
Experimental and Computational Investigation of the Oxime Bond Stereochemistry in c-Jun N-terminal Kinase 3 Inhibitors 11H-Indeno[1,2-b]quinoxalin-11-one Oxime and Tryptanthrin-6-oxime
 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. NMR and X-ray Diffraction Equipment
2.2. Computational Chemistry Details
2.2.1. NMR Chemical Shift Calculations
2.2.2. Thermodynamic Calculations for IQ-1 and Trp-Ox
2.3. Preparation of Single Crystals for X-ray Diffraction Analysis
2.3.1. 11H-Indeno[1,2-b]quinoxalin-11-one Oxime (IQ-1)
2.3.2. Tryptanthrin-6-oxime (Trp-Ox)
2.3.3. Tryptanthrin-6-oxime Pyridine Solvate (Trp-Ox·Py)
3. Results and Discussion
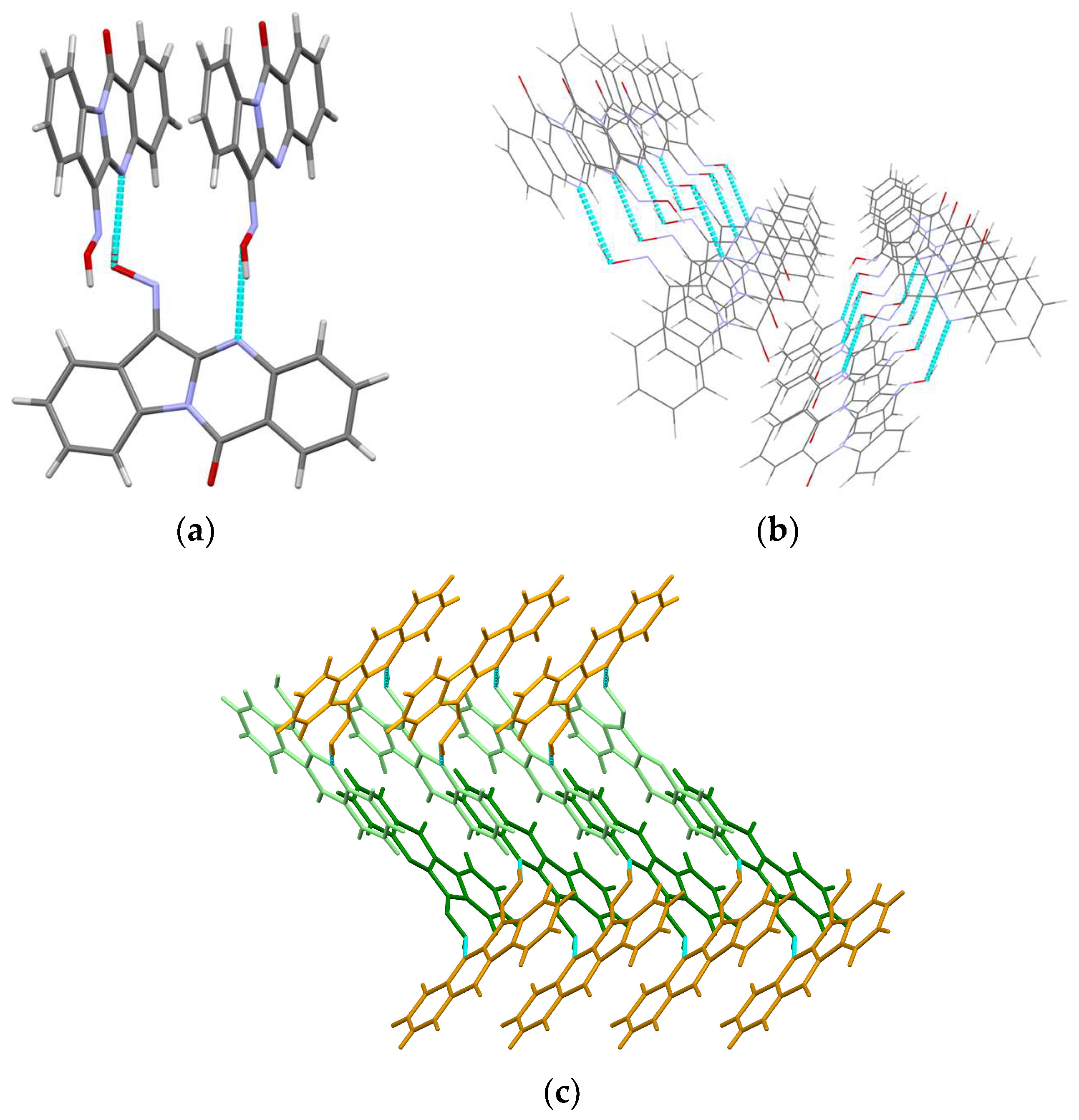
3.1. X-ray Crystal Structures
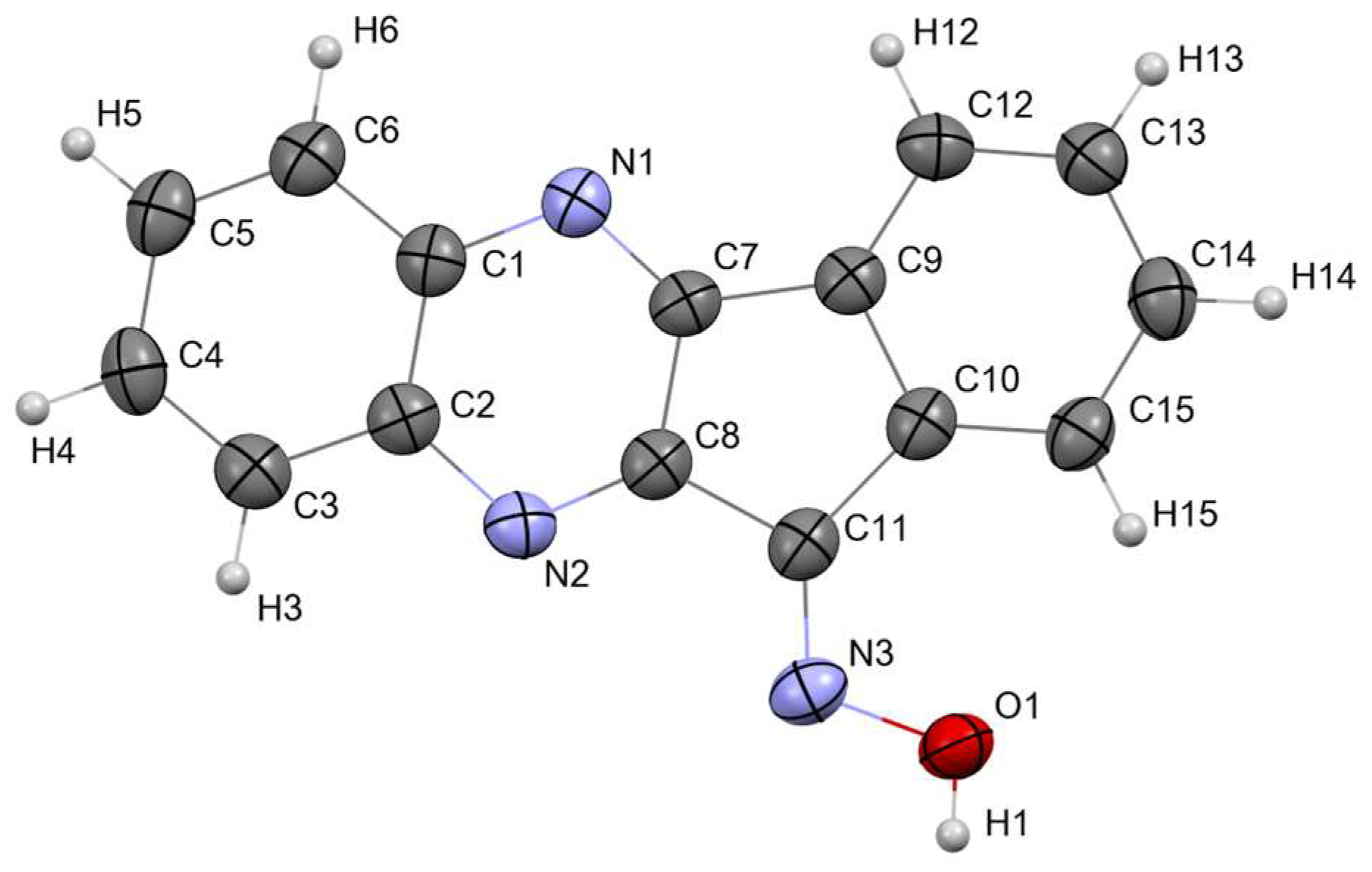
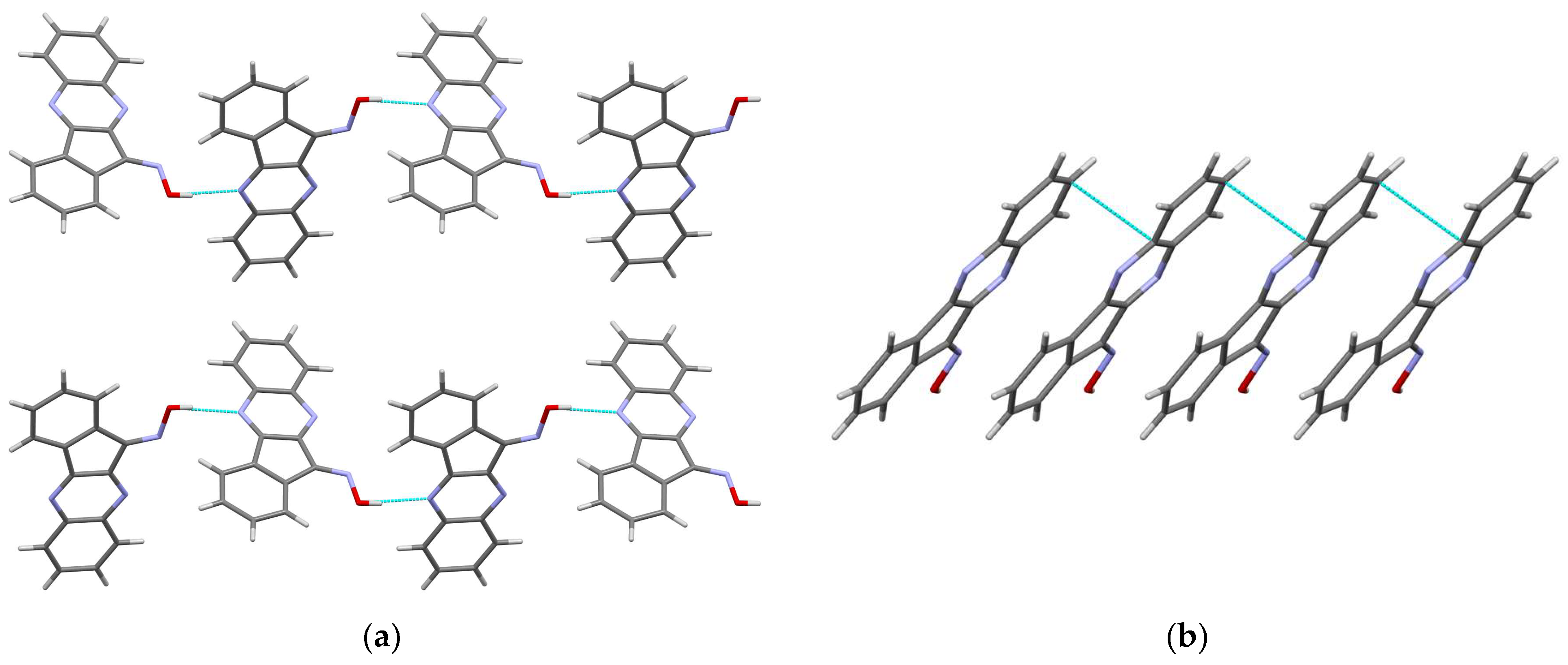
3.1.1. Crystal Structure of 11H-Indeno[1,2-b]quinoxalin-11-one Oxime (IQ-1)
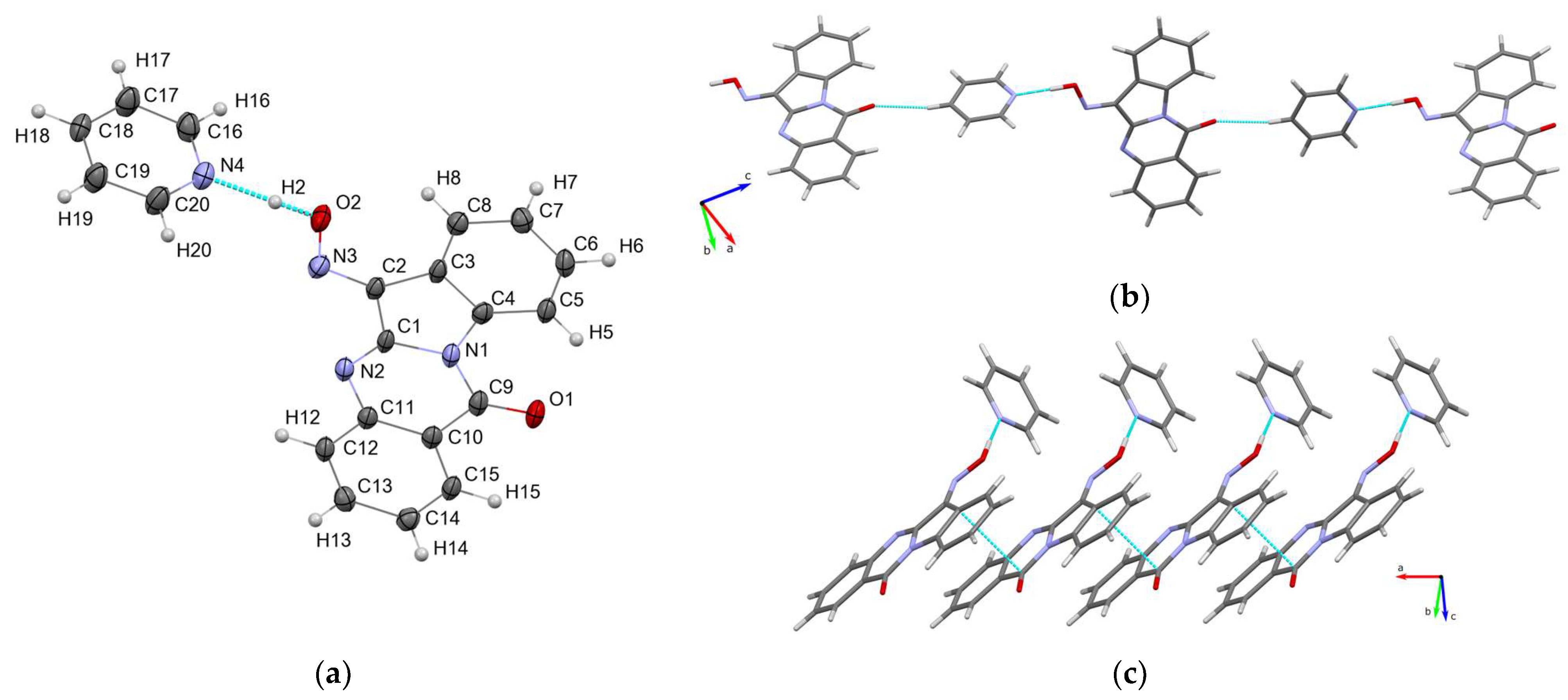
3.1.2. Crystal Structure of Tryptanthrin-6-oxime-pyridine Solvate (Trp-Ox·Py)
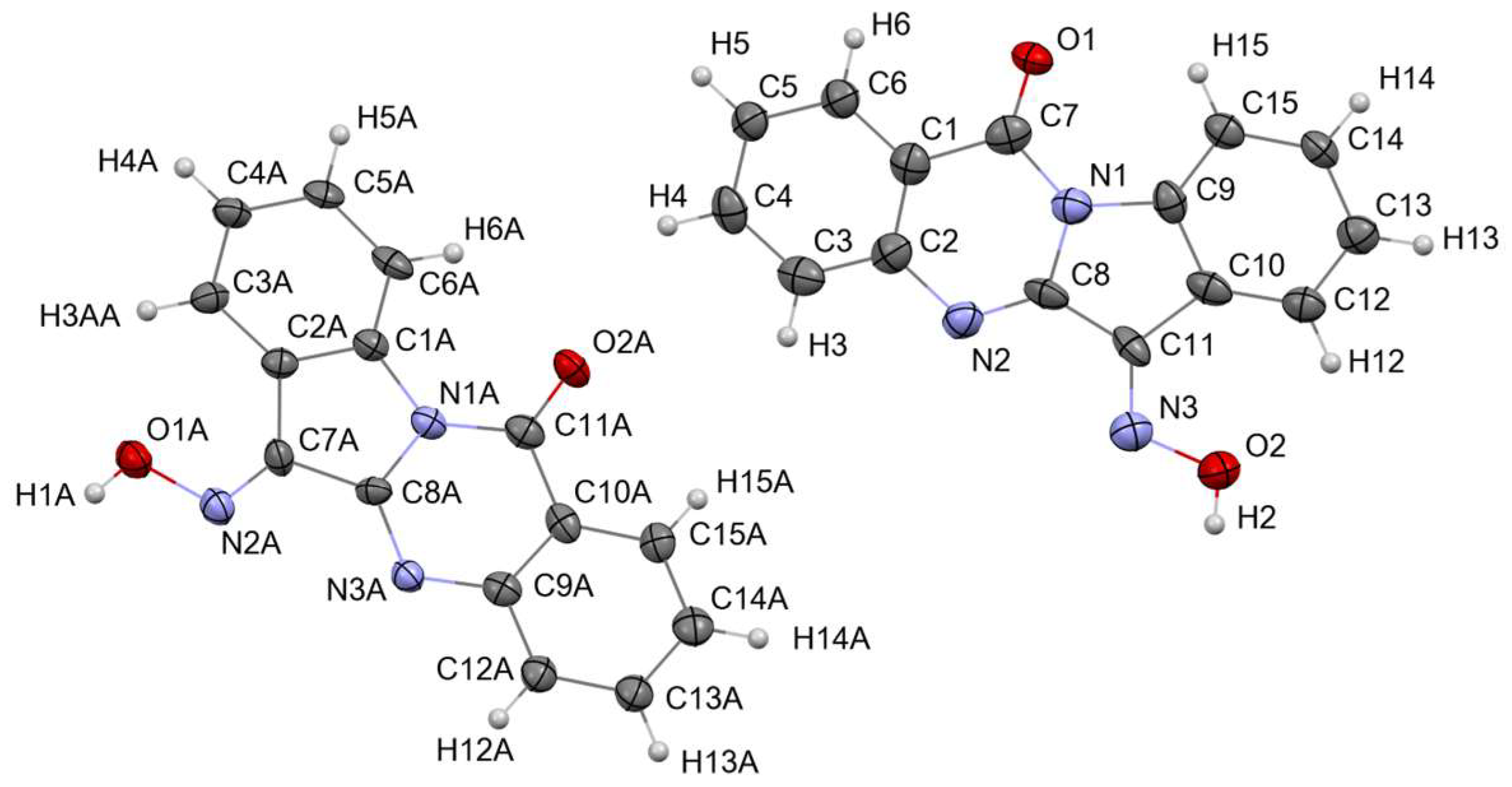
3.1.3. Crystal Structure of Tryptanthrin-6-oxime (Trp-Ox)
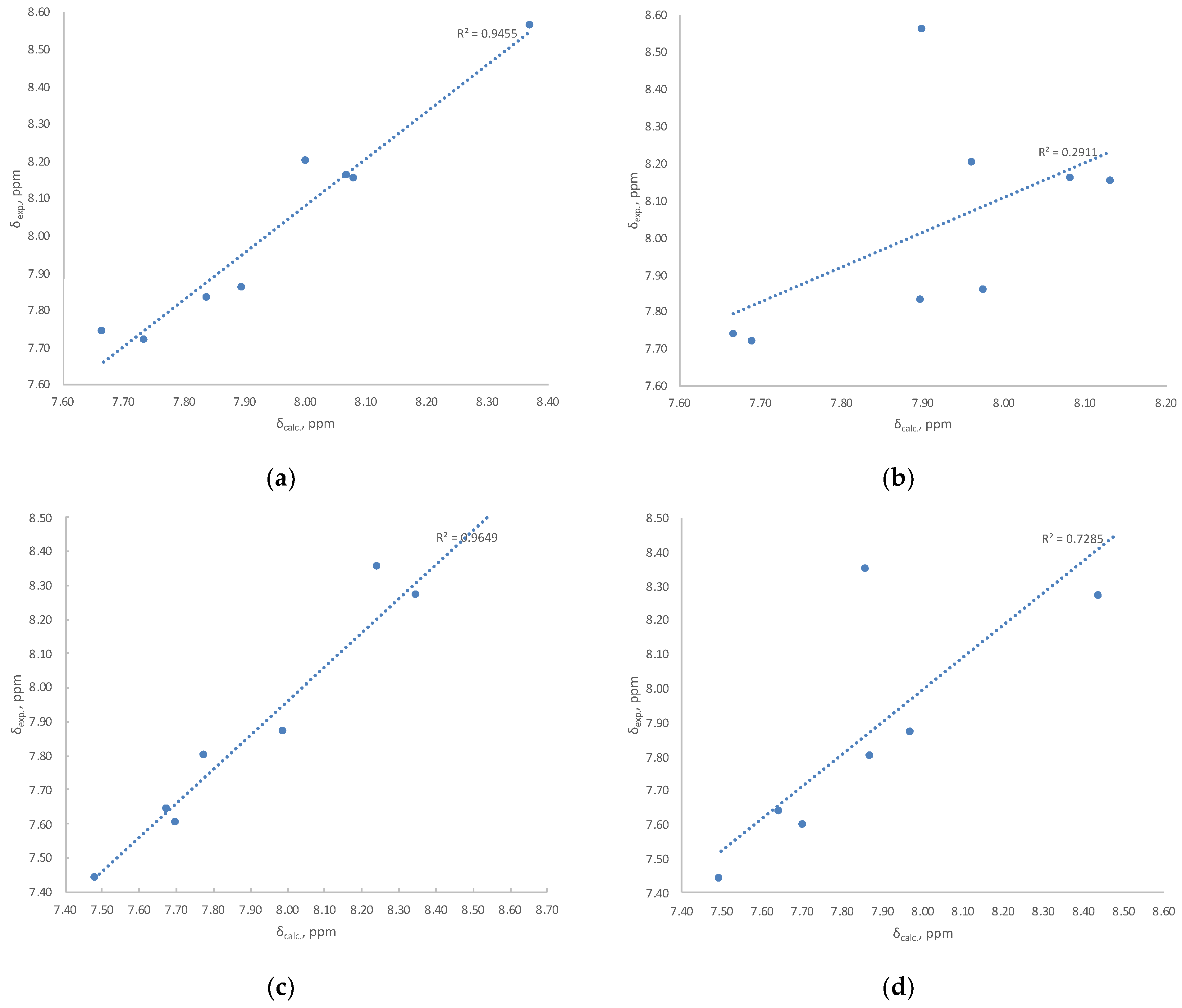
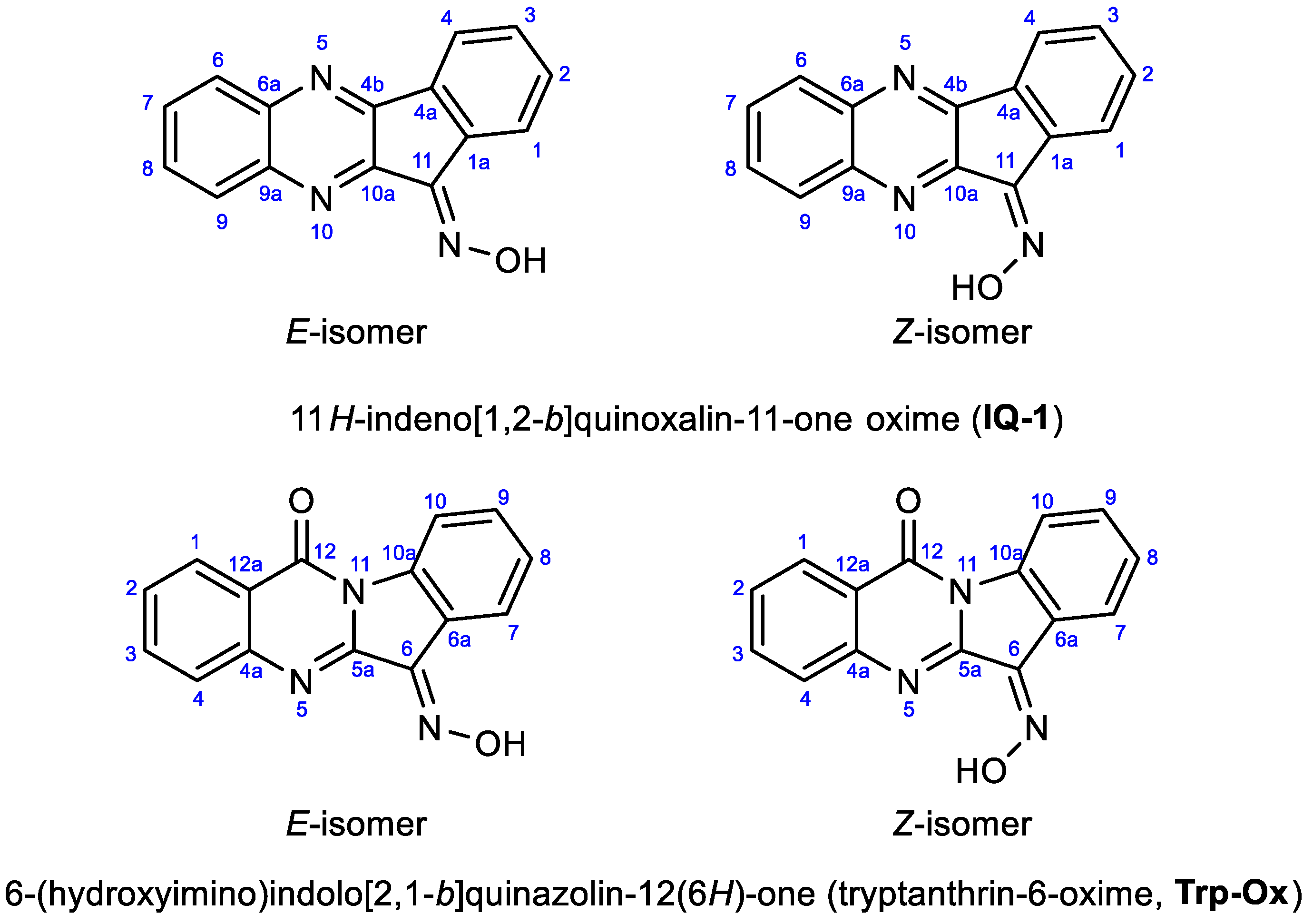
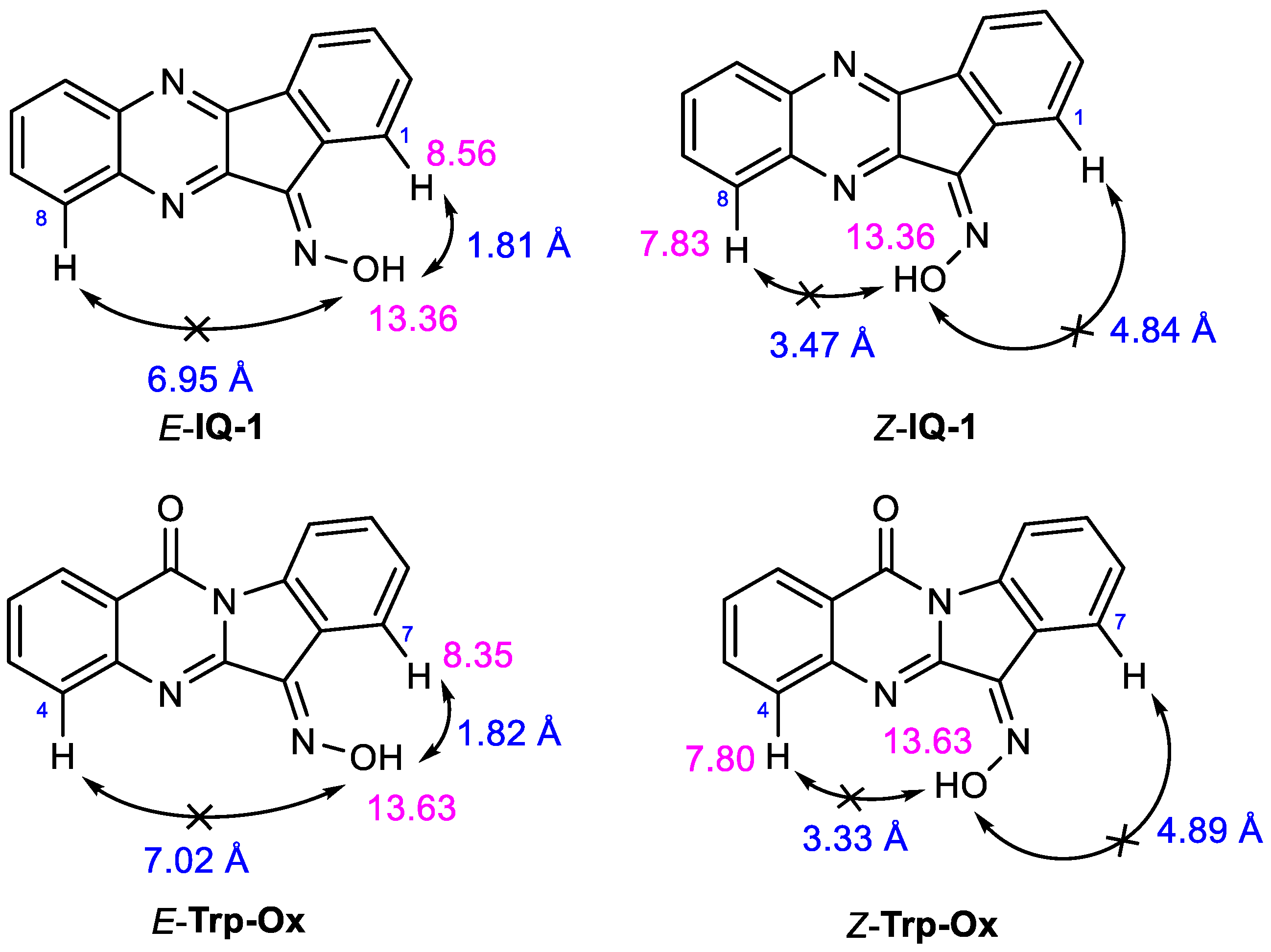
3.2. Evaluation of the Oxime Group Configuration in IQ-1 and Tpr-Ox in Solution by NMR Methods
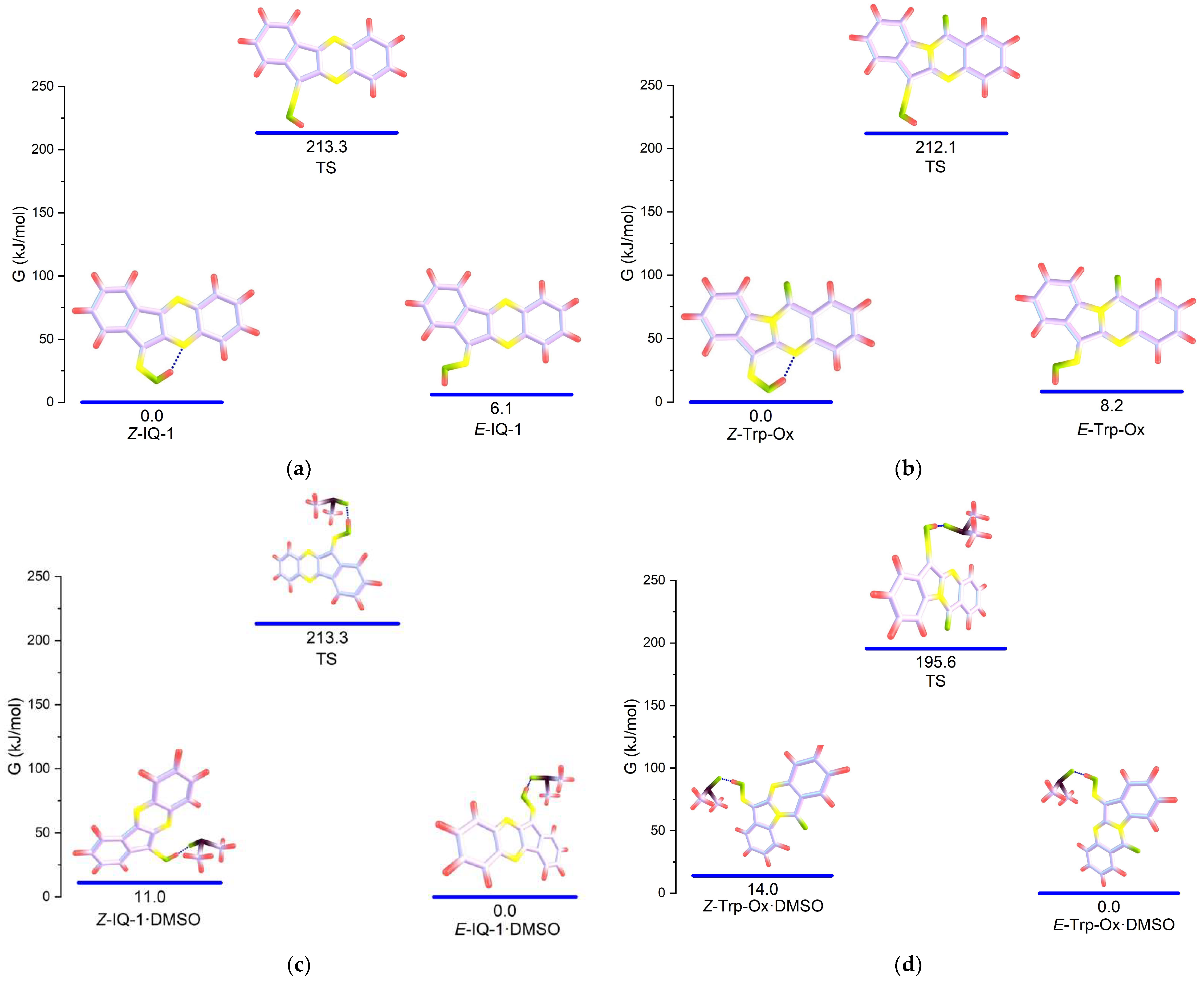
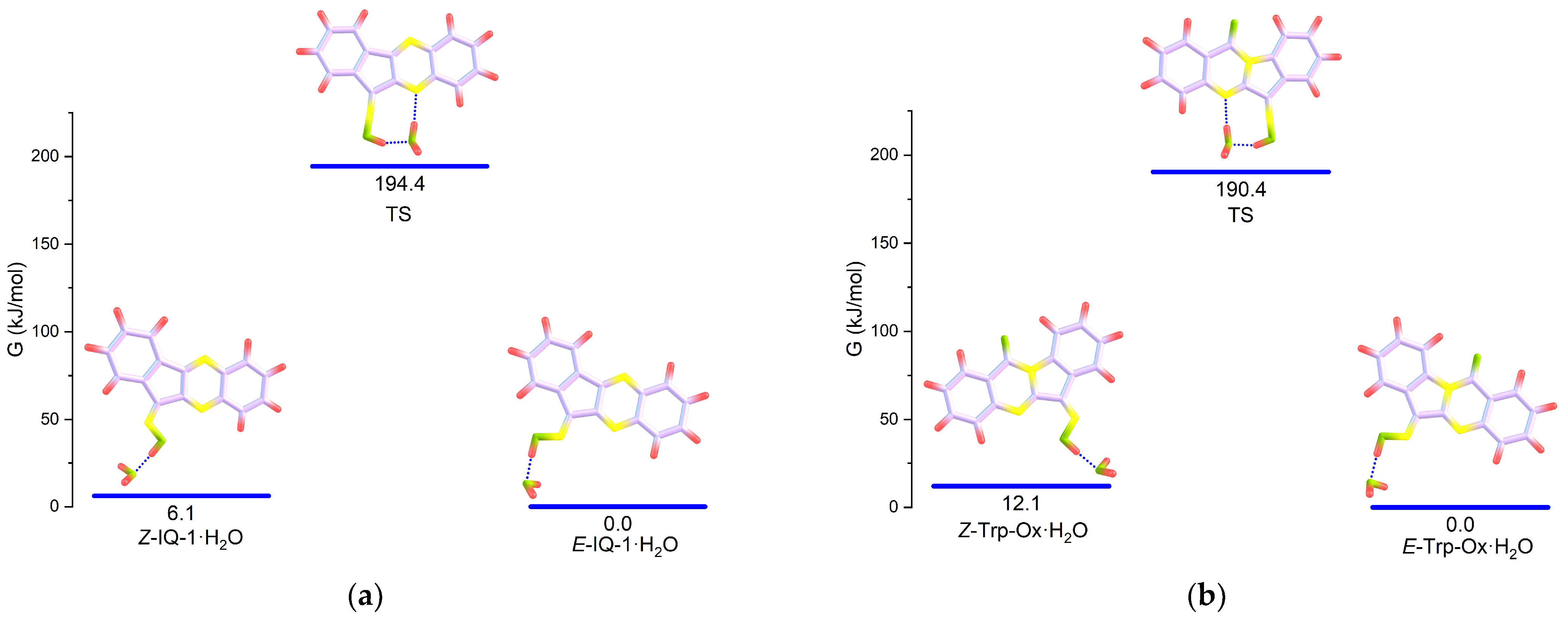
3.3. Calculation of Thermodynamic Parameters of the Isomeric IQ-1 and Trp-Ox
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bapat, S.P.; Whitty, C.; Mowery, C.T.; Liang, Y.; Yoo, A.; Jiang, Z.; Peters, M.C.; Zhang, L.; Vogel, I.; Zhou, C.; et al. Obesity alters pathology and treatment response in inflammatory disease. Nature 2022, 604, 337–342. [Google Scholar] [CrossRef]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-Activated Protein Kinase: Conservation of a Three-Kinase Module from Yeast to Human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [Green Version]
- Anfinogenova, N.D.; Quinn, M.T.; Schepetkin, I.A.; Atochin, D.N. Alarmins and c-Jun N-Terminal Kinase (JNK) Signaling in Neuroinflammation. Cells 2020, 9, 2350. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [Green Version]
- Shvedova, M.; Anfinogenova, Y.; Atochina-Vasserman, E.N.; Schepetkin, I.A.; Atochin, D.N. c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury. Front. Pharmacol. 2018, 9, 715. [Google Scholar] [CrossRef]
- Shvedova, M.V.; Anfinogenova, Y.D.; Shchepetkin, I.A.; Atochin, D.N. c-Jun N-Terminal Kinases and Their Pharmacological Modulation in Ischemic and Reperfusion Brain Injury. Neurosci. Behav. Physiol. 2018, 48, 721–728. [Google Scholar] [CrossRef]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2017, 6, 174–184. [Google Scholar] [CrossRef]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepetkin, I.A.; Plotnikov, M.B.; Khlebnikov, A.I.; Plotnikova, T.M.; Quinn, M.T. Oximes: Novel Therapeutics with Anticancer and Anti-Inflammatory Potential. Biomolecules 2021, 11, 777. