
Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives

1
Department of Medicine and Life Sciences, Universitat Pompeu Fabra, 08003 Barcelona, Spain
2
University of Rijeka, Department of Biotechnology, 51000 Rijeka, Croatia
*
Authors to whom correspondence should be addressed.
Pharmaceutics 2023, 15(2), 357; https://doi.org/10.3390/pharmaceutics15020357
Submission received: 7 December 2022
/
Revised: 16 January 2023
/
Accepted: 17 January 2023
/
Published: 20 January 2023
(This article belongs to the Special Issue Application of Peptide-Drug-Conjugates in the Treatment of Cancer and Microbial Infections)
Abstract
:Infectious diseases caused by microbial pathogens (bacteria, virus, fungi, parasites) claim millions of deaths per year worldwide and have become a serious challenge to global human health in our century. Viral infections are particularly notable in this regard, not only because humankind is facing some of the deadliest viral pandemics in recent history, but also because the arsenal of drugs to combat the high levels of mutation, and hence the antigenic variability of (mostly RNA) viruses, is disturbingly scarce. Therefore, the search for new antivirals able to successfully fight infection with minimal or no adverse effects on the host is a pressing task. Traditionally, antiviral therapies have relied on relatively small-sized drugs acting as proteases, polymerases, integrase inhibitors, etc. In recent decades, novel approaches involving targeted delivery such as that achieved by peptide–drug conjugates (PDCs) have gained attention as alternative (pro)drugs for tackling viral diseases. Antiviral PDC therapeutics typically involve one or more small drug molecules conjugated to a cell-penetrating peptide (CPP) carrier either directly or through a linker. Such integration of two bioactive elements into a single molecular entity is primarily aimed at achieving improved bioavailability in conditions where conventional drugs are challenged, but may also turn up novel unexpected functionalities and applications. Advances in peptide medicinal chemistry have eased the way to antiviral PDCs, but challenges remain on the way to therapeutic success. In this paper, we review current antiviral CPP–drug conjugates (antiviral PDCs), with emphasis on the types of CPP and antiviral cargo. We integrate the conjugate and the chemical approaches most often applied to combine both entities. Additionally, we comment on various obstacles faced in the design of antiviral PDCs and on the future outlooks for this class of antiviral therapeutics.
1. Introduction
The ongoing SARS-CoV-2 pandemic is a sobering reminder that viruses continue to pose critical challenges to public health worldwide. In addition to SARS-CoV (2002, 2019), the last two decades have witnessed other deadly viral pandemics, such as influenza A (2009), Middle East respiratory syndrome coronavirus (MERS-CoV, 2012), Ebola (2013) and Zika virus (ZIKV, 2016) [1]. In addition to—and because of—their overall health impacts, such outbreaks of emerging and/or re-emerging viral infections disrupt the global economy, often with devastating effects on public welfare. Altogether, their biological, environmental and socio-economic impacts make viruses and virus-related diseases one of the greatest challenges to humankind [2,3].
Dealing with viral disease requires addressing still unsolved issues, such as (i) a limited arsenal of antiviral drugs with often narrow activity (e.g., diverging effects on different virus subtypes) [4], (ii) in vivo delivery hurdles (toxicity, solubility, efficacy, safety, etc.) [5] or (iii) high mutation rates, leading to drug resistance and ensuing therapeutic failure [6]. In over half a century since the launch of the first antiviral drug, idoxuridine, in 1963, only 90 other approvals have been recorded, which have been catalogued into 13 groups for treating just 9 human viral infections [7]. They largely rely on relatively small molecules—e.g., proteases, polymerases or integrase inhibitors. Clearly, development of effective antiviral therapies remains a pressing need, in whose pursuit not only must viruses be targeted, but host cell processes in the virus life cycle as well [8].
Peptides are making steady inroads into diverse therapeutic categories, particularly as anti-infectives. Antimicrobial peptides (AMPs) have proven successful against various microbial infections; 11 AMPs are FDA-approved and 19 are currently under clinical trials [9], mostly targeting bacterial infections. In contrast, the number of peptides studied for antiviral action (AVPs) is much lower, roughly a third of those validated as antibacterial [10]. Even so, given the challenges posed by viral disease, it is no surprise that a number of peptide-inspired endeavors are underway, not only involving AVPs, but more attuned to this work, peptide conjugates with the ability to cross biological barriers and to exert their functions in specific and efficient ways.
The field of peptide-drug conjugates (PDCs) has grown considerably over recent decades, as a regular flow of candidate conjugates have entered clinical trials [11] aimed at cancer, diabetes, Alzheimer’s disease or microbial infections caused by Escherichia coli, Pseudomonas aeruginosa, Salmonella, etc. However, as with AVPs, antiviral PDCs are still scarcely represented in drug pipelines. In most of those few instances, the approach involves chemical linkage of an established (small molecule) antiviral to a cell-penetrating peptide (CPP) to achieve efficient delivery at a particular intracellular target.
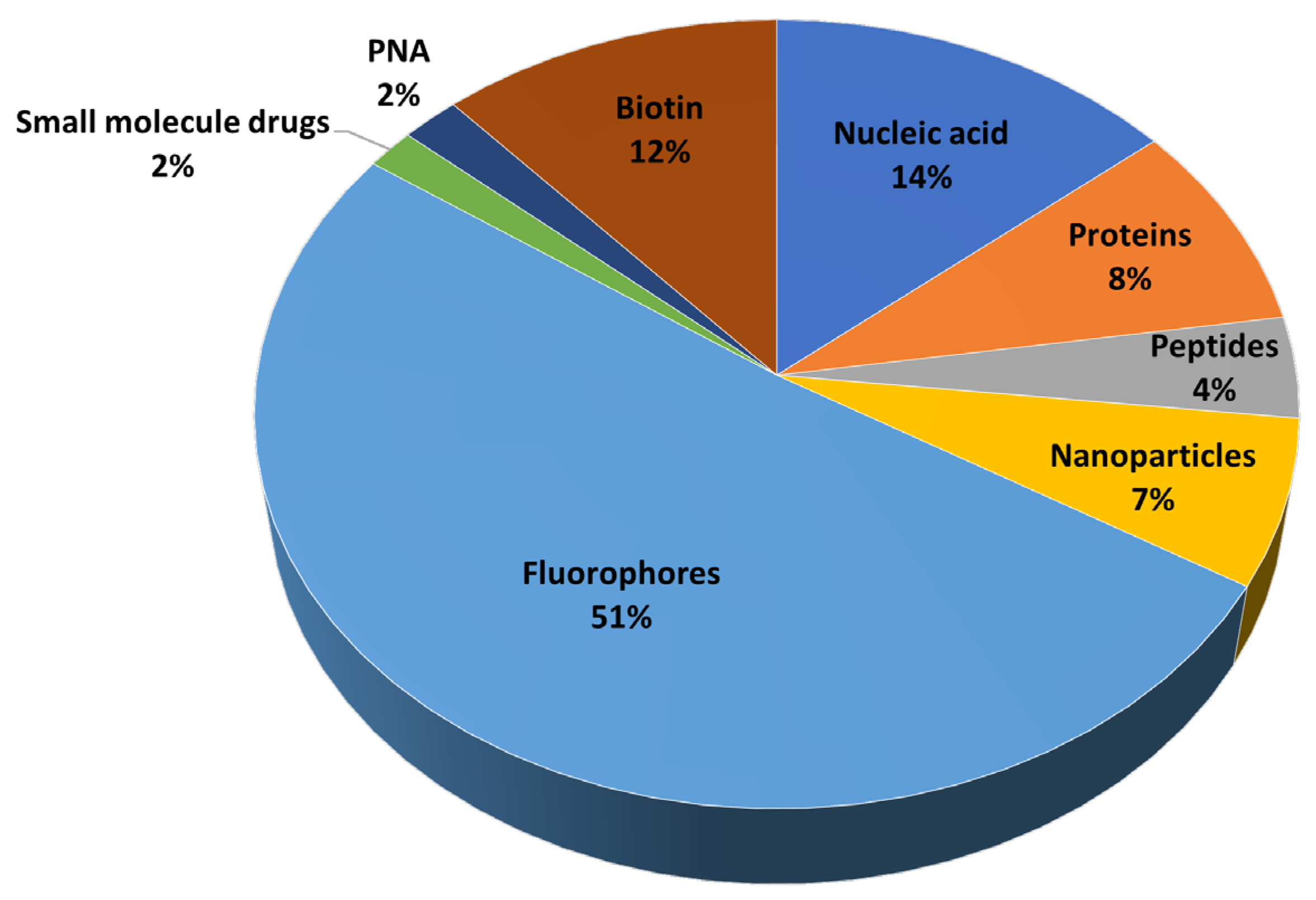
CPPs are typically 5–30 amino acid residues in size and structurally diverse but usually cationic. They have the ability to translocate bioactive payloads into living cells [12]. CPPs successfully deliver into cells diverse types of cargoes, often exceeding (~200 times) typical bioavailability size restrictions [13]. As shown in Figure 1 (data taken from [14]), the most frequent cargoes are fluorescent dyes used mainly for diagnostic purposes, but conjugates delivering nucleic acids, proteins, nanoparticles, therapeutic peptides or other payloads of biomedical relevance are also favored.
