCGGTATTGCCCAGAACTTT
4.3. Leaf Shape Phenotyping
Digitized leaf silhouettes of developed rosette leaves were obtained with the help of a flatbed scanner. The leaf silhouette area (A) and perimeter (P) were determined within FIJI [26] and used to calculate the leaf dissection index, P/sqrt(A).
4.4. Genome Sequencing and Off-Target Analysis
Genome DNA sequencing was performed by the Max Planck-Genome-centre Cologne, Germany (https://mpgc.mpipz.mpg.de/home/), on an Illumina HiSeq3000, and the data quality of fastq files was assessed using FastQC (v0.11.7) [27]. Multiple fastq files for each individual sample were combined, and clumpify.sh from the BBMap (v38.34) package [28] was used to remove duplicates before mapping. Trim_galore (v0.4.3) [29] was used to quality trim and remove adapters. Mapping was performed using bwa (v0.7.17-r1188) [30]. Picard (v2.20.8) (http://broadinstitute.github.io/picard/) was used to mark optical duplicates. Indel realignment was performed using the Genome Analysis Toolkit GATK (v3.8-0-ge9d806836) [31] and SNP and Indel calling done with VarScan (v2.4.2) [32]. The pileup of reads, which is an input for VarScan, was generated using samtools (v1.9) [33]. The resulting VCF (Variant Call Format) file for Indels was run through bcftools norm (v1.9) [34] to split multiallelic sites in separate entries. The off-target locations were determined using an offline version of Cas-OFFinder (v2.4) [25] with an edit distance of up to 4 bp. Bcftools (v1.9) was used to check for overlaps of entries in VCF files with off-target locations. The variants in the VCF files were annotated using snpEff (v4.4) [35]. The database for snpEff annotation was created from the annotation file available for C. hirsuta found at http://chi.mpipz.mpg.de/download/annotations/carhr38.gff. A fasta file with the reference C. hirsuta genome sequence can be found at http://chi.mpipz.mpg.de/download/sequences/chi_v1.fa.
Author Contributions
Conceptualization, M.T. and P.H.; methodology, C.L.A.K. and P.H.; validation, B.P. and S.L.; investigation, C.L.A.K. and P.H.; resources, M.T. and P.H.; writing, C.L.A.K., M.T. and P.H.; visualization, C.L.A.K., S.L. and P.H.; supervision, M.T. and P.H.; project administration, P.H.; funding acquisition, M.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a core grant from the Max Planck Society to M.T. and by grant 031B0189B from the German Federal Ministry of Education and Research (BMBF) to M.T. in the context of the project “Enhancing Crop Photosynthesis (EnCroPho)” carried out by B.P.
Acknowledgments
We thank Holger Puchta for providing the pDE-Cas9 vector construct, Saurabh Pophaly for technical assistance with bioinformatics analysis and the Max Planck-Genome-Centre Cologne for performing DNA sequencing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hay, A.; Tsiantis, M. Cardamine hirsuta: A comparative view. Curr. Opin. Genet. Dev. 2016, 39, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlad, D.; Kierzkowski, D.; Rast, M.I.; Vuolo, F.; Dello Ioio, R.; Galinha, C.; Gan, X.; Hajheidari, M.; Hay, A.; Smith, R.S.; et al. Leaf shape evolution through duplication, regulatory diversification, and loss of a homeobox gene. Science 2014, 343, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Vuolo, F.; Kierzkowski, D.; Runions, A.; Hajheidari, M.; Mentink, R.A.; Gupta, M.D.; Zhang, Z.; Vlad, D.; Wang, Y.; Pecinka, A.; et al. LMI1 homeodomain protein regulates organ proportions by spatial modulation of endoreduplication. Genes Dev. 2018, 32, 1361–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofhuis, H.; Moulton, D.; Lessinnes, T.; Routier-Kierzkowska, A.L.; Bomphrey, R.J.; Mosca, G.; Reinhardt, H.; Sarchet, P.; Gan, X.; Tsiantis, M.; et al. Morphomechanical innovation drives explosive seed dispersal. Cell 2016, 166, 222–233. [Google Scholar] [CrossRef] [Green Version]
- Monniaux, M.; Pieper, B.; McKim, S.M.; Routier-Kierzkowska, A.L.; Kierzkowski, D.; Smith, R.S.; Hay, A. The role of APETALA1 in petal number robustness. eLife 2018, 7, e39399. [Google Scholar] [CrossRef]
