
Tomato Domestication Affects Potential Functional Molecular Pathways of Root-Associated Soil Bacteria

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
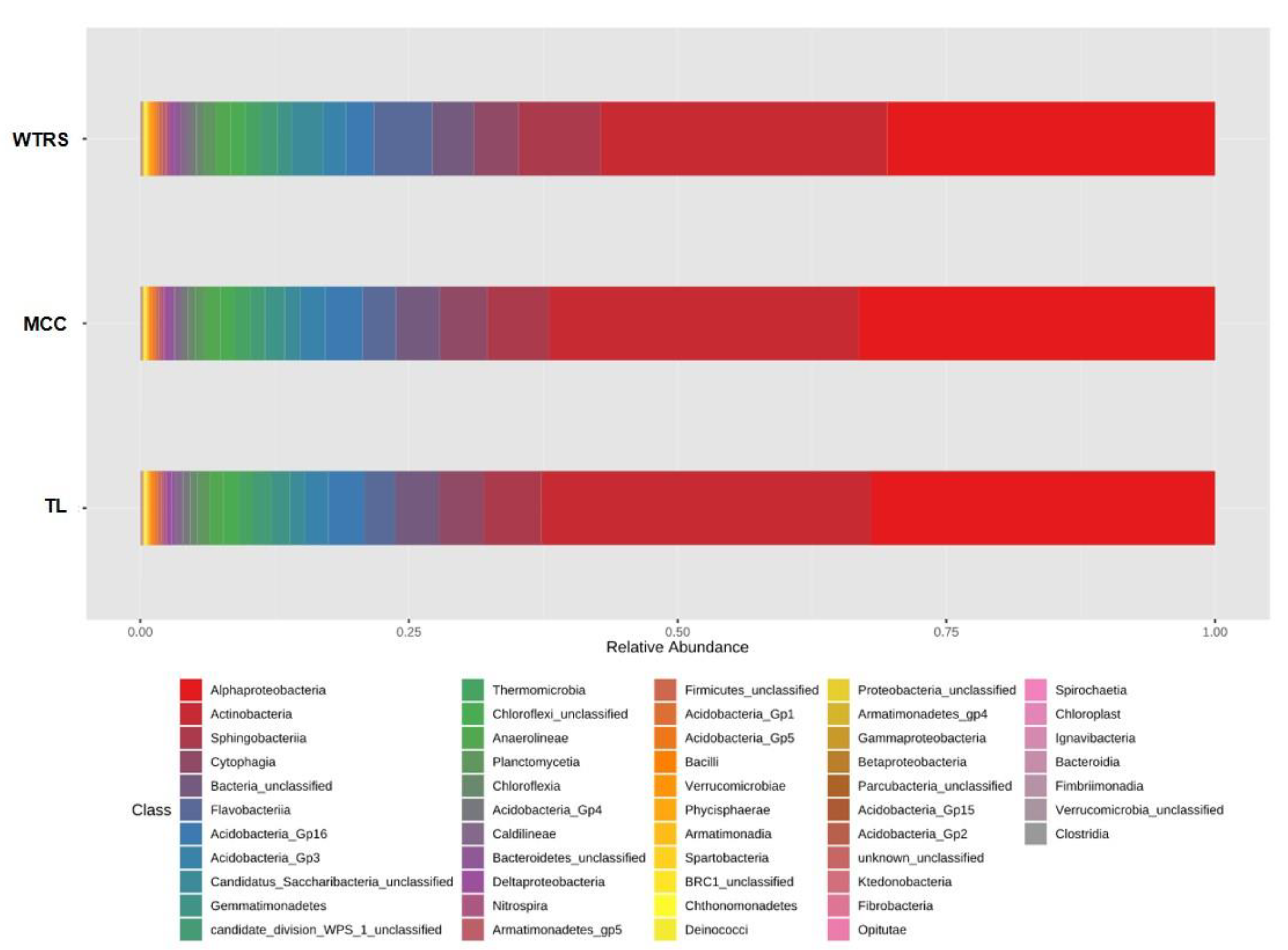
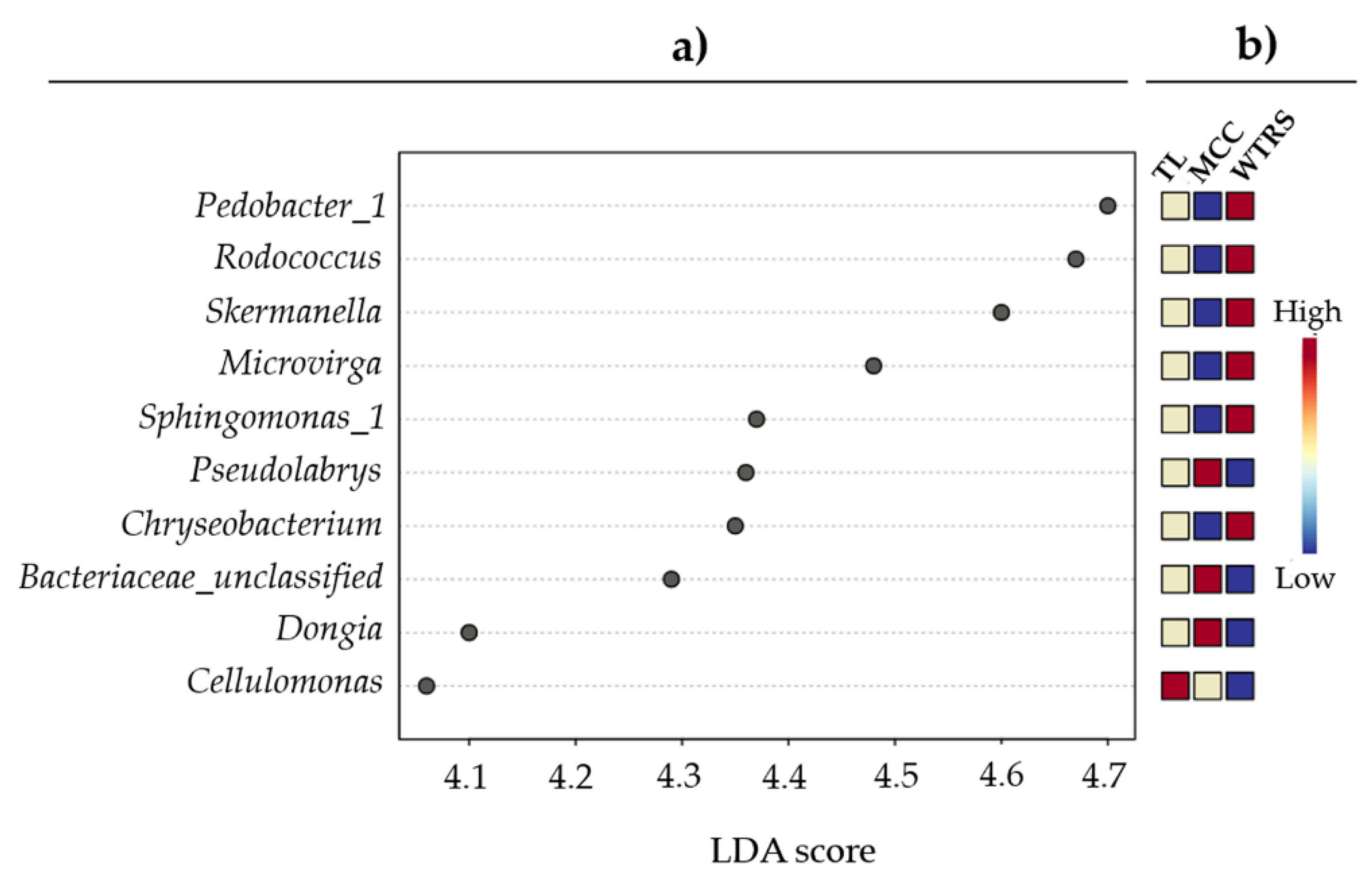
2.1. Bacterial Community Structure
2.2. Bacterial Community Functional Analysis
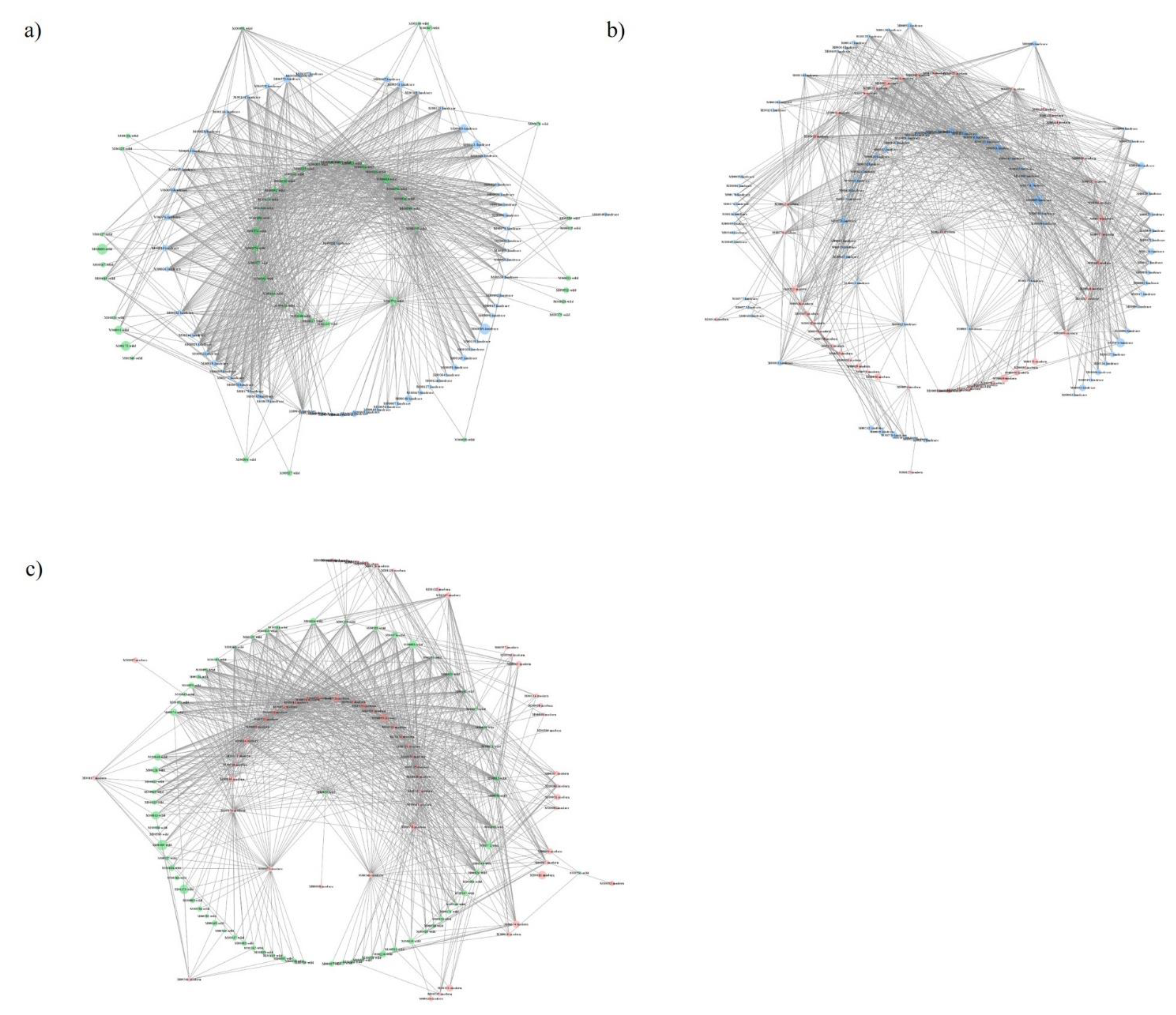
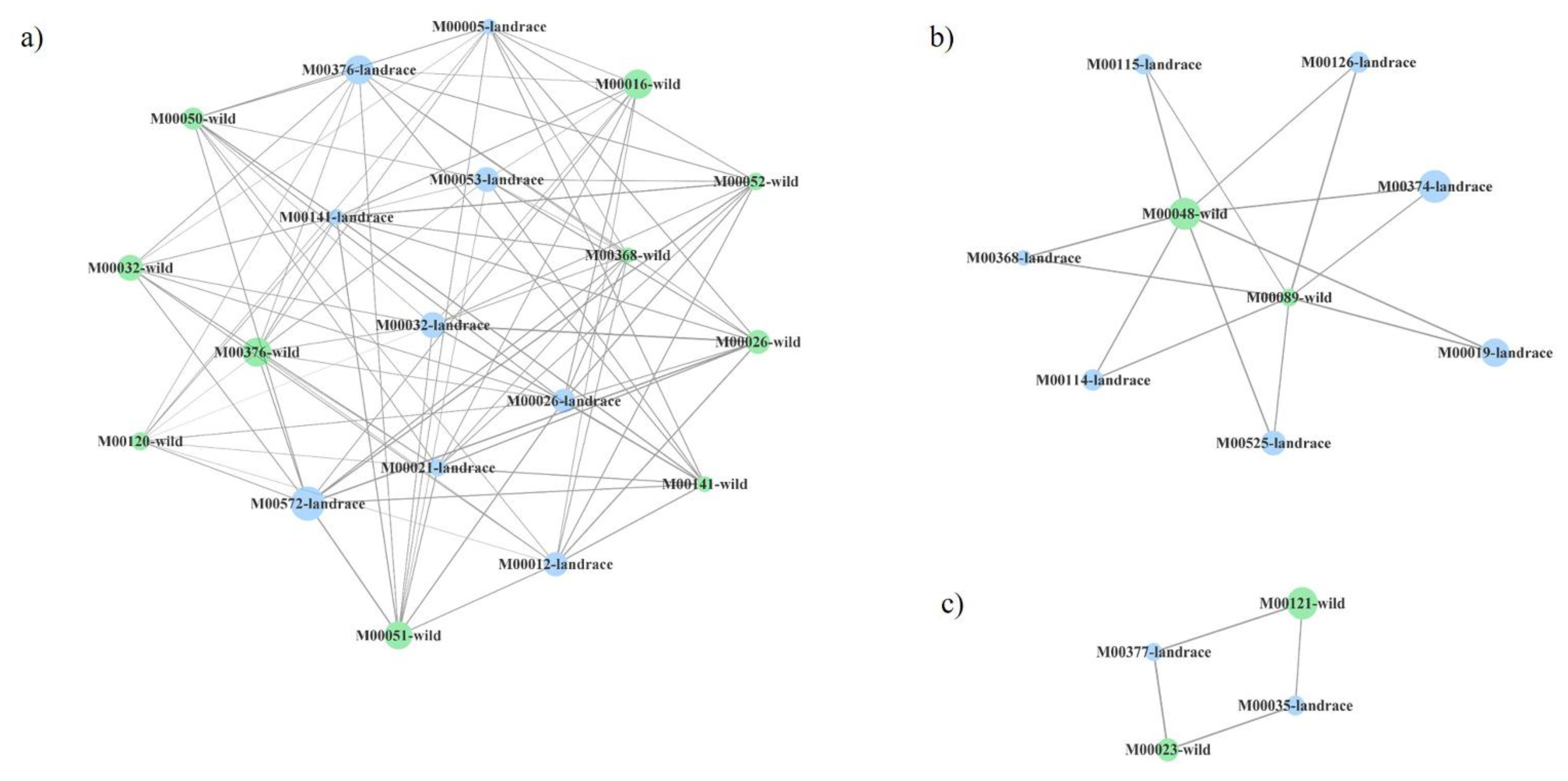
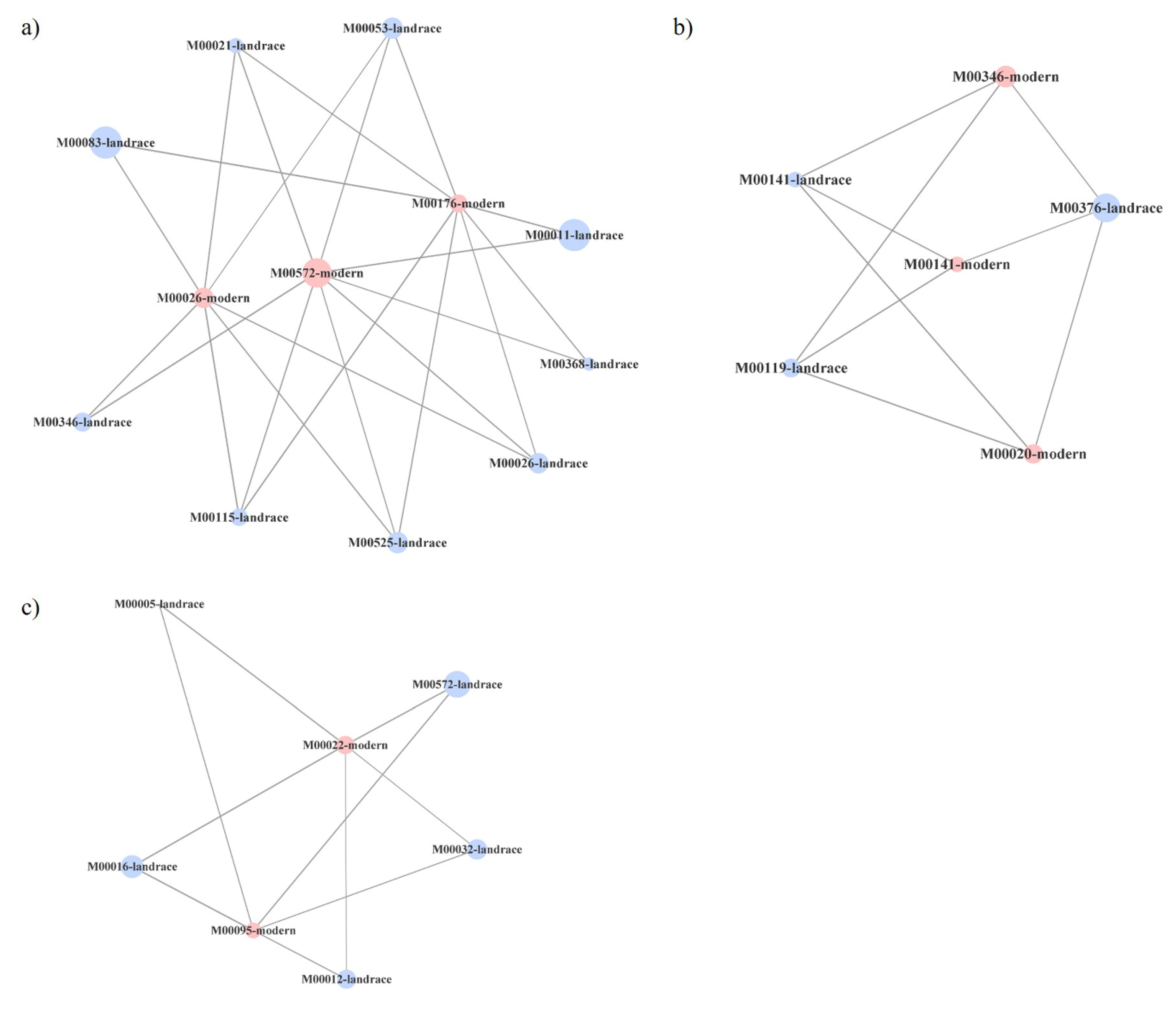
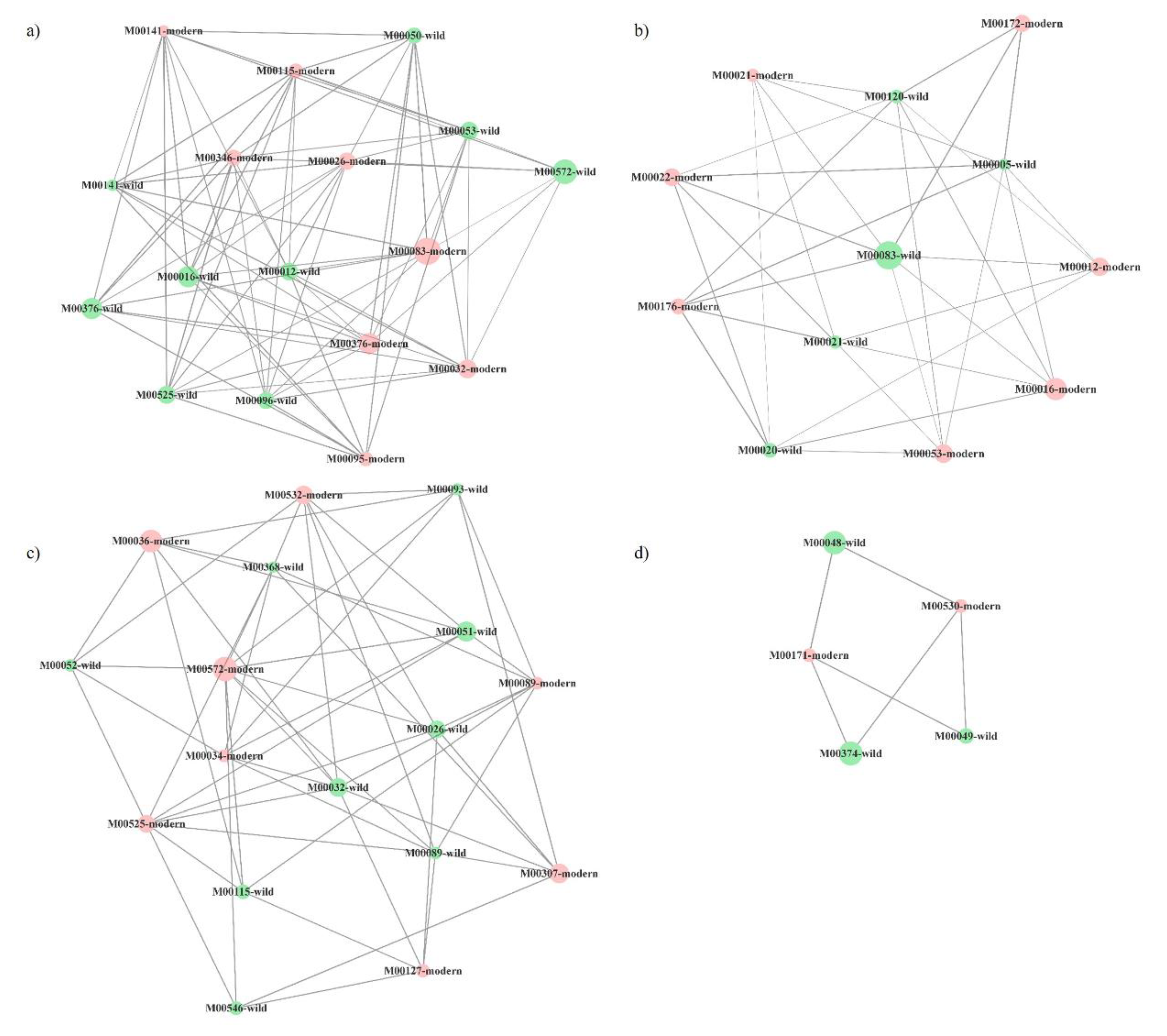
2.3. Functional Networks of KEGG Orthologous Groups
3. Discussion
4. Materials and Methods
4.1. Field Experiment
4.2. Chemical Characteristics of the Soil
4.3. Molecular Analyses of Soil Bacteria
4.4. Predictive Metagenomics Profiling
4.5. Functional Networks
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots shaping their microbiome, global hotspots for microbial activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef] [PubMed]
- Cordovez, V.; Dini-Andreote, F.; Carrión, V.J.; Raaijmakers, J.M. Ecology and Evolution of Plant Microbiomes. Annu. Rev. Microbiol. 2019, 73, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.H.; Bardgett, R.D.; Behan-Pelletier, V.; Herrick, J.E.; Jones, H.; Ritz, K.; Six, J.; Strong, D.R.; van der Putten, W.H. Soil Ecology and Ecosystem Services; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Houlden, A.; Timms-Wilson, T.M.; Day, M.J.; Bailey, M.J. Influence of plant developmental stage on microbial community structure and activity in the rhizosphere of three field crops. FEMS Microbiol. Ecol. 2008, 65, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Lei, S.; Xu, X.; Cheng, Z.; Xiong, J.; Ma, R.; Zhang, L.; Yang, X.; Zhu, Y.; Zhang, B.; Tian, B. Analysis of the community composition and bacterial diversity of the rhizosphere microbiome across different plant taxa. Microbiol. Open 2019, 8, e00762. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome, ecology; functions; and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant. Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions, from community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant. Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.E.; Bowles, T.M.; Gaudin, A.C.M. Using ancient traits to convert soil health into crop yield, impact of selection on maize root and rhizosphere function. Front. Plant. Sci. 2016, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Iannucci, A.; Fragasso, M.; Beleggia, R.; Nigro, F.; Papa, R. Evolution of the Crop Rhizosphere, Impact of Domestication on Root Exudates in Tetraploid Wheat (Triticum turgidum L.). Front. Plant. Sci. 2017, 8, 2124. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; de Hollander, M.; Raaijmakers, J.M. The wild side of plant microbiomes. Microbiome 2018, 6, 143. [Google Scholar] [CrossRef] [Green Version]