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kirpotina, L.N.; Khlebnikov, A.I.; Hanks, T.S.; Kochetkova, I.; Pascual, D.W.; Jutila, M.A.; Quinn, M.T. Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 2012, 81, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepetkin, I.A.; Kirpotina, L.N.; Hammaker, D.; Kochetkova, I.; Khlebnikov, A.I.; Lyakhov, S.A.; Firestein, G.S.; Quinn, M.T. Anti-inflammatory effects and joint protection in collagen-induced arthritis after treatment with IQ-1S, a selective c-Jun N-terminal kinase inhibitor. J. Pharmacol. Exp. Ther. 2015, 353, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Atochin, D.N.; Schepetkin, I.A.; Khlebnikov, A.I.; Seledtsov, V.I.; Swanson, H.; Quinn, M.T.; Huang, P.L. A novel dual NO-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia–reperfusion injury in mice. Neurosci. Lett. 2016, 618, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, M.B.; Chernysheva, G.A.; Aliev, O.I.; Smol’iakova, V.I.; Fomina, T.I.; Osipenko, A.N.; Rydchenko, V.S.; Anfinogenova, Y.J.; Khlebnikov, A.I.; Schepetkin, I.A.; et al. Protective Effects of a New C-Jun N-terminal Kinase Inhibitor in the Model of Global Cerebral Ischemia in Rats. Molecules 2019, 24, 1722. [Google Scholar] [CrossRef] [Green Version]
- Schepetkin, I.A.; Chernysheva, G.A.; Aliev, O.I.; Kirpotina, L.N.; Smol’yakova, V.I.; Osipenko, A.N.; Plotnikov, M.B.; Kovrizhina, A.R.; Khlebnikov, A.I.; Plotnikov, E.V.; et al. Neuroprotective Effects of the Lithium Salt of a Novel JNK Inhibitor in an Animal Model of Cerebral Ischemia–Reperfusion. Biomedicines 2022, 10, 2119. [Google Scholar] [CrossRef]
- Plotnikov, M.B.; Aliev, O.I.; Shamanaev, A.Y.; Sidekhmenova, A.V.; Anishchenko, A.M.; Fomina, T.I.; Rydchenko, V.S.; Khlebnikov, A.I.; Anfinogenova, Y.J.; Schepetkin, I.A.; et al. Antihypertensive activity of a new c-Jun N-terminal kinase inhibitor in spontaneously hypertensive rats. Hypertens. Res. 2020, 43, 1068–1078. [Google Scholar] [CrossRef]
- Liakhov, S.A.; Schepetkin, I.A.; Karpenko, O.S.; Duma, H.I.; Haidarzhy, N.M.; Kirpotina, L.N.; Kovrizhina, A.R.; Khlebnikov, A.I.; Bagryanskaya, I.Y.; Quinn, M.T. Novel c-Jun N-Terminal Kinase (JNK) Inhibitors with an 11H-Indeno[1,2-b]quinoxalin-11-one Scaffold. Molecules 2021, 26, 5688. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Xia, X.; Zhao, Y.; Zhang, Y.; Wang, J. JNK selective inhibitor, IQ-1S, protects the mice against lipopolysaccharides-induced sepsis. Bioorganic Med. Chem. 2021, 30, 115945. [Google Scholar] [CrossRef]
- Seledtsov, V.I.; Malashchenko, V.V.; Meniailo, M.E.; Atochin, D.N.; Seledtsova, G.V.; Schepetkin, I.A. Inhibitory effect of IQ-1S, a selective c-Jun N-terminal kinase (JNK) inhibitor, on phenotypical and cytokine-producing characteristics in human macrophages and T-cells. Eur. J. Pharmacol. 2020, 878, 173116. [Google Scholar] [CrossRef] [PubMed]