Since the discovery of the first CPPs (HIV-Tat, penetratin) [15,16], many other sequences have been documented, including synthetic and chimeric ones [17,18,19,20,21,22]. The CPPsite 2.0 database provides detailed updated information on hitherto reported CPPs and their categorization based on various criteria (Table 1).
In this review, we discuss current antiviral cell-penetrating peptide–drug conjugates (antiviral CPPDCs), focusing on the types of CPP and cargo involved, the conjugation chemistries used and their effects on conjugate performance. We close by discussing future perspectives of antiviral PDC application.
2. PDCs and Antiviral Cargoes
Drug conjugates are chemotherapeutic agents consisting of drug cargo bound to a carrier (antibody, CPP), either directly or through a linker unit. Most currently available PDCs are designed as new modalities of targeted therapy, with improved efficacy and reduced side effects, against various cancer types [23,24]. There are so far only two FDA-approved PDCs on the market, but several others are at various stages of clinical trials [23,24,25]. He et al. [26] have reviewed the technologies for conjugation of CPPs and small drugs and their outcomes as cancer therapeutics. Indeed, most entries in the comprehensive ConjuPepDB database of PDCs are antitumoral [27]. Even so, PDCs are receiving increasing attention as antimicrobials, as judged by the non-negligible number (224) of ConjuPepDB entries retrieved by the query “antimicrobial”. More importantly, and pertinently to this work, of those 224 PDCs, 118 are also retrieved by the query “antiviral”, 7.2% of total entries in the database [27].
Table 2 lists all antiviral PDCs reported so far, specifying the CPP sequence, antiviral cargo, the conjugation chemistry, the targeted virus and the experimental screen used for validation.
Peptide design in most of the PDCs presented above is mainly based on already-known CPP sequences (Tat, polyArg, transportan, penetratin) derived from natural protein sequences or resulting from de novo design. In other instances, the sequences are derived from a protease specific substrate (Table 2 entry 20) or from viral protein domain(s) (Table 2, entry 1–8, 21), or are a mix of CPPs with these previous categories (Table 2, some examples in entries 29 and 31). Moreover, in some cases (Table 2, entries 11–19), sequences with membrane-permeating characteristics are used as prodrugs/substrate recognition motifs for proteases. Finally, the majority of CPPs in Table 2 are linear, of L-chirality, cationic or amphipathic; to the best of our knowledge, only two reports on antiviral PDCs based on anionic CPPs have appeared [82,83].
The antiviral payloads are mainly small drug molecules or modified oligonucleotides. In the former case, conjugation to a CPP usually increases the concentration of the therapeutic molecule in body fluids due to improved conjugate solubility—a feature critical for in vivo applications. With modified oligonucleotides (PMO or PNA) as antiviral payloads, conjugation is achieved either through an amide bond or by thiol-ene chemistry via a suitable linker. In addition to their wide-spectrum antiviral activity, these conjugates (Table 2) have been also used as diagnostic tools for the detection of viral nucleic acids [84,85].
3. PDC Design Considerations
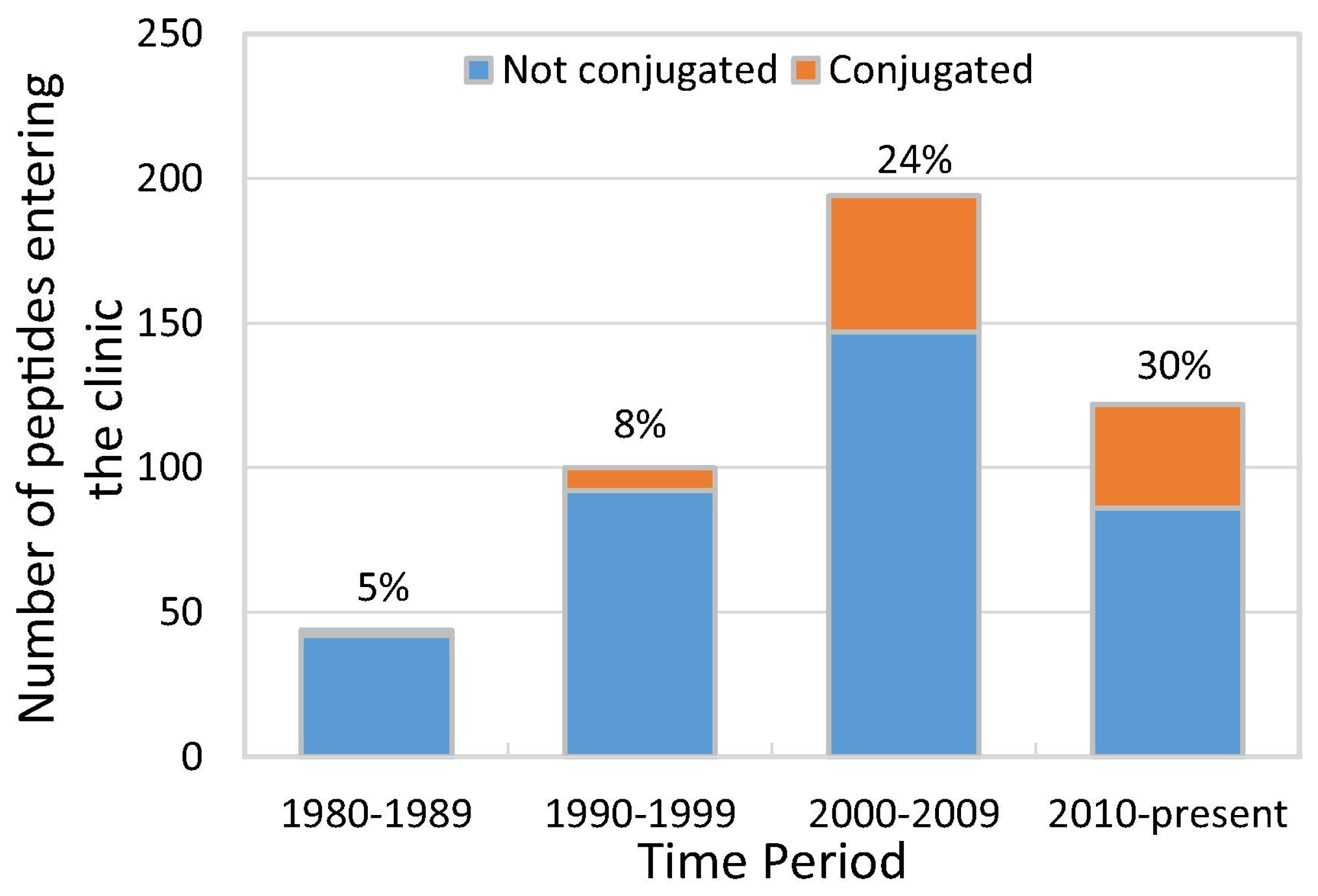
While peptide-based medicines remained for years a small fraction of pharmaceutical business—and PDCs an even smaller one—the situation changed in the 1990s (Figure 2) when a number of PDCs entered clinical trials. The upward trend continued to the point where between 2010 and 2018, about 30% of peptides in clinical development were conjugates.
While recent, steady advances in peptide chemistry allow relatively straightforward access to PDCs, there are still issues to be addressed in the quest for pharmaceutical success, particularly the following:
- (i)
- A robust enough biological rationale endorsing the combination of the two (peptide + antiviral) or three (peptide + antiviral drug + linker) components of the conjugate is desirable.
- (ii)
- A CPP moiety should be chosen that warrants tissue-specific delivery and hence reduces off-target adverse effects. This ideal scenario is very often ignored, as can be seen in Table 2, where typical CPP sequences with broad spectra of membrane permeability are the ones used in the design of the PDCs.
- (iii)
- Antiviral cargoes active at concentrations commensurate with those of the CPP are desirable. Peptides can act at rather low (e.g., nM) concentrations not met by antiviral molecules. These, in turn, can bind plasma proteins (e.g., albumin) with high affinities that can significantly alter pharmacokinetics and/or pharmacodynamics of the conjugate. Those issues should ideally be considered and harmonized.