- Shenton, M.; Iwamoto, C.; Kurata, N.; Ikeo, K. Effect of Wild and Cultivated Rice Genotypes on Rhizosphere Bacterial Community Composition. Rice 2016, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Carrillo, J.; Ingwell, L.L.; Li, X.; Kaplan, I. Domesticated tomatoes are more vulnerable to negative plant–soil feedbacks than their wild relatives. J. Ecol. 2019, 107, 1753–1766. [Google Scholar] [CrossRef]
- Terrazas, R.A.; Balbirnie-Cumming, K.; Morris, J.; Hedley, P.E.; Russell, J.; Paterson, E.; Baggs, E.M.; Fridman, E.; Bulgarelli, D.A. A footprint of plant eco-geographic adaptation on the composition of the barley rhizosphere bacterial microbiota. Sci. Rep. 2020, 10, 12916. [Google Scholar] [CrossRef]
- Jia, Y.; Whalen, J.K. A new perspective on functional redundancy and phylogenetic niche conservatism in soil microbial communities. Pedosphere 2020, 30, 18–24. [Google Scholar] [CrossRef]
- Spor, A.; Roucou, A.; Mounier, A.; Bru, D.; Breuil, M.C.; Fort, F.; Vile, D.; Roumet, P.; Philippot, L.; Violle, C. Domestication-driven changes in plant traits associated with changes in the assembly of the rhizosphere microbiota in tetraploid wheat. Sci. Rep. 2020, 10, 12234. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome, significance of plant beneficial; plant pathogenic; and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Romero, E.; Aguirre-Noyola, J.L.; Taco-Taype, N.; Martínez-Romero, J.; Zuñiga-Dávila, D. Plant microbiota modified by plant domestication. Syst. Appl. Microbiol. 2020, 43, 126106. [Google Scholar] [CrossRef]
- Milla, R.; García-Palacios, P.; Matesanz, S. Looking at past domestication to secure ecosystem services of future croplands. J. Ecol. 2017, 105, 885–889. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, F.J. Humus Chemistry, Genesis, Composition, Reactions, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1994. [Google Scholar]
- Zhang, Y.; Yue, D.; Ma, H. Darkening mechanism and kinetics of humification process in catechol-Maillard system. Chemosphere 2015, 130, 40–45. [Google Scholar] [CrossRef]
- Tiessen, H.; Cuevas, E.; Chacon, P. The role of soil organic matter in sustaining soil fertility. Nature 1994, 371, 783–785. [Google Scholar] [CrossRef]