- Zhdankina, A.A.; Tikhonov, D.I.; Logvinov, S.V.; Plotnikov, M.B.; Khlebnikov, A.I.; Kolosova, N.G. Suppression of Age-Related Macular Degeneration-like Pathology by c-Jun N-Terminal Kinase Inhibitor IQ-1S. Biomedicines 2023, 11, 395. [Google Scholar] [CrossRef]
- Tseng, C.-H.; Chen, Y.-R.; Tzeng, C.-C.; Liu, W.; Chou, C.-K.; Chiu, C.-C.; Chen, Y.-L. Discovery of indeno[1,2-b]quinoxaline derivatives as potential anticancer agents. Eur. J. Med. Chem. 2016, 108, 258–273. [Google Scholar] [CrossRef]
- Eldeken, G.A.; El-Samahy, F.A.; Zayed, E.M.; Osman, F.H.; Elgemeie, G.E.H. Synthesis, Biological Activities and Molecular Docking analysis of a Novel Series of 11H-Indeno[1,2-b]quinoxalin-11-one Derivatives. J. Mol. Struct. 2022, 1261, 132929. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Sukhikh, T.S.; Gushchin, A.L.; Ivanov, A.Y.; Tennikova, T.B.; Sharoyko, V.V.; Baykov, S.V.; Benassi, E.; Potapov, A.S. Arene-Ruthenium(II) Complexes Containing 11H-Indeno[1,2-b]quinoxalin-11-one Derivatives and Tryptanthrin-6-oxime: Synthesis, Characterization, Cytotoxicity, and Catalytic Transfer Hydrogenation of Aryl Ketones. ACS Omega 2020, 5, 11167–11179. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Samsonenko, D.G.; Bonfili, L.; Cuccioloni, M.; Benassi, E.; Pettinari, R.; Potapov, A.S. Arene-ruthenium(II) complexes with tetracyclic oxime derivatives: Synthesis, structure and antiproliferative activity against human breast cancer cells. Inorganica Chim. Acta 2022, 535, 120879. [Google Scholar] [CrossRef]
- Jahng, Y. Progress in the studies on tryptanthrin, an alkaloid of history. Arch. Pharm. Res. 2013, 36, 517–535. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Manjal, S.K.; Rawal, R.K.; Kumar, K. Recent synthetic and medicinal perspectives of tryptanthrin. Bioorg. Med. Chem. 2017, 25, 4533–4552. [Google Scholar] [CrossRef]
- Tsai, Y.-C.; Lee, C.-L.; Yen, H.-R.; Chang, Y.-S.; Lin, Y.-P.; Huang, S.-H.; Lin, C.-W. Antiviral Action of Tryptanthrin Isolated from Strobilanthes cusia Leaf against Human Coronavirus NL63. Biomolecules 2020, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-Y.; Zhang, T.; He, Y.-N.; Huang, S.-J.; Deng, X.; Han, L.; Xie, C.-G. From natural dye to herbal medicine: A systematic review of chemical constituents, pharmacological effects and clinical applications of indigo naturalis. Chin. Med. 2020, 15, 127. [Google Scholar]
- Schepetkin, I.A.; Khlebnikov, A.I.; Potapov, A.S.; Kovrizhina, A.R.; Matveevskaya, V.V.; Belyanin, M.L.; Atochin, D.N.; Zanoza, S.O.; Gaidarzhy, N.M.; Lyakhov, S.A.; et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019, 161, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Kirpotina, L.N.; Schepetkin, I.A.; Hammaker, D.; Kuhs, A.; Khlebnikov, A.I.; Quinn, M.T. Therapeutic Effects of Tryptanthrin and Tryptanthrin-6-Oxime in Models of Rheumatoid Arthritis. Front. Pharmacol. 2020, 11, 1145. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Kovrizhina, A.R.; Stankevich, K.S.; Khlebnikov, A.I.; Kirpotina, L.N.; Quinn, M.T.; Cook, M.J. Design, synthesis and biological evaluation of novel O-substituted tryptanthrin oxime derivatives as c-Jun N-terminal kinase inhibitors. Front. Pharmacol. 2022, 13, 958687. [Google Scholar] [CrossRef]