- (iv)
- For linker-containing conjugates, the linker should if possible be chosen while bearing in mind factors such as the desirable circulation time for the conjugate to reach its target or the specific location where the drug needs to be released.
- (v)
- Another important consideration is the position where the payload is placed. While the N-terminus of the CPP—elongated or not via an intervening spacer unit—is rather usual, alternative approaches, e.g., by way of an extra residue (often Lys or Cys) at either (N- or C-) end of the proper CPP sequence are also favored. Recent work has shown that whichever of these attachment modes is used can have a significant impact on conjugate performance [82,83].
- (vi)
- The conjugate end-product should ideally be non-cytotoxic, non-immunogenic and have minimal interference (hence adverse reactions) with other drugs in multi-therapy schedules.
- (vii)
- Finally, uptake mechanisms ensuring successful release of the antiviral drug from the PDC need to be elucidated. CPP internalization (with or without cargo) is a complex process with multiple factors (positive charge, amphipathicity, folding ability, cargo structure or cell internalization, through active (energy-dependent) or passive (energy-independent) penetration pathways) influencing the peptide–membrane interaction, which is crucial for successful outcomes [86,87,88,89,90].
Despite the complexities of the design process and the high number of factors influencing conjugate performance, by carefully considering the above-listed aspects, it is possible to produce effective antiviral PDCs. A suitable algorithm guiding conjugate design based on activity, serum stability and penetration capability would be desirable to speed up the development process. While algorithms derived from random peptide library design [91], machine learning-based predictive models [92] and natural sequence scanning [93] have been proposed for predicting peptide bioactivity, unfortunately, no such tool is yet available for peptide–antiviral-drug conjugate activity. Future studies should also focus on PDCs with various therapeutic profiles for further development in stability and performance.
4. Conjugation Chemistry
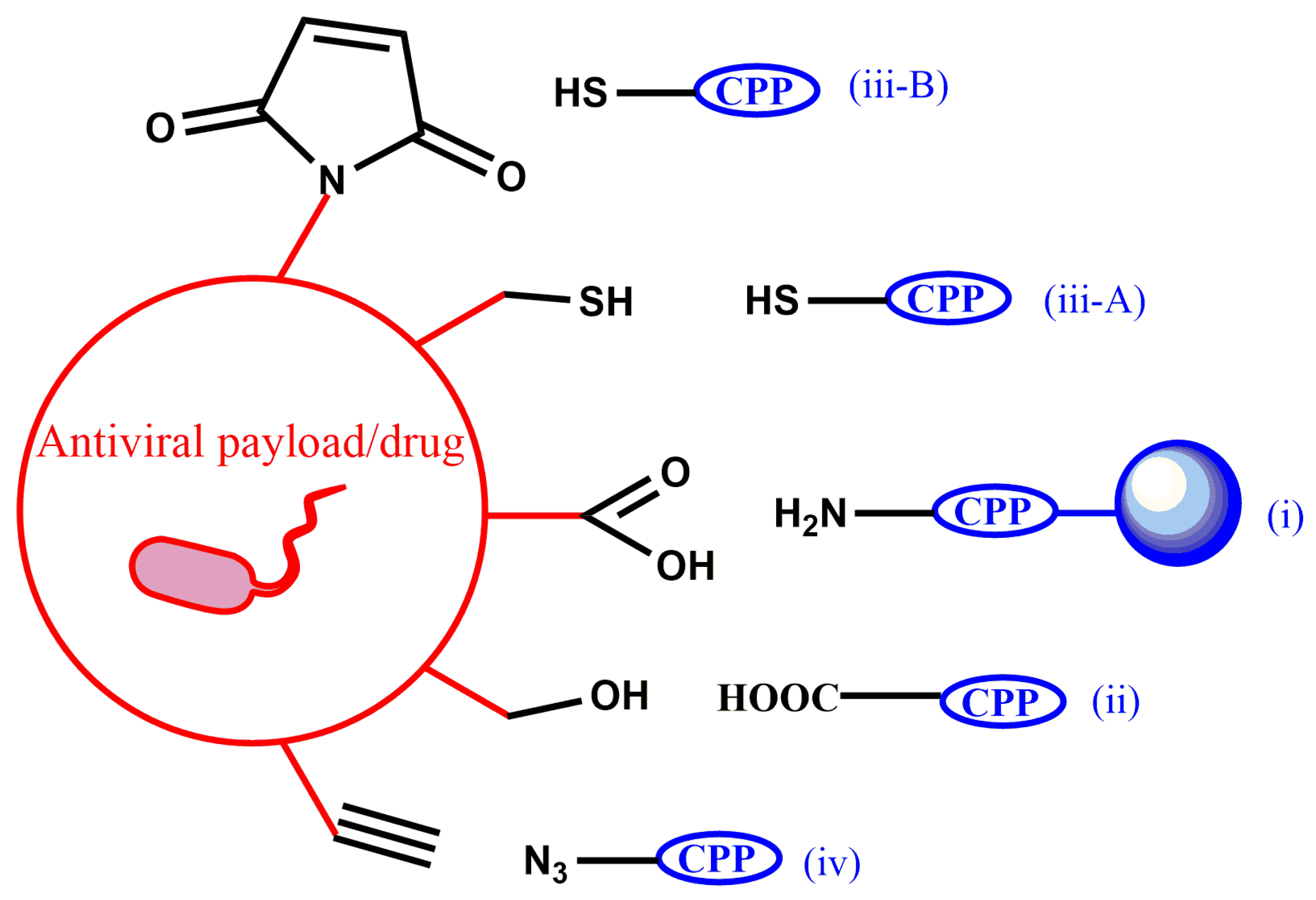
Although joining two different chemical entities into a single molecule while preserving their physico-chemical properties can be challenging, synthetic strategies to obtain antiviral PDCs, either in solution or on solid supports, are relatively well established. The latter approach exploits the advantages of solid-phase peptide synthesis plus the chemoselectivity provided by specific amino-acid side chains. In all examples in Table 2, conjugation between CPP and antiviral cargo is achieved through a covalent bond whose formation has as main requirement that the CPP, the cargo and the resulting conjugate are stable throughout the synthesis and purification steps. In practice, five types of conjugation chemistries are favored: (i) amide bond formation, (ii) ester bond formation, (iii) thiol-based chemistries that comprise disulfide, (iii-A) thioether bond formation (including thiol-ene coupling, iii-B) and (iv) click reaction (Figure 3).
Usually, the methods based on amide bond formation employ peptide N-terminal conjugation or lysine-residue-selective activation and coupling while the peptide is still anchored to the solid support. In the other three coupling strategies, the activation and corresponding chemical bond formation are predominantly performed in solution.
4.1. Conjugation Based on Amide Bond Formation
Amide bond formation, a straightforward and versatile way to attach a drug molecule to a peptide [11,94,95,96,97], is a preferred conjugation option whenever possible, affording substantial stability towards acid or basic media, temperature, etc. Their chemical stability is, however, counterbalanced by their often rather short lifespans in biological fluids and cell compartments (e.g., lysosomes) owing to peptide proteolytic susceptibility [98,99,100,101]. To mitigate this limitation, integration into various platforms (polypeptide carriers, liposomes, nanocarriers, etc.) has been proposed as providing steric protection and increased in vitro or in vivo lifetimes [101,102,103]. A few PDCs with the antiviral cargo directly attached to the CPP through an amide bond have so far been described [29,32,34]. For example, Wang et al. [29] and Nitsche et al. [34] reported conjugation through the CPP N-terminus, and Diez-Torrubia et al. [32] attached the antiviral payload by direct reaction with the C-terminal carboxyl group. This latter study also showed that amide-based conjugates acting as acyclovir carriers provided faster release of the antiviral compared to ester-linked ones. For its part, in the study of Nitsche et al. describing a new generation of PDCs acting as dengue virus protease inhibitors, the position at which the 5-arylidenerhodamine or 5-thiazolidinedione cargo was linked by amide bond to the CPP was consequential [34,104], and a structural change in the payload (e.g., S by O exchange; thiazolidinone vs. rhodamine, respectively) likewise altered the interaction profile of the conjugate [34].