- Gougoulias, C.; Clark, J.M.; Shaw, L.J. The role of soil microbes in the global carbon cycle, tracking the below-ground microbial processing of plant-derived carbon for manipulating carbon dynamics in agricultural systems. J. Sci. Food Agric. 2014, 94, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Six, J.; Frey, S.D.; Thiet, R.K.; Batten, K.M. Bacterial and Fungal Contributions to Carbon Sequestration in Agroecosystems. Soil Sci. Soc. Am. J. 2006, 70, 555–569. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; de Hollander, M.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M.; Carrión, V.J. Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 2019, 7, 114. [Google Scholar] [CrossRef] [Green Version]
- Vries, F.; Wallenstein, M.D. Below-ground connections underlying above-ground food production: A framework for optimising ecological connections in the rhizosphere. J. Ecol. 2017, 105, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity, microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, T.M.; Ge, T.; Yuan, H.; Wei, X.; Wu, X.; Xiao, K.; Kumaresan, D.; Yu, S.S.; Wu, J.; Whiteley, A.S.; et al. Soil Carbon-Fixation Rates and Associated Bacterial Diversity and Abundance in Three Natural Ecosystems. Microb. Ecol. 2017, 73, 645–657. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieterse, C.M.; Leon-Reyes, A.; Van der Ent, S.; Van Wees, S.C. Networking by small-molecule hormones in plant immunity. Nat. Chem. Biol. 2009, 5, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Bari, R.; Jones, J.D. Role of plant hormones in plant defence responses. Plant. Mol. Biol. 2009, 69, 473–488. [Google Scholar] [CrossRef]
- Kenrick, P.; Crane, P. The origin and early evolution of plants on land. Nature 1997, 389, 33–39. [Google Scholar] [CrossRef]
- Pozo, M.J.; López-Ráez, J.A.; Azcón-Aguilar, C.; García-Garrido, J.M. Phytohormones as integrators of environmental signals in the regulation of mycorrhizal symbioses. New Phytol. 2015, 205, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Gruden, K.; Lidoy, J.; Petek, M.; Podpečan, V.; Flors, V.; Papadopoulou, K.K.; Pappas, M.L.; Martinez-Medina, A.; Bejarano, E.; Biere, A.; et al. Ménage à Trois, Unraveling the Mechanisms Regulating Plant-Microbe-Arthropod Interactions. Trends Plant. Sci. 2020, 25, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Eng, F.; Marin, J.E.; Zienkiewicz, K.; Gutiérrez-Rojas, M.; Favela-Torres, E.; Feussner, I. Jasmonic acid biosynthesis by fungi, derivatives; first evidence on biochemical pathways and culture conditions for production. PeerJ 2021, 9, e10873. [Google Scholar] [CrossRef] [PubMed]