- Nikolov, L.A.; Shushkov, P.; Nevado, B.; Gan, X.; Al-Shehbaz, I.A.; Filatov, D.; Bailey, C.D.; Tsiantis, M. Resolving the backbone of the Brassicaceae phylogeny for investigating trait diversity. New Phytol. 2019, 222, 1638–1651. [Google Scholar] [CrossRef] [Green Version]
- Hay, A.; Tsiantis, M. The genetic basis for differences in leaf form between Arabidopsis thaliana and its wild relative Cardamine hirsuta. Nat. Genet. 2006, 38, 942–947. [Google Scholar] [CrossRef]
- Blein, T.; Pulido, A.; Vialette-Guiraud, A.; Nikovics, K.; Morin, H.; Hay, A.; Johansen, I.E.; Tsiantis, M.; Laufs, P. A conserved molecular framework for compound leaf development. Science 2008, 322, 1835–1839. [Google Scholar] [CrossRef] [Green Version]
- Rast-Somssich, M.I.; Broholm, S.; Jenkins, H.; Canales, C.; Vlad, D.; Kwantes, M.; Bilsborough, G.; Dello Ioio, R.; Ewing, R.M.; Laufs, P.; et al. Alternate wiring of a KNOXI genetic network underlies differences in leaf development of A. thaliana and C. hirsuta. Genes Dev. 2015, 29, 2391–2404. [Google Scholar] [CrossRef] [Green Version]
- Vuolo, F.; Mentink, R.A.; Hajheidari, M.; Bailey, C.D.; Filatov, D.A.; Tsiantis, M. Coupled enhancer and coding sequence evolution of a homeobox gene shaped leaf diversity. Genes Dev. 2016, 30, 2370–2375. [Google Scholar] [CrossRef]
- Kierzkowski, D.; Runions, A.; Vuolo, F.; Strauss, S.; Lymbouridou, R.; Routier-Kierzkowska, A.L.; Wilson-Sánchez, D.; Jenke, H.; Galinha, C.; Mosca, G.; et al. A Growth-Based Framework for Leaf Shape Development and Diversity. Cell 2019, 177, 1405–1418. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Hay, A.; Kwantes, M.; Haberer, G.; Hallab, A.; Dello Ioio, R.; Hofhuis, H.; Pieper, B.; Cartolano, M.; Neumann, U.; et al. The Cardamine hirsuta genome offers insight into the evolution of morphological diversity. Nat. Plants 2016, 2, 16167–16171. [Google Scholar] [CrossRef] [PubMed]
- Cartolano, M.; Pieper, B.; Lempe, J.; Tattersall, A.; Huijser, P.; Tresch, A.; Darrah, P.R.; Hay, A.; Tsiantis, M. Heterochrony underpins natural variation in Cardamine hirsuta leaf form. Proc. Natl. Acad. Sci. USA 2015, 112, 10539–10544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, A.S.; Pieper, B.; Cooke, E.; Mandáková, T.; Cartolano, M.; Tattersall, A.D.; Dello Ioio, R.; McGowan, S.J.; Barkoulas, M.; Galinha, C.; et al. Cardamine hirsuta: A versatile genetic system for comparative studies. Plant. J. 2014, 78, 1–5. [Google Scholar] [CrossRef]
- Gilles, A.F.; Averof, M. Functional genetics for all: Engineered nucleases, CRISPR and the gene editing revolution. EvoDevo 2014, 5, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.; Gruetzner, R.; Werner, S.; Engler, C.; Marillonnet, S. Assembly of designer TAL effectors by Golden Gate cloning. PLoS ONE 2011, 6, e19722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauser, F.; Schiml, S.; Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant. J. 2014, 79, 348–359. [Google Scholar] [CrossRef]
- Callis, J.; Raasch, J.A.; Vierstra, R.D. Ubiquitin extension proteins of Arabidopsis thaliana. Structure, localization, and expression of their promoters in transgenic tobacco. J. Biol. Chem. 1990, 265, 12486–12493. [Google Scholar]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant. J. 1998, 16, 735–743. [Google Scholar] [CrossRef] [Green Version]
- TIDE. Available online: https://tide.nki.nl/ (accessed on 1 December 2019).
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Poly Peak Parser. Available online: http://yosttools.genetics.utah.edu/PolyPeakParser/ (accessed on 1 December 2019).
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Su, Y.C.; Smith, M.; Yost, H.J. Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev. Dynam 2014, 243, 1632–1636. [Google Scholar] [CrossRef] [Green Version]
- Cas-OFFinder. Available online: http://www.rgenome.net/cas-offinder/ (accessed on 1 December 2019).