In a study by Moulton et al., it was observed that the CPP used is quite relevant for successful delivery of PMO conjugates [105]. These findings were in tune with those of Abes et al., who showed that a particular CPP (R-Ahx-R)4 was much superior in delivering PMOs to the nucleus [106]. In recent reports on antiviral-peptide–porphyrin conjugates, Mendonça et al. and Todorovski et al. evaluated various amide-based conjugation schemes (N-terminal, C-terminal via an extra Lys, with or without an intervening PEG-based linker) and showed that the linker between the CPP and the cargo can influence aspects such as cell penetration, toxicity, antiviral activity [82,83]. Finally, it is worth mentioning here that in all currently described PDCs with PMO or PNA as antiviral cargo, a suitable linker is present between both moieties (Table 2, entries 22–30).
4.2. Conjugation Based on Ester Bond Formation
Although, in comparison to other covalent linkages, ester bonds do not provide high chemical or plasma stability, they are nonetheless widely used for conjugating drugs to peptides given their relatively simple synthesis and their well-characterized cleavage mechanisms, either by esterases or under acidic conditions [26]. Ester bond-linked antiviral PDCs have already been mentioned above as anti-herpes agents [32], and Liotard et al. reported higher chemical stability (e.g., hydrolysis resistance) of HIV-targeting ester-conjugated prodrugs compared to phosphoramidate-based analogs [33]. In most cases, the antiviral activity of these conjugates is correlated to their hydrolysis rates. Prodrug conjugates with Boc or Fmoc protection at the CPP N-terminus were generally less stable than those with Z- or Qnc protecting groups [33]. Despite the authors’ conclusion that the data obtained do not validate their initial hypothesis for designing HIV RT prodrugs, the work offers helpful insights on the importance of the synthetic chemistry used, the position where the conjugation is performed and the overall stability.
4.3. Conjugation Based on Thiol Chemistry
The ability of thiols to act as nucleophiles in a variety of reactions with nearly quantitative yields has long made them attractive groups for conjugation. Among the various reaction types reported (thiol-halogen, thiol-maleimide, thiol-ene, etc.) [107], only antiviral PDCs based on disulfide, thioether (including thiol-maleimide) or thiol-halogen linkages have been reported so far (Table 2).
Disulfide formation is most often achieved in a directed, efficient manner by reaction of a Cys residue in the peptide with an activated (electrophilic) thiol group in the antiviral drug. Intracellular release of disulfide-linked payloads is glutathione-trigged; in contrast, for thioether or thiol-maleimide PDCs, there are no specific enzymes able to release the payload, although decomposition by oxidation or β-elimination has been reported for long plasma exposure [108,109]. A large number of PMO- and PNA-based antiviral PDCs are based on either of these approaches (Table 2, entries 23–30), following Moulton et al. [100]. These reports also show that linker chemistries used for antisense PMO delivery did not significantly affect activity but did influence uptake and intracellular distribution of the conjugate [105]. Turner et al. compared the antiviral activities of PNA-CPP conjugates formed by either amide or disulfide linkages and demonstrated better performance for the latter ones [67]. Additionally, Kumar et al. reported the systemic delivery of antiviral siRNAs to T cells by way of a single-chain antibody conjugated to a poly-Arg CPP through a disulfide linkage [76]. For their part, Meng et al. (Table 2, entry 31) were able to successfully conjugate siRNA to HIV-1 TAT47-57, using thiol-maleimide coupling, and to show that the corresponding conjugate could effectively block hepatitis C virus replication in the cells [75]. Another report based on thiol-maleimide conjugation was by Zeng et al. (Table 2, entry 30), where a PNA conjugate exerted considerable inhibitory effects against hepatitis B virus in vitro and in vivo [72]. To the best of our knowledge, these two reports, along with that by Moulton et al., are the only examples where thiol-maleimide chemistry is used to produce the antiviral agent. However, further research in this area is needed to clarify whether and how cleavable or stable linkages influence antiviral activity by their stability (or lack thereof) in specific microenvironments.
4.4. Conjugation Based on Click Chemistry
The recently awarded Nobel prize in Chemistry for the “development of click chemistry and bioorthogonal chemistry” testifies about the importance of efficient reactions that allow the formation of functional bioconjugates. In particular, the Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) takes place under mild conditions and does not interfere with the Cys or Lys residues that, as discussed above, are frequent players in conjugation chemistries. The triazole ring resulting from CuAAC is stable against enzymatic degradation, reduction, hydrolysis and oxidation. Somehow, surprisingly, examples of antiviral conjugates made by this approach are mainly limited to the paper by Liang et al., where PDCs able to inhibit HIV-1 mediated cell fusion and infection are described [28]. Two of the described conjugates (those with indole and Gls, Table 2, entries 1 and 2, correspondingly) showed 6-fold improved inhibitory activity compared to the clinically used fusion inhibitor. Additionally, Zhou et al. compared the ability of various antiviral PDCs, obtained by either click chemistry and thiol-chloroacetyl reaction, to activate human RNase. The latter ones were 3-fold more potent [30], which in turn led to more efficient rRNA cleavage.
5. Future Perspectives
PDCs combining a CPP and a relatively small drug rely with few exceptions on a covalent bond between both moieties to create a single entity that achieves efficient cell penetration and antiviral activity. Despite the promising outlook, the modest success rate indicates that important drawbacks, more of biological than chemical nature, still need to be overcome.
(1) Selectivity remains a substantial challenge in that it is rare to find extracellular targets (i.e., receptors recognized by the CPP) expressed at a distinct tissue. Thus, antiviral PDCs may access any cell or tissue where a particular receptor is expressed, with the risk of causing off-target toxicity in healthy cells. To address this issue, three solutions, well researched and applied in the anticancer CPP-drug conjugate field, can be advanced: (i) cell- and tissue-specifically designed CPPs, (ii) conjugation of existing CPPs to suitable targeting entities and (iii) modulation of CPP uptake by a stimulus-sensitive signal. In the first strategy, the target-specific CPPs are usually isolated through phage-display [22,110,111,112]. This technology, awarded a Nobel Prize in Chemistry in 2018, allows high-affinity target-binding peptides to be selected from a complex pool of billions of peptides displayed on a phage and has become one of the most common methods for the identification of specific peptide ligands to virtually any target [112]. In the second approach, improved targeting by the PDC is typically achieved by endowing the conjugate with an additional, tissue-specific homing unit such as folic acid, transferrin and antibodies [113,114,115]. In the third strategy, the most used in targeted delivery of anticancer PDCs, CPP uptake is impaired until a specific stimulus (pH, temperature, light, proteolysis) triggers cell membrane penetration [22]. pH-responsive anticancer PDCs are most favored, due to the significant pH differences between cancer and healthy cells [116]. Unfortunately, virus-infected cells do not display such a pH imbalance; therefore, a next-best approach would be to develop antiviral PDCs that can be activated on demand by virus-specific enzymes.
(2) Another challenge to obtaining stable conjugates is proteolytic instability [117,118]. As CPPs (made up of L-amino acids) are prone to degradation in biological fluids, approaches such as switching to all- or partial-D configuration, or modifications to specific residues or the backbone, or using cyclic CPPs, have shown that this obstacle can be successfully addressed. Additionally, the linkage between the CPP and the drug is another vulnerable element easily recognized and cleaved (ester and amide-bond based linkages) by certain proteases in biological fluids. This is not so with thiol-maleimide and triazole-based junctions, which are protease-stable and should be preferred when a long circulation and stability of the PDC is required, or when the PDC unit has better antiviral activity than the drug alone. In contrast, if a well-controlled intracellular release of the conjugated drug is required, disulfide and ester-based conjugation chemistries should be employed. In those instances when an intervening linker unit is used to connect CPP and cargo, additional issues may be considered. For example, a particular type of CPP–cargo junction, receiving significant attention over recent decades within the PDC field [119], is that of so-called self-immolative linkers (SILs), designed to degrade spontaneously in response to specific stimuli [120]. Unfortunately, to the best of our knowledge, no antiviral PDCs have been reported so far using this technology for selective and targeted drug delivery within the infected cells. The approach is mainly used in cancer therapeutics, but any progress in identifying specific viral enzymes/proteins that can recognize and cleave certain SILs would become a real breakthrough within the field of antiviral PDCs.
(3) A further challenge has to do with our limited knowledge of intracellular biochemistry—i.e., after endocytosis of a conjugate, it is not yet clear how the internalized molecule avoids lysosomal degradation—a step critical for successful release into the cytosol. Furthermore, even when endosomal escape and intracellular delivery are successful, the mechanism of final transport to the preferred intracellular destination (mitochondria, nucleus, other sites) is largely unknown and constitutes an active field of study. Moreover, once the desired biological effect is achieved, issues such as its switch-off and the related question of possible reverse extracellular transport still stand open. On a related tune, unintended PDC effects on tissues/cells—avoided at the entry but unknown at the exit phase—need to be explored.