- Dagorn, A.; Chapalain, A.; Mijouin, L.; Hillion, M.; Duclairoir-Poc, C.; Chevalier, S.; Taupin, L.; Orange, N.; Feuilloley, M.G. Effect of GABA, a bacterial metabolite; on Pseudomonas fluorescens surface properties and cytotoxicity. Int. J. Mol. Sci. 2013, 14, 12186–12204. [Google Scholar] [CrossRef] [Green Version]
- Somorin, Y.; Abram, F.; Brennan, F.; O’Byrne, C. The General Stress Response Is Conserved in Long-Term Soil-Persistent Strains of Escherichia coli. Appl. Environ. Microbiol. 2016, 82, 4628–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, Z.; He, P.; Ma, S.; Du, J.; Jiang, R. Construction and Analysis of Functional Networks in the Gut Microbiome of Type 2 Diabetes Patients. Genom. Proteom. Bioinform. 2016, 14, 314–324. [Google Scholar] [CrossRef] [Green Version]
- FAO. World Reference Base for Soil Resources 2014: International Soil Classification System for Naming Soils and Creating Legends for Soil Maps; Food and Agriculture Organization of the United Nations: Roma, Italy, 2015. [Google Scholar]
- Ferrero, V.; Baeten, L.; Blanco-Sanchez, L.; Planello, R.; Diaz-Pendon, J.A.; Rodríguez-Echeverría, S.; Haegeman, A.; de la Peña, E. Complex patterns in tolerance and resistance to pests and diseases underpin the domestication of tomato. New Phytol. 2020, 226, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneous analysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef]
- Vetrovský, T.; Baldrian, P.; Morais, D. SEED 2, a user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 2018, 34, 2292–2294. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE, highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur, open-source; platform-independent; community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project, improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical; functional; and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst, a web-based tool for comprehensive statistical; visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, 180–188. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun, predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet app, inference of biological association networks using Cytoscape. F1000Research 2016, 5, 1519. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape, a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.; Weisberg, S. An R Companion to Applied Regression, 3rd ed.; Sage: Thousand Oaks, CA, USA, 2019. [Google Scholar]