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics-FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 December 2019).
- BBMap. SourceForge. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 19 December 2019).
- Babraham Bioinformatics - Trim Galore! Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 19 December 2019).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Schematic representation of the ChRCO locus targeted for mutagenesis. The two sgRNA complementary sequences are shown in red, and the PAM site is shown in blue. The yellow asterisk in exon 2 marks the position of the premature stop codon causing the first described rco mutant allele.
Figure 1.
Schematic representation of the ChRCO locus targeted for mutagenesis. The two sgRNA complementary sequences are shown in red, and the PAM site is shown in blue. The yellow asterisk in exon 2 marks the position of the premature stop codon causing the first described rco mutant allele.

Figure 2.
TIDE analysis of T1 Cardamine plants to estimate CRISPR heritable mutations in the sgRNA 1 target site. Plants with 50% or more wild-type sequence traces are classified either as carrying targeted mutations with ‘low heredity probability’, or as ‘wild type’ if indels are present with less than 15%.
Figure 2.
TIDE analysis of T1 Cardamine plants to estimate CRISPR heritable mutations in the sgRNA 1 target site. Plants with 50% or more wild-type sequence traces are classified either as carrying targeted mutations with ‘low heredity probability’, or as ‘wild type’ if indels are present with less than 15%.

Figure 3.
Leaf shape comparison between wild-type and rco mutant lines. (A) Representative silhouettes of the fifth rosette leaf of the C. hirsuta wild type, the original rco mutant [2], and of two independently CRISPR-derived rco mutant lines. Scale bar: 1 cm; (B) Dissection index of the fifth rosette leaf. A red dot indicates the mean and error bars ± 1SD. Significance groups are determined based on ANOVA and Tukey’s HSD test for multiple pairwise comparisons. Only pairwise comparisons involving different groups, labeled a-b, are significantly different below the 5% level; (C) a schematic representation of the RCO locus. Mutations due to single nucleotide insertions in three independently CRISPR-derived alleles are indicated in lower case magenta. The previously known ‘non-CRISPR’ mutation in the second exon is shown in lower case green.
Figure 3.
Leaf shape comparison between wild-type and rco mutant lines. (A) Representative silhouettes of the fifth rosette leaf of the C. hirsuta wild type, the original rco mutant [2], and of two independently CRISPR-derived rco mutant lines. Scale bar: 1 cm; (B) Dissection index of the fifth rosette leaf. A red dot indicates the mean and error bars ± 1SD. Significance groups are determined based on ANOVA and Tukey’s HSD test for multiple pairwise comparisons. Only pairwise comparisons involving different groups, labeled a-b, are significantly different below the 5% level; (C) a schematic representation of the RCO locus. Mutations due to single nucleotide insertions in three independently CRISPR-derived alleles are indicated in lower case magenta. The previously known ‘non-CRISPR’ mutation in the second exon is shown in lower case green.

Figure 4.
Simple flow diagram of the CRISPR/Cas9-mediated mutagenesis process, as followed in C. hirsuta in order to generate novel rco mutant alleles.
Figure 4.
Simple flow diagram of the CRISPR/Cas9-mediated mutagenesis process, as followed in C. hirsuta in order to generate novel rco mutant alleles.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
T2 analysis to identify homozygous CRISPR mutants lacking Cas9.
| Construct | Segregating Lines Identified | Plants Screened | Cas9 Free Homozygous Mutants |
|---|---|---|---|
| p35S::SpCas9 | 5 | 478 | 1 (0.2%) |
| pPcUbi4-2::SpCas9 | 2 | 159 | 6 (3.8%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Alvim Kamei, C.L.; Pieper, B.; Laurent, S.; Tsiantis, M.; Huijser, P. CRISPR/Cas9-Mediated Mutagenesis of RCO in Cardamine hirsuta. Plants 2020, 9, 268. https://doi.org/10.3390/plants9020268
AMA Style
Alvim Kamei CL, Pieper B, Laurent S, Tsiantis M, Huijser P. CRISPR/Cas9-Mediated Mutagenesis of RCO in Cardamine hirsuta. Plants. 2020; 9(2):268. https://doi.org/10.3390/plants9020268
Chicago/Turabian StyleAlvim Kamei, Claire Lessa, Bjorn Pieper, Stefan Laurent, Miltos Tsiantis, and Peter Huijser. 2020. "CRISPR/Cas9-Mediated Mutagenesis of RCO in Cardamine hirsuta" Plants 9, no. 2: 268. https://doi.org/10.3390/plants9020268
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.