(4) An equally open challenge, as novel PDCs continue to be developed, is the fine-tuning of activities between the CPP component and its small molecule cargo, to avoid potency disparities.
(5) In vivo trials of PDCs into the pipeline must include not only mandatory toxicity and immunogenicity tests, but also, for antiviral PDCs, ensure that viral inactivation does not entail any deleterious effects on the host cell’s machinery [121].
(6) Finally, given the high genetic and antigenic heterogeneity of viruses, multivalency screens on different strains/isolates must be performed to avoid that conjugates with promise against one particular virus are inefficient against other types or subtypes.
The speed at which these challenges are addressed will determine, to a great extent, the future of antiviral therapies using PDCs and their ability to compete/replace existing unmodified drug formulations.
Author Contributions
Conceptualization, T.T. and D.A.; validation, T.T., D.K. and D.A.; resources, T.T., D.K. and D.A.; writing—original draft preparation, T.T.; writing—review and editing, T.T, D.K. and D.A.; supervision, D.A.; funding acquisition, D.K. and D.A. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the La Caixa Health Foundation (project HR17_00409, ID 100010434, agreement LCF/PR/HR17/52150011) and by the European Union (H2020-FETOPEN-2018-2019-2020-01 grant no. 828774). DK was supported by the Croatian Science Foundation (UIP-2019-04-7999). The Department of Medicine and Life Sciences, Pompeu Fabra University, belongs to the María de Maeztu network of Units of Excellence, funded by the Spanish MICINN and AEI (CEX2018-000792-M).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roychoudhury, S.; Das, A.; Sengupta, P.; Dutta, S.; Roychoudhury, S.; Choudhury, A.P.; Ahmed, A.B.F.; Bhattacharjee, S.; Slama, P. Viral Pandemics of the Last Four Decades: Pathophysiology, Health Impacts and Perspectives. Int. J. Environ. Res. Pub. Health 2020, 17, 9411. [Google Scholar] [CrossRef] [PubMed]
- Falcinelli, S.D.; Chertow, D.S.; Kindrachuk, J. Integration of Global Analyses of Host Molecular Responses with Clinical Data to Evaluate Pathogenesis and Advance Therapies for Emerging and Re-Emerging Viral Infections. ACS Infect. Dis. 2016, 2, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Ka-Wai Hui, E. Reasons for the Increase in Emerging and Re-Emerging Viral Infectious Diseases. Microbes. Infect. 2006, 8, 905–916. [Google Scholar] [CrossRef]
- Nováková, L.; Pavlík, J.; Chrenková, L.; Martinec, O.; Červený, L. Current Antiviral Drugs and Their Analysis in Biological Materials—Part II: Antivirals against Hepatitis and HIV Viruses. J. Pharm. Biomed. Anal. 2018, 147, 378–399. [Google Scholar] [CrossRef]
- Szunerits, S.; Barras, A.; Khanal, M.; Pagneux, Q.; Boukherroub, R. Nanostructures for the Inhibition of Viral Infections. Molecules 2015, 20, 14051–14081. [Google Scholar] [CrossRef]
- Irwin, K.K.; Renzette, N.; Kowalik, T.F.; Jensen, J.D. Antiviral Drug Resistance as an Adaptive Process. Virus Evol. 2016, 2, vew014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Lou, Z.; Sun, Y.; Rao, Z. Current Progress in Antiviral Strategies. Trends Pharmacol. Sci. 2014, 35, 86–102. [Google Scholar] [CrossRef]
- Divyashree, M.; Mani, M.K.; Reddy, D.; Kumavath, R.; Ghosh, P.; Azevedo, V.; Barh, D. Clinical Applications of Antimicrobial Peptides (AMPs): Where Do We Stand Now? Protein Pept. Lett. 2020, 27, 120–134. [Google Scholar] [CrossRef]
- Jhong, J.-H.; Chi, Y.-H.; Li, W.-C.; Lin, T.-H.; Huang, K.-Y.; Lee, T.-Y. DbAMP: An Integrated Resource for Exploring Antimicrobial Peptides with Functional Activities and Physicochemical Properties on Transcriptome and Proteome Data. Nucleic Acids Res. 2019, 47, D285–D297. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Järver, P.; Mäger, I.; Langel, Ü. In Vivo Biodistribution and Efficacy of Peptide Mediated Delivery. Trends Pharmacol. Sci. 2010, 31, 528–535. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein Transduction: Unrestricted Delivery into All Cells? Trends Cell Biol. 2000, 10, 290–295. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A Repository of Experimentally Validated Cell-Penetrating Peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [Green Version]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell Penetration by Transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef]
- Lindgren, M.; Hällbrink, M.; Prochiantz, A.; Langel, Ü. Cell-Penetrating Peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell Penetrating Peptides: A Concise Review with Emphasis on Biomedical Applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, Ü. Cell-Penetrating Peptides: Design, Synthesis, and Applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Nilvebrant, J.; Nygren, P.-Å.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- al Shaer, D.; al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef]
- He, R.; Finan, B.; Mayer, J.P.; DiMarchi, R.D. Peptide Conjugates with Small Molecules Designed to Enhance Efficacy and Safety. Molecules 2019, 24, 1855. [Google Scholar] [CrossRef] [Green Version]
- Balogh, B.; Ivánczi, M.; Nizami, B.; Beke-Somfai, T.; Mándity, I.M. ConjuPepDB: A Database of Peptide–Drug Conjugates. Nucleic Acids Res. 2021, 49, D1102–D1112. [Google Scholar] [CrossRef]
- Liang, G.; Wang, H.; Chong, H.; Cheng, S.; Jiang, X.; He, Y.; Wang, C.; Liu, K. An Effective Conjugation Strategy for Designing Short Peptide-Based HIV-1 Fusion Inhibitors. Org. Biomol. Chem. 2016, 14, 7875–7882. [Google Scholar] [CrossRef]
- Wang, C.; Shi, W.; Cai, L.; Lu, L.; Wang, Q.; Zhang, T.; Li, J.; Zhang, Z.; Wang, K.; Xu, L.; et al. Design, Synthesis, and Biological Evaluation of Highly Potent Small Molecule–Peptide Conjugates as New HIV-1 Fusion Inhibitors. J. Med. Chem. 2013, 56, 2527–2539. [Google Scholar] [CrossRef]
- Zhou, L.; Thakur, C.S.; Molinaro, R.J.; Paranjape, J.M.; Hoppes, R.; Jeang, K.-T.; Silverman, R.H.; Torrence, P.F. Delivery of 2-5A Cargo into Living Cells Using the Tat Cell Penetrating Peptide: 2-5A-Tat. Bioorg. Med. Chem. 2006, 14, 7862–7874. [Google Scholar] [CrossRef]
- García-Aparicio, C.; Diez-Torrubia, A.; Balzarini, J.; Lambeir, A.-M.; Velázquez, S.; Camarasa, M.-J. Efficient Conversion of Tetrapeptide-Based TSAO Prodrugs to the Parent Drug by Dipeptidyl-Peptidase IV (DPPIV/CD26). Antivir. Res. 2007, 76, 130–139. [Google Scholar] [CrossRef]
- Diez-Torrubia, A.; Cabrera, S.; de Castro, S.; García-Aparicio, C.; Mulder, G.; de Meester, I.; Camarasa, M.-J.; Balzarini, J.; Velázquez, S. Novel Water-Soluble Prodrugs of Acyclovir Cleavable by the Dipeptidyl-Peptidase IV (DPP IV/CD26) Enzyme. Eur. J. Med. Chem. 2013, 70, 456–468. [Google Scholar] [CrossRef]
- Liotard, J.-F.; Mehiri, M.; di Giorgio, A.; Boggetto, N.; Reboud-Ravaux, M.; Aubertin, A.-M.; Condom, R.; Patino, N. AZT and AZT-Monophosphate Prodrugs Incorporating HIV-Protease Substrate Fragment: Synthesis and Evaluation as Specific Drug Delivery Systems. Antivir. Chem. Chemother. 2006, 17, 193–213. [Google Scholar] [CrossRef]