- Lenth, R.V. Least-squares means, The R package lsmeans. J. Stat. Soft 2016, 69, 1–33. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R, A Language and Environment for Statistical Computing; R Development Core Team: Vienna, Austria, 2017. [Google Scholar]
- RStudio Team. RStudio, Integrated Development Environment for R; RStudio Team: Boston, MA, USA, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WTRS | TL | MCC | |
|---|---|---|---|
| Shannon–Wiener Diversity Index | 5.79 ± 0.26 | 6.23 ± 0.06 | 6.18 ± 0.09 |
| Shannon Entropy | 8.35 ± 0.37 | 8.98 ± 0.08 | 8.92 ± 0.12 |
| Species Richness | 3260 ± 346 | 3727 ± 155 | 3471 ± 231 |
| Total Abundance | 52,838 ± 3338 | 59,144 ± 1702 | 57,100 ± 2964 |
| Simpson Diversity Index | 0.031 ± 0.013 | 0.010 ± 0.001 | 0.011 ± 0.002 |
| Evenness | 0.719 ± 0.021b | 0.759 ± 0.004a | 0.764 ± 0.005a |
| Chao-1 | 4453 ± 450 | 4983 ± 241 | 4619 ± 329 |
| Pathway Modules | WTRS | TL | MCC | p-Value | |
|---|---|---|---|---|---|
| Amino acid metabolism; Arginine and proline metabolism | M00015_Proline biosynthesis, glutamate =>proline | 1656a | 1609b | 1626b | 0.02086 |
| M00023_Tryptophan biosynthesis, chorismate => tryptophan | 3173b | 3234a | 3205a | 0.008801 | |
| M00037_Melatonin biosynthesis, tryptophan => serotonin => melatonin | 69b | 77a | 74ab | 0.04182 | |
| M00040_Tyrosine biosynthesis, chorismate => arogenate => tyrosine | 508a | 464b | 467b | 5.579 × 10−6 | |
| M00042_Catecholamine biosynthesis, tyrosine => dopamine => noradrenaline => adrenaline | 95b | 106a | 104ab | 0.02289 | |
| M00533_Homoprotocatechuate degradation, homoprotocatechuate => 2-oxohept-3-enedioate | 491a | 474b | 473ab | 0.04715 | |
| Amino acid metabolism; Aromatic amino acid metabolism | M00545_Trans-cinnamate degradation, trans-cinnamate => acetyl-CoA | 1048a | 1004b | 1014b | 0.001853 |
| Amino acid metabolism; Branched-chain amino acid metabolism | M00036_Leucine degradation, leucine => acetoacetate + acetyl-CoA | 7057a | 6938b | 6874b | 6.597 × 10−5 |
| Amino acid metabolism; Cysteine and methionine metabolism | M00017_Methionine biosynthesis, apartate => homoserine => methionine | 5393b | 5464a | 5422a | 0.006857 |
| M00035_Methionine degradation | 2031b | 2101a | 2065b | 6.836 × 10−5 | |
| M00338_Cysteine biosynthesis, homocysteine + serine => cysteine | 233c | 276a | 257b | 9.38 × 10−11 | |
| Amino acid metabolism; Lysine metabolism | M00031_Lysine biosynthesis, mediated by LysW, 2-aminoadipate => lysine | 81b | 106a | 98b | 1.069 × 10−5 |