- Lakeev, A.P.; Frelikh, G.A.; Yanovskaya, E.A.; Kovrizhina, A.R.; Udut, V. V Quantification of a promising JNK inhibitor and nitrovasodilator IQ-1 and its major metabolite in rat plasma by LC–MS/MS. Bioanalysis 2022, 14, 1423–1441. [Google Scholar] [CrossRef]
- Dhuguru, J.; Zviagin, E.; Skouta, R. FDA-Approved Oximes and Their Significance in Medicinal Chemistry. Pharmaceuticals 2022, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Gergely, A.; Gyimesi-Forras, K.; Horvath, P.; Hosztafi, S.; Kokosi, J.; Nagy, I.P.; Szasz, G.; Szentesi, A. 6-Oxo-Morphinane Oximes: Pharmacology, Chemistry and Analytical Application. Curr. Med. Chem. 2004, 11, 2555–2564. [Google Scholar] [CrossRef]
- Claassen, V.; Davies, J.E.; Hertting, G.; Placheta, P. Fluvoxamine, a specific 5-hydroxytryptamine uptake inhibitor. Br. J. Pharmacol. 1977, 60, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA Diffraction Beamline for Studying Crystalline Samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-throughput small-molecule crystallography at the ‘Belok’ beamline of the Kurchatov synchrotron radiation source: Transition metal complexes with azomethine ligands as a case study. Crystals 2017, 7, 325. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Wiitala, K.W.; Hoye, T.R.; Cramer, C.J. Hybrid Density Functional Methods Empirically Optimized for the Computation of 13C and 1H Chemical Shifts in Chloroform Solution. J. Chem. Theory Comput. 2006, 2, 1085–1092. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Pierens, G.K. 1H and 13C NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J. Comput. Chem. 2014, 35, 1388–1394. [Google Scholar] [CrossRef]
- Benassi, E. Benchmarking of density functionals for a soft but accurate prediction and assignment of 1H and 13C NMR chemical shifts in organic and biological molecules. J. Comput. Chem. 2017, 38, 87–92. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 34108. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.J.; Byrd, J.N.; Montgomery, J.A., Jr. Benchmark comparison of dual-basis double-hybrid density functional theory and a neural-network-optimized method for intermolecular interactions. J. Mol. Spectrosc. 2021, 376, 111406. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem.–A Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, D.; Williamson, M.P. The Nuclear Overhauser Effect in Structural and Conformational Analysis; Methods in Stereochemical Analysis; Wiley: Hoboken, NJ, USA, 2000; ISBN 9780471246756. [Google Scholar]