- Nitsche, C.; Schreier, V.N.; Behnam, M.A.M.; Kumar, A.; Bartenschlager, R.; Klein, C.D. Thiazolidinone–Peptide Hybrids as Dengue Virus Protease Inhibitors with Antiviral Activity in Cell Culture. J. Med. Chem. 2013, 56, 8389–8403. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, Y.; Lei, Y.; Wang, R.; Zhan, M.; Liu, J.; An, Y.; Zhou, Y.; Zhan, J.; Yin, F.; et al. Design and Evaluation of a Novel Peptide–Drug Conjugate Covalently Targeting SARS-CoV-2 Papain-like Protease. J. Med. Chem. 2022, 65, 876–884. [Google Scholar] [CrossRef]
- Lan, Q.; Wang, C.; Zhou, J.; Wang, L.; Jiao, F.; Zhang, Y.; Cai, Y.; Lu, L.; Xia, S.; Jiang, S. 25-Hydroxycholesterol-Conjugated EK1 Peptide with Potent and Broad-Spectrum Inhibitory Activity against SARS-CoV-2, Its Variants of Concern, and Other Human Coronaviruses. Int. J. Mol. Sci. 2021, 22, 11869. [Google Scholar] [CrossRef]
- Lan, Q.; Chan, J.F.-W.; Xu, W.; Wang, L.; Jiao, F.; Zhang, G.; Pu, J.; Zhou, J.; Xia, S.; Lu, L.; et al. A Palmitic Acid-Conjugated, Peptide-Based Pan-CoV Fusion Inhibitor Potently Inhibits Infection of SARS-CoV-2 Omicron and Other Variants of Concern. Viruses 2022, 14, 549. [Google Scholar] [CrossRef]
- Rosenke, K.; Leventhal, S.; Moulton, H.M.; Hatlevig, S.; Hawman, D.; Feldmann, H.; Stein, D.A. Inhibition of SARS-CoV-2 in Vero Cell Cultures by Peptide-Conjugated Morpholino Oligomers. J. Antimicrob. Chemother. 2021, 76, 413–417. [Google Scholar] [CrossRef]
- Deas, T.S.; Binduga-Gajewska, I.; Tilgner, M.; Ren, P.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kauffman, E.B.; Kramer, L.D.; Shi, P.-Y. Inhibition of Flavivirus Infections by Antisense Oligomers Specifically Suppressing Viral Translation and RNA Replication. J. Virol. 2005, 79, 4599–4609. [Google Scholar] [CrossRef] [Green Version]
- Deas, T.S.; Bennett, C.J.; Jones, S.A.; Tilgner, M.; Ren, P.; Behr, M.J.; Stein, D.A.; Iversen, P.L.; Kramer, L.D.; Bernard, K.A.; et al. In Vitro Resistance Selection and In Vivo Efficacy of Morpholino Oligomers against West Nile Virus. Antimicrob. Agents Chemother. 2007, 51, 2470–2482. [Google Scholar] [CrossRef]
- Stone, J.K.; Rijnbrand, R.; Stein, D.A.; Ma, Y.; Yang, Y.; Iversen, P.L.; Andino, R. A Morpholino Oligomer Targeting Highly Conserved Internal Ribosome Entry Site Sequence Is Able To Inhibit Multiple Species of Picornavirus. Antimicrob. Agents Chemother. 2008, 52, 1970–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrer, R.; Neuman, B.W.; Ting, J.P.C.; Stein, D.A.; Moulton, H.M.; Iversen, P.L.; Kuhn, P.; Buchmeier, M.J. Antiviral Effects of Antisense Morpholino Oligomers in Murine Coronavirus Infection Models. J. Virol. 2007, 81, 5637–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Stein, D.A.; Lim, T.; Qiu, D.; Coughlin, S.; Liu, Z.; Wang, Y.; Blouch, R.; Moulton, H.M.; Iversen, P.L.; et al. Inhibition of Coxsackievirus B3 in Cell Cultures and in Mice by Peptide-Conjugated Morpholino Oligomers Targeting the Internal Ribosome Entry Site. J. Virol. 2006, 80, 11510–11519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paessler, S.; Rijnbrand, R.; Stein, D.A.; Ni, H.; Yun, N.E.; Dziuba, N.; Borisevich, V.; Seregin, A.; Ma, Y.; Blouch, R.; et al. Inhibition of Alphavirus Infection in Cell Culture and in Mice with Antisense Morpholino Oligomers. Virology 2008, 376, 357–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.-H.; Stein, D.A.; Guerrero-Plata, A.; Liao, S.-L.; Ivanciuc, T.; Hong, C.; Iversen, P.L.; Casola, A.; Garofalo, R.P. Inhibition of Respiratory Syncytial Virus Infections With Morpholino Oligomers in Cell Cultures and in Mice. Mol. Ther. 2008, 16, 1120–1128. [Google Scholar] [CrossRef]
- Sleeman, K.; Stein, D.A.; Tamin, A.; Reddish, M.; Iversen, P.L.; Rota, P.A. Inhibition of Measles Virus Infections in Cell Cultures by Peptide-Conjugated Morpholino Oligomers. Virus Res. 2009, 140, 49–56. [Google Scholar] [CrossRef]
- Ge, Q.; Pastey, M.; Kobasa, D.; Puthavathana, P.; Lupfer, C.; Bestwick, R.K.; Iversen, P.L.; Chen, J.; Stein, D.A. Inhibition of Multiple Subtypes of Influenza A Virus in Cell Cultures with Morpholino Oligomers. Antimicrob. Agents Chemother. 2006, 50, 3724–3733. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, G.; Nordmann, A.; Stein, D.A.; Iversen, P.L.; Klenk, H.-D. Morpholino Oligomers Targeting the PB1 and NP Genes Enhance the Survival of Mice Infected with Highly Pathogenic Influenza A H7N7 Virus. J. Gen. Virol. 2008, 89, 939–948. [Google Scholar] [CrossRef]
- Lupfer, C.; Stein, D.A.; Mourich, D.v.; Tepper, S.E.; Iversen, P.L.; Pastey, M. Inhibition of Influenza A H3N8 Virus Infections in Mice by Morpholino Oligomers. Arch. Virol. 2008, 153, 929–937. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Bonaparte, R.S.; Patel, D.; Stein, D.A.; Iversen, P.L. Blockade of Viral Interleukin-6 Expression of Kaposi’s Sarcoma–Associated Herpesvirus. Mol. Cancer Ther. 2008, 7, 712–720. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Stein, D.A.; Eide, K.; Blouch, R.E.; Bildfell, R.; Iversen, P.; Jin, L. Inhibition of HSV-1 Ocular Infection with Morpholino Oligomers Targeting ICP0 and ICP27. Antivir. Res. 2009, 84, 131–141. [Google Scholar] [CrossRef]
- Kinney, R.M.; Huang, C.Y.-H.; Rose, B.C.; Kroeker, A.D.; Dreher, T.W.; Iversen, P.L.; Stein, D.A. Inhibition of Dengue Virus Serotypes 1 to 4 in Vero Cell Cultures with Morpholino Oligomers. J. Virol. 2005, 79, 5116–5128. [Google Scholar] [CrossRef] [Green Version]
- Holden, K.L.; Stein, D.A.; Pierson, T.C.; Ahmed, A.A.; Clyde, K.; Iversen, P.L.; Harris, E. Inhibition of Dengue Virus Translation and RNA Synthesis by a Morpholino Oligomer Targeted to the Top of the Terminal 3′ Stem–Loop Structure. Virology 2006, 344, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.A.; Huang, C.Y.-H.; Silengo, S.; Amantana, A.; Crumley, S.; Blouch, R.E.; Iversen, P.L.; Kinney, R.M. Treatment of AG129 Mice with Antisense Morpholino Oligomers Increases Survival Time Following Challenge with Dengue 2 Virus. J. Antimicrob. Chemother. 2008, 62, 555–565. [Google Scholar] [CrossRef]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Churchill, M.J.; Kim, A.M.; Kuhn, P.; Dawson, P.; Moulton, H.M.; Bestwick, R.K.; Iversen, P.L.; et al. Inhibition, Escape, and Attenuated Growth of Severe Acute Respiratory Syndrome Coronavirus Treated with Antisense Morpholino Oligomers. J. Virol. 2005, 79, 9665–9676. [Google Scholar] [CrossRef] [Green Version]
- van den Born, E.; Stein, D.A.; Iversen, P.L.; Snijder, E.J. Antiviral Activity of Morpholino Oligomers Designed to Block Various Aspects of Equine Arteritis Virus Amplification in Cell Culture. J. Gen. Virol. 2005, 86, 3081–3090. [Google Scholar] [CrossRef]
- Vagnozzi, A.; Stein, D.A.; Iversen, P.L.; Rieder, E. Inhibition of Foot-and-Mouth Disease Virus Infections in Cell Cultures with Antisense Morpholino Oligomers. J. Virol. 2007, 81, 11669–11680. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Stein, D.A.; Kroeker, A.D.; Paulino, A.D.; Moulton, H.M.; Iversen, P.L.; Buchmeier, M.J. Antisense Morpholino-Oligomers Directed against the 5′ End of the Genome Inhibit Coronavirus Proliferation and Growth. J. Virol. 2004, 78, 5891–5899. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Stein, D.A.; Fan, S.-M.; Wang, K.-Y.; Kroeker, A.D.; Meng, X.-J.; Iversen, P.L.; Matson, D.O. Suppression of Porcine Reproductive and Respiratory Syndrome Virus Replication by Morpholino Antisense Oligomers. Vet. Microbiol. 2006, 117, 117–129. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Wang, K.-Y.; Stein, D.A.; Patel, D.; Watkins, R.; Moulton, H.M.; Iversen, P.L.; Matson, D.O. Inhibition of Replication and Transcription Activator and Latency-Associated Nuclear Antigen of Kaposi’s Sarcoma-Associated Herpesvirus by Morpholino Oligomers. Antivir. Res. 2007, 73, 12–23. [Google Scholar] [CrossRef]