| Amino acid metabolism; Other amino acid metabolism | M00118_Glutathione biosynthesis, glutamate => glutathione | 944a | 876b | 880b | 7.781 × 10−8 |
| M00027_GABA (gamma-Aminobutyrate) shunt | 2018a | 1942b | 1921b | 1.037 × 10−5 | |
| Amino acid metabolism; Polyamine biosynthesis | M00133_Polyamine biosynthesis, arginine => agmatine => putrescine => spermidine | 865b | 895a | 879ab | 0.01599 |
| M00134_Polyamine biosynthesis, arginine => ornithine => putrescine | 872a | 838b | 841b | 0.008463 | |
| M00136_GABA biosynthesis, prokaryotes, putrescine => GABA | 677a | 620b | 636b | 6.265 × 10−9 | |
| Amino acid metabolism; Serine and threonine metabolism | M00555_Betaine biosynthesis, choline => betaine | 1473a | 1377b | 1383b | 4.206 × 10−11 |
| Carbohydrate metabolism; Central carbohydrate metabolism | M00006_Pentose phosphate pathway, oxidative phase, glucose 6P => ribulose 5P | 1535ab | 1523b | 1546a | 0.04953 |
| M00077_Chondroitin sulfate degradation | 105b | 118a | 123a | 6.408 × 10−5 | |
| M00008_Entner–Doudoroff pathway, glucose-6P => glyceraldehyde-3P + pyruvate | 1993a | 1905c | 1928b | 2.838 × 10−6 | |
| M00009_Citrate cycle (TCA cycle, Krebs cycle) | 12,529a | 12,667b | 12,563a | 0.0001383 | |
| M00011_Citrate cycle, second carbon oxidation, 2-oxoglutarate => oxaloacetate | 9185b | 9286a | 9207b | 0.001811 | |
| M00003_Gluconeogenesis, oxaloacetate => fructose-6P | 5474b | 5544a | 5498b | 0.008293 | |
| M00633_Semi-phosphorylative Entner–Doudoroff pathway, gluconate/galactonate => glycerate-3P | 85b | 91ab | 95a | 0.03837 | |
| Carbohydrate metabolism; Other carbohydrate metabolism | M00061_D-Glucuronate degradation, D-glucuronate => pyruvate + D-glyceraldehyde 3P | 1694a | 1654b | 1680ab | 0.01455 |
| M00081_Pectin degradation | 113b | 129a | 132a | 6.318 × 10−5 | |
| M00114_Ascorbate biosynthesis, plants, glucose-6P => ascorbate | 2958ab | 2995a | 2949b | 0.0271 | |
| M00131_Inositol phosphate metabolism, Ins(1,3,4,5)P4 => Ins(1,3,4)P3 => myo-inositol | 1003a | 969b | 968ab | 0.02686 | |
| M00550_Ascorbate degradation, ascorbate => D-xylulose-5P | 27a | 19b | 18b | 1.195 × 10−5 | |
| M00554_Nucleotide sugar biosynthesis, galactose => UDP-galactose | 199b | 207ab | 217a | 0.005133 | |
| M00565_Trehalose biosynthesis, D-glucose 1P => trehalose | 3380b | 3603a | 3666a | 2.2 × 10−16 | |
| Energy metabolism; Carbon fixation | M00170_C4-dicarboxylic acid cycle, phosphoenolpyruvate carboxykinase type | 1302c | 1357a | 1321bc | 1.883 × 10−5 |
| M00172_C4-dicarboxylic acid cycle, NADP—malic enzyme type | 3505a | 3444b | 3445b | 0.03007 | |