- Rodrigues, C.H.P.; Leite, V.B.P.; Bruni, A.T. Can NMR spectroscopy discriminate between NPS amphetamines and cathinones? An evaluation by in silico studies and chemometrics. Chemom. Intell. Lab. Syst. 2021, 210, 104265. [Google Scholar] [CrossRef]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2014, 9, 643–660. [Google Scholar] [CrossRef]
- Iron, M.A. Evaluation of the Factors Impacting the Accuracy of 13C NMR Chemical Shift Predictions using Density Functional Theory—The Advantage of Long-Range Corrected Functionals. J. Chem. Theory Comput. 2017, 13, 5798–5819. [Google Scholar] [CrossRef]
- Cohen, R.D.; Wood, J.S.; Lam, Y.-H.; Buevich, A.V.; Sherer, E.C.; Reibarkh, M.; Williamson, R.T.; Martin, G.E. DELTA50: A Highly Accurate Database of Experimental 1H and 13C NMR Chemical Shifts Applied to DFT Benchmarking. Molecules 2023, 28, 2449. [Google Scholar] [CrossRef] [PubMed]
- Palivec, V.; Pohl, R.; Kaminský, J.; Martinez-Seara, H. Efficiently Computing NMR 1H and 13C Chemical Shifts of Saccharides in Aqueous Environment. J. Chem. Theory Comput. 2022, 18, 4373–4386. [Google Scholar] [CrossRef]
- Nguyen, T.T. 1H/13C chemical shift calculations for biaryls: DFT approaches to geometry optimization. R. Soc. Open Sci. 2021, 8, 210954. [Google Scholar] [CrossRef]
- Weiss, K.; Warren, C.H.; Wettermark, G. cis-trans isomerization about the carbon-nitrogen double bond. Structures of the isomers of N-benzylideneaniline. J. Am. Chem. Soc. 1971, 93, 4658–4663. [Google Scholar] [CrossRef]
- Blanco, F.; Alkorta, I.; Elguero, J. Barriers about Double Carbon-Nitrogen Bond in Imine Derivatives (Aldimines, Oximes, Hydrazones, Azines). Croat. Chem. Acta 2009, 82, 173–183. [Google Scholar]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. J. Chem. Phys. 2015, 143, 54107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolmacheva, I.A.; Nazarov, A.V.; Dmitriev, M.V.; Boreko, E.I.; Grishko, V. V Synthesis of 1,10-seco-triterpenoids by the Beckmann fragmentation from allobetulin. Tetrahedron 2017, 73, 6448–6455. [Google Scholar] [CrossRef]
- Chand, D.; Parrish, D.A.; Shreeve, J.M. Di(1H-tetrazol-5-yl)methanone oxime and 5,5′-(hydrazonomethylene)bis(1H-tetrazole) and their salts: A family of highly useful new tetrazoles and energetic materials. J. Mater. Chem. A 2013, 1, 15383–15389. [Google Scholar] [CrossRef]
- Xu, J.; Yang, H.; He, L.; Huang, L.; Shen, J.; Li, W.; Zhang, P. Synthesis of (E)-Quinoxalinone Oximes through a Multicomponent Reaction under Mild Conditions. Org. Lett. 2021, 23, 195–201. [Google Scholar] [CrossRef]
- Boone, L.L.; Mroz, A.E.; VanDerveer, D.G.; Hancock, R.D. Metal Ion Coordinating Properties of the Highly Preorganized Tetradentate Ligand 1,10-Phenanthroline-2,9-dicarboxaldehyde-2,9-dioxime. Eur. J. Inorg. Chem. 2011, 2011, 2706–2711. [Google Scholar] [CrossRef]
- Voloshin, Y.Z.; Varzatskii, O.A.; Vorontsov, I.I.; Antipin, M.Y.; Tkachenko, E.Y. Potentially hexadentate bisazine dioximate ligands: “correct” synthetic procedure and encapsulation reactions of the iron(II) ion. Russ. Chem. Bull. 2002, 51, 1015–1019. [Google Scholar] [CrossRef]