- Swenson, D.L.; Warfield, K.L.; Warren, T.K.; Lovejoy, C.; Hassinger, J.N.; Ruthel, G.; Blouch, R.E.; Moulton, H.M.; Weller, D.D.; Iversen, P.L.; et al. Chemical Modifications of Antisense Morpholino Oligomers Enhance Their Efficacy against Ebola Virus Infection. Antimicrob. Agents Chemother. 2009, 53, 2089–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enterlein, S.; Warfield, K.L.; Swenson, D.L.; Stein, D.A.; Smith, J.L.; Gamble, C.S.; Kroeker, A.D.; Iversen, P.L.; Bavari, S.; Mühlberger, E. VP35 Knockdown Inhibits Ebola Virus Amplification and Protects against Lethal Infection in Mice. Antimicrob. Agents Chemother. 2006, 50, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, N.; Basu, A.; Palumbo, P.; Myers, R.L.; Pandey, V.N. Anti-TAR Polyamide Nucleotide Analog Conjugated with a Membrane-Permeating Peptide Inhibits Human Immunodeficiency Virus Type 1 Production. J. Virol. 2002, 76, 3881–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaubey, B.; Tripathi, S.; Ganguly, S.; Harris, D.; Casale, R.A.; Pandey, V.N. A PNA-Transportan Conjugate Targeted to the TAR Region of the HIV-1 Genome Exhibits Both Antiviral and Virucidal Properties. Virology 2005, 331, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, S. Anti-HIV-1 Activity of Anti-TAR Polyamide Nucleic Acid Conjugated with Various Membrane Transducing Peptides. Nucleic Acids Res. 2005, 33, 4345–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, S.; Chaubey, B.; Barton, B.E.; Pandey, V.N. Anti HIV-1 Virucidal Activity of Polyamide Nucleic Acid-Membrane Transducing Peptide Conjugates Targeted to Primer Binding Site of HIV-1 Genome. Virology 2007, 363, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.J. Cell-Penetrating Peptide Conjugates of Peptide Nucleic Acids (PNA) as Inhibitors of HIV-1 Tat-Dependent Trans-Activation in Cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, A.; Ponzio, N.M.; Pandey, V.N. Immunological Response to Peptide Nucleic Acid and Its Peptide Conjugate Targeted to Transactivation Response (TAR) Region of HIV-1 RNA Genome. Oligonucleotides 2008, 18, 329–335. [Google Scholar] [CrossRef]
- Chaubey, B.; Tripathi, S.; Pandey, V.N. Single Acute-Dose and Repeat-Doses Toxicity of Anti-HIV-1 PNA TAR –Penetratin Conjugate after Intraperitoneal Administration to Mice. Oligonucleotides 2008, 18, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, S.; Chaubey, B.; Tripathi, S.; Upadhyay, A.; Neti, P.V.S.V.; Howell, R.W.; Pandey, V.N. Pharmacokinetic Analysis of Polyamide Nucleic-Acid-Cell Penetrating Peptide Conjugates Targeted against HIV-1 Transactivation Response Element. Oligonucleotides 2008, 18, 277–286. [Google Scholar] [CrossRef]
- Yoo, J.-S.; Kim, C.-M.; Kim, J.-H.; Kim, J.-Y.; Oh, J.-W. Inhibition of Japanese Encephalitis Virus Replication by Peptide Nucleic Acids Targeting Cis-Acting Elements on the plus- and Minus-Strands of Viral RNA. Antivir. Res. 2009, 82, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Han, S.; Hong, W.; Lang, Y.; Li, F.; Liu, Y.; Li, Z.; Wu, Y.; Li, W.; Zhang, X.; et al. A Tat-Conjugated Peptide Nucleic Acid Tat-PNA-DR Inhibits Hepatitis B Virus Replication In Vitro and In Vivo by Targeting LTR Direct Repeats of HBV RNA. Mol. Ther. Nucleic Acids 2016, 5, e295. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.-G.; Shim, S.-B.; Moon, J.-E.; Kim, J.-H.; Kim, S.-J.; Oh, J.-W. Interference of Hepatitis C Virus Replication in Cell Culture by Antisense Peptide Nucleic Acids Targeting the X-RNA. J. Viral Hepat. 2011, 18, e298–e306. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.-G.; Lee, W.; Choi, J.-K.; Kim, S.-J.; Plant, E.P.; Almazán, F.; Taylor, D.R.; Enjuanes, L.; Oh, J.-W. Interference of Ribosomal Frameshifting by Antisense Peptide Nucleic Acids Suppresses SARS Coronavirus Replication. Antivir. Res. 2011, 91, 1–10. [Google Scholar] [CrossRef]
- Meng, S.; Wei, B.; Xu, R.; Zhang, K.; Wang, L.; Zhang, R.; Li, J. TAT Peptides Mediated Small Interfering RNA Delivery to Huh-7 Cells and Efficiently Inhibited Hepatitis C Virus RNA Replication. Intervirology 2009, 52, 135–140. [Google Scholar] [CrossRef]
- Kumar, P.; Ban, H.-S.; Kim, S.-S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; et al. T Cell-Specific SiRNA Delivery Suppresses HIV-1 Infection in Humanized Mice. Cell 2008, 134, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Bivalkar-Mehla, S.; Mehla, R.; Chauhan, A. Chimeric Peptide-Mediated SiRNA Transduction to Inhibit HIV-1 Infection. J. Drug Target 2017, 25, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ren, W.; Liu, Q.; Tan, Z.; Li, J.; Tong, C. Transportan-Derived Cell-Penetrating Peptide Delivers SiRNA to Inhibit Replication of Influenza Virus in Vivo. Drug Des. Devel. Ther. 2019, 13, 1059. [Google Scholar] [CrossRef] [Green Version]
- Mino, T.; Mori, T.; Aoyama, Y.; Sera, T. Cell-Permeable Artificial Zinc-Finger Proteins as Potent Antiviral Drugs for Human Papillomaviruses. Arch. Virol. 2008, 153, 1291–1298. [Google Scholar] [CrossRef]
- Chu, X.; Wu, B.; Fan, H.; Hou, J.; Hao, J.; Hu, J.; Wang, B.; Liu, G.; Li, C.; Meng, S. PTD-Fused P53 as a Potential Antiviral Agent Directly Suppresses HBV Transcription and Expression. Antivir. Res. 2016, 127, 41–49. [Google Scholar] [CrossRef]
- Jung, H.; Oh, J.; Lee, H. Cell-Penetrating Mx1 Enhances Anti-Viral Resistance against Mucosal Influenza Viral Infection. Viruses 2019, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, D.A.; Bakker, M.; Cruz-Oliveira, C.; Neves, V.; Jiménez, M.A.; Defaus, S.; Cavaco, M.; Veiga, A.S.; Cadima-Couto, I.; Castanho, M.A.R.B.; et al. Penetrating the Blood-Brain Barrier with New Peptide–Porphyrin Conjugates Having Anti-HIV Activity. Bioconjug. Chem. 2021, 32, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Todorovski, T.; Mendonça, D.A.; Fernandes-Siqueira, L.O.; Cruz-Oliveira, C.; Guida, G.; Valle, J.; Cavaco, M.; Limas, F.I.V.; Neves, V.; Cadima-Couto, Í.; et al. Targeting Zika Virus with New Brain- and Placenta-Crossing Peptide–Porphyrin Conjugates. Pharmaceutics 2022, 14, 738. [Google Scholar] [CrossRef] [PubMed]
- Saarbach, J.; Sabale, P.M.; Winssinger, N. Peptide Nucleic Acid (PNA) and Its Applications in Chemical Biology, Diagnostics, and Therapeutics. Curr. Opin. Chem. Biol. 2019, 52, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; McQuistan, T.J.; Stanek, J.W.; Summerton, J.E.; Mata, J.E.; Squier, T.C. Detection of Unique Ebola Virus Oligonucleotides Using Fluorescently-Labeled Phosphorodiamidate Morpholino Oligonucleotide Probe Pairs. Anal. Biochem. 2018, 557, 84–90. [Google Scholar] [CrossRef]
- Pärn, K.; Eriste, E.; Langel, Ü. The Antimicrobial and Antiviral Applications of Cell-Penetrating Peptides. Methods Mol. Biol. 2015, 1324, 223–245. [Google Scholar]