| M00173_Reductive citrate cycle (Arnon-Buchanan cycle) | 10,778b | 10,891a | 10,850ab | 0.004089 | |
| M00374_Dicarboxylate-hydroxybutyrate cycle | 7259b | 7345a | 7333a | 0.01178 | |
| M00620_Incomplete reductive citrate cycle, acetyl-CoA => oxoglutarate | 2168b | 2224a | 2231a | 0.0007637 | |
| Energy metabolism;; Methane metabolism | M00344_Formaldehyde assimilation, xylulose monophosphate pathway | 913b | 944a | 942ab | 0.03158 |
| M00345_Formaldehyde assimilation, ribulose monophosphate pathway | 749b | 808a | 800a | 6.137 × 10−7 | |
| M00346_Formaldehyde assimilation, serine pathway | 3166b | 3234a | 3226ab | 0.006593 | |
| M00356_Methanogenesis, methanol => methane | 22b | 26ab | 27a | 0.03877 | |
| M00358_Coenzyme M biosynthesis | 177b | 190a | 198a | 0.0004887 | |
| M00378_F420 biosynthesis | 82b | 93a | 89ab | 0.05467 | |
| M00563_Methanogenesis, methylamine/dimethylamine/trimethylamine => methane | 465a | 434b | 464a | 3.724 × 10−6 | |
| Energy metabolism; Nitrogen metabolism | M00530_Dissimilatory nitrate reduction, nitrate => ammonia | 1864a | 1823b | 1848a | 0.01764 |
| Energy metabolism; Sulfur metabolism | M00176_Assimilatory sulfate reduction, sulfate => H2S | 2814a | 2741b | 2766ab | 0.006395 |
| Glycan metabolism; Glycosaminoglycan metabolis | M00076_Dermatan sulfate degradation | 115b | 129a | 135a | 2.073 × 10−5 |
| M00077_Chondroitin sulfate degradation | 105b | 118a | 123a | 6.408 × 10−5 | |
| M00078_Heparan sulfate degradation | 191b | 215a | 224a | 2.272 × 10−7 | |
| M00079_Keratan sulfate degradation | 475b | 526a | 547a | 1.002 × 10−12 | |
| Glycan metabolism; Lipopolysaccharide metabolism | M00060_KDO2-lipid A biosynthesis, Raetz pathway, LpxL-LpxM type | 3058b | 3132a | 3124a | 0.001684 |
| M00064_ADP-L-glycero-D-manno-heptose biosynthesis | 692b | 743a | 771a | 3.151 × 10−7 | |
| Lipid metabolism; Fatty acid metabolism | M00082_Fatty acid biosynthesis, initiation | 3785b | 3861a | 3842ab | 0.01467 |
| M00083_Fatty acid biosynthesis, elongation | 9121b | 9218a | 9214a | 0.01719 | |
| M00086_beta-Oxidation, acyl-CoA synthesis | 1699b | 1743a | 1746ab | 0.01575 | |
| Lipid metabolism; Lipid metabolism | M00113_Jasmonic acid biosynthesis | 428b | 454a | 438b | 0.002276 |
| Metabolism of cofactors and vitamins; Cofactor and vitamin metabolism | M00116_Menaquinone biosynthesis, chorismate => menaquinol | 943b | 1026a | 977b | 1.104 × 10−10 |
| M00117_Ubiquinone biosynthesis, prokaryotes, chorismate => ubiquinone | 2772a | 2703b | 2707ab | 0.01037 | |