- Hoogesteger, F.J.; Jenneskens, L.W.; Kooijman, H.; Veldman, N.; Spek, A.L. Self-complementary hydrogen bonding of 1,1′-bicyclohexylidene-4,4′-dione dioxixe. Formation of a non-covalent polymer. Tetrahedron 1996, 52, 1773–1784. [Google Scholar] [CrossRef]
- Tien, C.-H.; Lough, A.J.; Yudin, A.K. Iminologous epoxide ring-closure. Chem. Sci. 2022, 13, 12175–12179. [Google Scholar] [CrossRef]
- Angeloff, A.; Daran, J.-C.; Bernadou, J.; Meunier, B. The Ligand 1,10-Phenanthroline-2,9-dicarbaldehyde Dioxime can Act Both as a Tridentate and as a Tetradentate Ligand−Synthesis, Characterization and Crystal Structures of its Transition Metal Complexes. Eur. J. Inorg. Chem. 2000, 2000, 1985–1996. [Google Scholar] [CrossRef]
- Curtis, S.; Ilkun, O.; Brown, A.; Silchenko, S.; Gerasimchuk, N. Synthesis, spectroscopic and structural characterization of the first phenyl bis-cyanoximes: Non-chelating extended ionisable building block ligands for new MOFs. CrystEngComm 2013, 15, 152–159. [Google Scholar] [CrossRef]
- Plutenko, M.V.; Moroz, Y.S.; Sliva, T.Y.; Haukka, M.; Fritsky, I.O. A square-planar NiII complex with an asymmetric coordination of a novel polynucleative 2,6-diacetylpyridine bis-{[2-(hydroxyimino)propanoyl]hydrazone} ligand. Acta Crystallogr. Sect. C 2008, 64, m137–m139. [Google Scholar] [CrossRef]
- Otto, S.; Chanda, A.; Samuleev, P.V.; Ryabov, A.D. DFT-Verified Crystallographic Mechanism of Cycloplatination. Eur. J. Inorg. Chem. 2006, 2006, 2561–2565. [Google Scholar] [CrossRef]
- Walton, I.; Davis, M.; Munro, L.; Catalano, V.J.; Cragg, P.J.; Huggins, M.T.; Wallace, K.J. A Fluorescent Dipyrrinone Oxime for the Detection of Pesticides and Other Organophosphates. Org. Lett. 2012, 14, 2686–2689. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Demakova, M.Y.; Legin, A.A.; Suslonov, V.V.; Nazarov, A.A.; Jakupec, M.A.; Keppler, B.K.; Kukushkin, V.Y. Amidoxime platinum(ii) complexes: PH-dependent highly selective generation and cytotoxic activity. New J. Chem. 2017, 41, 6840–6848. [Google Scholar] [CrossRef] [Green Version]
- Pandya, S.U.; Moss, K.C.; Bryce, M.R.; Batsanov, A.S.; Fox, M.A.; Jankus, V.; Al Attar, H.A.; Monkman, A.P. Luminescent Platinum(II) Complexes Containing Cyclometallated Diaryl Ketimine Ligands: Synthesis, Photophysical and Computational Properties. Eur. J. Inorg. Chem. 2010, 2010, 1963–1972. [Google Scholar] [CrossRef]
- Bakir, M. The first pyridyl N,N’-coordinated di-2-pyridyl ketone oxime, fac-tricarbonylchloro(di-2-pyridyl ketone oxime)rhenium(I) dimethyl sulfoxide solvate. Acta Crystallogr. Sect. C 2001, 57, 1154–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | IQ-1 | Trp-Ox·Py | Trp-Ox |
|---|---|---|---|
| Chemical formula | C15H9N3O | C15H9N3O2·C5H5N | C15H9N3O2 |
| Mr | 247.26 | 342.35 | 263.25 |
| Crystal system, space group | Monoclinic, Ia | Monoclinic, P21/n | Monoclinic, P21 |