| M00122_Cobalamin biosynthesis, cobinamide => cobalamin | 2143a | 2105b | 2153a | 0.00244 | |
| M00128_Ubiquinone biosynthesis, eukaryotes, 4-hydroxybenzoate => ubiquinone | 74a | 64b | 67ab | 0.01119 | |
| Nucleotide metabolism; Purine metabolism | M00546_Purine degradation, xanthine => urea | 2126a | 2089b | 2125a | 0.01079 |
| Xenobiotics biodegradation; Aromatics degradation | M00537_Xylene degradation, xylene => methylbenzoate | 215a | 199b | 200ab | 0.01542 |
| M00541_Benzoyl-CoA degradation, benzoyl-CoA => 3-hydroxypimeloyl-CoA | 59b | 67a | 67ab | 0.02392 | |
| M00548_Benzene degradation, benzene => catechol | 27a | 20b | 21b | 0.0002254 | |
| M00551_Benzoate degradation, benzoate => catechol/methylbenzoate => methylcatechol | 124a | 108b | 110b | 0.003958 | |
| M00568_Catechol ortho-cleavage, catechol => 3-oxoadipate | 445a | 421b | 433ab | 0.01165 | |
| M00569_Catechol meta-cleavage, catechol => acetyl-CoA/4-methylcatechol => propanoyl-CoA | 466a | 441b | 430b | 0.001843 | |
| M00637_Anthranilate degradation, anthranilate => catechol | 90a | 73b | 82b | 1.726 × 10−5 | |
| MCC:WTRS | TL:WTRS | MCC:TL | |
|---|---|---|---|
| Number of nodes | 133 | 116 | 132 |
| Number of edges | 1005 | 1003 | 1001 |
| Average number of neighbors | 15,113 | 17,293 | 15,167 |
| Network diameter | 6 | 6 | 7 |
| Network radius | 3 | 3 | 4 |
| Characteristic path length | 2.542 | 2.371 | 2.577 |
| Clustering coefficient | 0.000 | 0.000 | 0.000 |
| Network density | 0.114 | 0.150 | 0.116 |
| Network heterogeneity | 0.850 | 0.780 | 0.869 |
| Network centralization | 0.230 | 0.325 | 0.309 |
| Connected components | 1 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smulders, L.; Benítez, E.; Moreno, B.; López-García, Á.; Pozo, M.J.; Ferrero, V.; de la Peña, E.; Alcalá Herrera, R. Tomato Domestication Affects Potential Functional Molecular Pathways of Root-Associated Soil Bacteria. Plants 2021, 10, 1942. https://doi.org/10.3390/plants10091942
Smulders L, Benítez E, Moreno B, López-García Á, Pozo MJ, Ferrero V, de la Peña E, Alcalá Herrera R. Tomato Domestication Affects Potential Functional Molecular Pathways of Root-Associated Soil Bacteria. Plants. 2021; 10(9):1942. https://doi.org/10.3390/plants10091942
Chicago/Turabian StyleSmulders, Lisanne, Emilio Benítez, Beatriz Moreno, Álvaro López-García, María J. Pozo, Victoria Ferrero, Eduardo de la Peña, and Rafael Alcalá Herrera. 2021. "Tomato Domestication Affects Potential Functional Molecular Pathways of Root-Associated Soil Bacteria" Plants 10, no. 9: 1942. https://doi.org/10.3390/plants10091942