| Temperature (K) | 150 | 92 | 100 |
| a, b, c (Å) | 4.4961 (1), 23.1804 (4), 14.3978 (3) | 5.0745 (1), 12.3974 (2), 25.5587 (4) | 13.614 (3), 4.898 (1), 17.322 (4) |
| β (°) | 98.812 (1) | 92.126 (1) | 90.12 (3) |
| V (Å3) | 1482.85 (5) | 1606.81 (5) | 1155.1 (4) |
| Z | 4 | 4 | 4 |
| Radiation type | Cu Kα | Cu Kα | Synchrotron, 0.7527 Å |
| μ (mm−1) | 0.59 | 0.77 | 0.12 |
| Crystal size (mm) | 0.17 × 0.10 × 0.05 | 0.24 × 0.16 × 0.1 | 0.15 × 0.01 × 0.01 |
| Tmin, Tmax | 0.689, 0.753 | 0.689, 0.754 | 0.806, 1.000 |
| No. of measured, independent and observed (I > 2σ(I)) reflections | 4268, 2054, 1983 | 16,109, 3131, 2209 | 7050, 4249, 3075 |
| Rint | 0.023 | 0.061 | 0.076 |
| (sin θ/λ)max (Å−1) | 0.595 | 0.625 | 0.610 |
| R[F2 > 2σ(F2)], wR(F2), S | 0.037, 0.106, 1.06 | 0.046, 0.125, 1.03 | 0.066, 0.171, 1.03 |
| No. of reflections | 2054 | 3131 | 4249 |
| No. of parameters | 175 | 238 | 542 |
| No. of restraints | 2 | – | 1075 |
| Δ〉max, Δ〉min (e Å−3) | 0.14, −0.17 | 0.25, −0.30 | 0.61, −0.24 |
IQ-1 | Trp-Ox | ||
|---|---|---|---|
| Atom Label | δ, ppm | Atom Label | δ, ppm |
| H-1 (d) | 8.56 | H-1 (d) | 8.27 |
| H-2 (t) | 7.74 | H-2 (t) | 7.60 |
| H-3 (t) | 7.72 | H-3 (t) | 7.87 |
| H-4 (d) | 8.20 | H-4 (d) | 7.80 |
| H-6 (d) | 8.15 | H-7 (d) | 8.35 |
| H-7 (t) | 7.86 | H-8 (t) | 7.44 |
| H-8 (t) | 7.83 | H-9 (t) | 7.64 |
| H-9 (d) | 8.16 | H-10 (d) | 8.52 |
| OH | 13.37 | OH | 13.63 |
| C-1 | 128.58 | C-1 | 126.52 |
| C-1a | 135.9 | C-2 | 127.48 |
| C-2 | 131.81 | C-3 | 134.65 |
| C-3 | 132.28 | C-4 | 128.08 |
| C-4 | 122.02 | C-4a | 146.96 |
| C-4a | 132.94 | C-5a | 148.3 |
| C-4b | 152.73 | C-6 | 144.21 |
| C-6 | 129.72 | C-6a | 118.83 |
| C-6a | 141.73 | C-7 | 127.36 |
| C-7 | 130.66 | C-8 | 126.62 |
| C-8 | 129.69 | C-9 | 132.04 |
| C-9 | 129.18 | C-10 | 116.23 |
| C-9a | 141.42 | C-10a | 139.28 |
| C-10a | 150.66 | C-12 | 158.45 |
| C-11 | 147.01 | C-12a | 121.54 |
| Optimization Functional/Basis Set a | NMR Calculation Functional/Basis Set a,b | 1H Slope/Intercept c | 13C Slope/Intercept c | RMSD 1H | RMSD 13C | Method/Reference |
|---|---|---|---|---|---|---|
| B3LYP[GD3BJ]/6-31+g(d,p), | WP04/aug-cc-pVDZ | −1.0138/31.7867 | −0.9862/184.1552 | 0.0789 | 3.0255 | A/[63] |
| B3LYP[GD3BJ]/6-311+g(2d,p) | mPW1PW91/6-311+g(2d,p) | −1.0942/31.9142 | −1.0466/186.1689 | 0.1592 | 2.4351 | B/[64] |
| B3LYP[GD3BJ]/6-311+g(2d,p) | mPW1LYP/6-311+g(2d,p) | −1.0804/31.9940 | −1.0507/181.9422 | 0.1584 | 2.7796 | C/[64] |
| B3LYP[GD3BJ]/6-311+g(2d,p) | PBE0/6-311+g(2d,p) | −1.0962/31.8652 | −1.0464/186.9614 | 0.1622 | 2.4284 | D/[64] |
| B3LYP[GD3BJ]/6-311+g(2d,p) | mPW1LYP/Def2TZVP | −1.0786/31.9641 | −1.0508/181.8634 | 0.1684 | 2.852 | E/[64] |
| Method A | Method B | Method C | Method D | Method E | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| E-Isomer | Z-Isomer | E-Isomer | Z-Isomer | E-Isomer | Z-Isomer | E-Isomer | Z-Isomer | E-Isomer | Z-Isomer | |
| IQ-1 | ||||||||||
| RMSD 1H/13C | 0.11/2.8 | 0.26/2.9 | 0.18/2.1 | 0.32/3.4 | 0.22/2.3 | 0.35/3.6 | 0.18/2.0 | 0.32/3.4 | 0.20/2.1 | 0.33/3.4 |
| r2 1H/13C | 0.9455/0.9546 | 0.2911/0.9413 | 0.9937/0.9702 | 0.4464/0.9134 | 0.9892/0.9656 | 0.4742/0.9101 | 0.9930/0.9708 | 0.4382/0.9120 | 0.9906/0.9675 | 0.4657/0.9106 |
| Trp-Ox | ||||||||||
| RMSD 1H/13C | 0.08/2.5 | 0.19/2.6 | 0.13/1.6 | 0.25/3.1 | 0.16/1.7 | 0.27/3.2 | 0.13/1.6 | 0.25/3.1 | 0.15/1.4 | 0.26/3.0 |
| r2 1H/13C | 0.9649/0.9813 | 0.7285/0.9633 | 0.9851/0.9951 | 0.7620/0.9659 | 0.9834/0.9950 | 0.7662/0.9619 | 0.9855/0.9953 | 0.7653/0.9650 | 0.9886/0.9949 | 0.7808/0.9613 |
| Structure | B3LYP[G]/6-31+G(d)/CPCM(DMSO) | RI-B2PLYP/def2-SVP/SMD(DMSO) | ||
|---|---|---|---|---|
| H298, kJ/mol | G298, kJ/mol | H298, kJ/mol | G298, kJ/mol | |
| E-IQ-1 (in) | 25.8 | 24.2 | 32.1 | 29.6 |
| E-IQ-1 (out) | 0 | 0 | 7.20 | 6.10 |
| Z-IQ-1 (in) | 1.15 | 2.05 | 0 | 0 |
| Z-IQ-1 (out) | 6.24 | 5.94 | 13.7 | 12.3 |
| IQ-1_TS | 214 | 214 | 214 | 213 |
| E-Trp-Ox (in) | 26.0 | 27.7 | 34.5 | 30.7 |
| E-Trp-Ox (out) | 0.13 | 0 | 9.56 | 8.19 |
| Z-Trp-Ox (in) | 0 | 0.92 | 0 | 0 |
| Z-Trp-Ox (out) | 9.31 | 8.79 | 19.5 | 18.0 |
| Trp-Ox_TS | 211 | 210 | 213 | 212 |
| Structure | H298, kJ/mol | G298, kJ/mol | r(OH∙∙∙O), Å | ∠(H∙∙∙O=S), ° |
|---|---|---|---|---|
| E-IQ-1·DMSO (out) | 0 | 0 | 1.525 | 123.0 |
| Z-IQ-1·DMSO (out) | 12.5 | 10.9 | 1.539 | 121.8 |
| IQ-1_TS·DMSO | 199 | 198 | 1.493 | 123.7 |
| E-Trp-Ox-1·DMSO (out) | 0 | 0 | 1.511 | 122.7 |
| Z-Trp-Ox·DMSO (out) | 15.7 | 14.0 | 1.523 | 122.7 |
| Trp-Ox_TS·DMSO | 196 | 196 | 1.479 | 123.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matveevskaya, V.V.; Pavlov, D.I.; Kovrizhina, A.R.; Sukhikh, T.S.; Sadykov, E.H.; Dorovatovskii, P.V.; Lazarenko, V.A.; Khlebnikov, A.I.; Potapov, A.S. Experimental and Computational Investigation of the Oxime Bond Stereochemistry in c-Jun N-terminal Kinase 3 Inhibitors 11H-Indeno[1,2-b]quinoxalin-11-one Oxime and Tryptanthrin-6-oxime. Pharmaceutics 2023, 15, 1802. https://doi.org/10.3390/pharmaceutics15071802
Matveevskaya VV, Pavlov DI, Kovrizhina AR, Sukhikh TS, Sadykov EH, Dorovatovskii PV, Lazarenko VA, Khlebnikov AI, Potapov AS. Experimental and Computational Investigation of the Oxime Bond Stereochemistry in c-Jun N-terminal Kinase 3 Inhibitors 11H-Indeno[1,2-b]quinoxalin-11-one Oxime and Tryptanthrin-6-oxime. Pharmaceutics. 2023; 15(7):1802. https://doi.org/10.3390/pharmaceutics15071802
Chicago/Turabian StyleMatveevskaya, Vladislava V., Dmitry I. Pavlov, Anastasia R. Kovrizhina, Taisiya S. Sukhikh, Evgeniy H. Sadykov, Pavel V. Dorovatovskii, Vladimir A. Lazarenko, Andrei I. Khlebnikov, and Andrei S. Potapov. 2023. "Experimental and Computational Investigation of the Oxime Bond Stereochemistry in c-Jun N-terminal Kinase 3 Inhibitors 11H-Indeno[1,2-b]quinoxalin-11-one Oxime and Tryptanthrin-6-oxime" Pharmaceutics 15, no. 7: 1802. https://doi.org/10.3390/pharmaceutics15071802