- Sadiq, I.Z.; Muhammad, A.; Mada, S.B.; Ibrahim, B.; Umar, U.A. Biotherapeutic Effect of Cell-Penetrating Peptides against Microbial Agents: A Review. Tissue Barriers 2022, 10, 1995285. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-Penetrating Peptides: Design Strategies beyond Primary Structure and Amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; Defaus, S.; Andreu, D. 1988–2018: Thirty Years of Drug Smuggling at the Nano Scale. Challenges and Opportunities of Cell-Penetrating Peptides in Biomedical Research. Arch. Biochem. Biophys. 2019, 661, 74–86. [Google Scholar] [CrossRef]
- Sánchez-Navarro, M.; Giralt, E. Peptide Shuttles for Blood–Brain Barrier Drug Delivery. Pharmaceutics 2022, 14, 1874. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Mauša, G.; Todorovski, T.; Giralt, E. Algorithm-Supported, Mass and Sequence Diversity-Oriented Random Peptide Library Design. J. Cheminform. 2019, 11, 25. [Google Scholar] [CrossRef]
- Otović, E.; Njirjak, M.; Kalafatovic, D.; Mauša, G. Sequential Properties Representation Scheme for Recurrent Neural Network-Based Prediction of Therapeutic Peptides. J. Chem. Inf. Model 2022, 62, 2961–2972. [Google Scholar] [CrossRef]
- Torrent, M.; di Tommaso, P.; Pulido, D.; Nogués, M.V.; Notredame, C.; Boix, E.; Andreu, D. AMPA: An Automated Web Server for Prediction of Protein Antimicrobial Regions. Bioinformatics 2012, 28, 130–131. [Google Scholar] [CrossRef] [Green Version]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug. Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, A.A.; Reichert, J.M. Future Directions for Peptide Therapeutics Development. Drug. Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef]
- Böhme, D.; Beck-Sickinger, A.G. Controlling Toxicity of Peptide-Drug Conjugates by Different Chemical Linker Structures. Chem. Med. Chem. 2015, 10, 804–814. [Google Scholar] [CrossRef]
- Mahesh, S.; Tang, K.-C.; Raj, M. Amide Bond Activation of Biological Molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [Green Version]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Müller, T.D.; Yi, C.-X.; Habegger, K.; Schriever, S.C.; García-Cáceres, C.; Kabra, D.G.; et al. Targeted Estrogen Delivery Reverses the Metabolic Syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, T.R.; Dai, Z.; Zhang, Y.; Julian, R.R. A Two-Trick Pony: Lysosomal Protease Cathepsin B Possesses Surprising Ligase Activity. RSC Chem. Biol. 2021, 2, 606–611. [Google Scholar] [CrossRef]
- Tugyi, R.; Mezõ, G.; Gitta, S.; Fellinger, E.; Andreu, D.; Hudecz, F. Effect of Conjugation with Polypeptide Carrier on the Enzymatic Degradation of Herpes Simplex Virus Glycoprotein D Derived Epitope Peptide. Bioconjug. Chem. 2008, 19, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Marqués-Gallego, P.; de Kroon, A.I.P.M. Ligation Strategies for Targeting Liposomal Nanocarriers. Biomed. Res. Int. 2014, 2014, 129458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spicer, C.D.; Jumeaux, C.; Gupta, B.; Stevens, M.M. Peptide and Protein Nanoparticle Conjugates: Versatile Platforms for Biomedical Applications. Chem. Soc. Rev. 2018, 47, 3574–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitsche, C.; Behnam, M.A.M.; Steuer, C.; Klein, C.D. Retro Peptide-Hybrids as Selective Inhibitors of the Dengue Virus NS2B-NS3 Protease. Antivir. Res. 2012, 94, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299. [Google Scholar] [CrossRef]
- Abes, S.; Moulton, H.M.; Clair, P.; Prevot, P.; Youngblood, D.S.; Wu, R.P.; Iversen, P.L.; Lebleu, B. Vectorization of Morpholino Oligomers by the (R-Ahx-R)4 Peptide Allows Efficient Splicing Correction in the Absence of Endosomolytic Agents. J. Control. Release 2006, 116, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Stenzel, M.H. Bioconjugation Using Thiols: Old Chemistry Rediscovered to Connect Polymers with Nature’s Building Blocks. ACS Macro Lett. 2013, 2, 14–18. [Google Scholar] [CrossRef]
- Fishkin, N.; Maloney, E.K.; Chari, R.V.J.; Singh, R. A Novel Pathway for Maytansinoid Release from Thioether Linked Antibody–Drug Conjugates (ADCs) under Oxidative Conditions. Chem. Commun. 2011, 47, 10752–10754. [Google Scholar] [CrossRef]
- Baldwin, A.D.; Kiick, K.L. Tunable Degradation of Maleimide–Thiol Adducts in Reducing Environments. Bioconjug. Chem. 2011, 22, 1946–1953. [Google Scholar] [CrossRef] [Green Version]
- Andrieu, J.; Re, F.; Russo, L.; Nicotra, F. Phage-Displayed Peptides Targeting Specific Tissues and Organs. J. Drug Target 2019, 27, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Saw, P.E.; Song, E.-W. Phage Display Screening of Therapeutic Peptide for Cancer Targeting and Therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and Applications of Peptide Phage Display Technology in Biomedical Science. J. Biomed. Sci. 2016, 23, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Sun, Y.; Lee, R.J.; Wang, G.; Zheng, X.; Zhang, H.; Fu, Y.; Yan, G.; Wang, Y.; Deng, W.; et al. Folate Receptor-Targeted Albumin Nanoparticles Based on Microfluidic Technology to Deliver Cabazitaxel. Cancers 2019, 11, 1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yang, S.; Chai, H.; Yang, Z.; Lee, R.J.; Liao, W.; Teng, L. A Novel Isoquinoline Derivative Anticancer Agent and Its Targeted Delivery to Tumor Cells Using Transferrin-Conjugated Liposomes. PLoS ONE 2015, 10, e0136649. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shin, M.C.; Liang, Q.; He, H.; Yang, V.C. 15 Years of ATTEMPTS: A Macromolecular Drug Delivery System Based on the CPP-Mediated Intracellular Drug Delivery and Antibody Targeting. J. Control. Release 2015, 205, 58–69. [Google Scholar] [CrossRef]
- Orange, J.S.; May, M.J. Cell Penetrating Peptide Inhibitors of Nuclear Factor-Kappa B. Cellular and Molecular Life Sci. 2008, 65, 3564–3591. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent Progress of Cell-Penetrating Peptides as New Carriers for Intracellular Cargo Delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef]
- Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Gavriel, A.G.; Sambrook, M.R.; Russell, A.T.; Hayes, W. Recent Advances in Self-Immolative Linkers and Their Applications in Polymeric Reporting Systems. Polym. Chem. 2022, 13, 3188–3269. [Google Scholar] [CrossRef]
- Dal Corso, A.; Arosio, S.; Arrighetti, N.; Perego, P.; Belvisi, L.; Pignataro, L.; Gennari, C. A Trifunctional Self-Immolative Spacer Enables Drug Release with Two Non-Sequential Enzymatic Cleavages. Chem. Commun. 2021, 57, 7778–7781. [Google Scholar] [CrossRef]
- Viru, L.; Heller, G.; Lehto, T.; Pärn, K.; el Andaloussi, S.; Langel, Ü.; Merits, A. Novel Viral Vectors Utilizing Intron Splice-Switching to Activate Genome Rescue, Expression and Replication in Targeted Cells. Virol. J. 2011, 8, 243. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Types of cargoes delivered into cells by CPPs (data from [14]).
Figure 1.
Types of cargoes delivered into cells by CPPs (data from [14]).

Figure 2.
Peptides and peptide conjugates entering clinical trials since 1980s (data from [11]).
Figure 2.
Peptides and peptide conjugates entering clinical trials since 1980s (data from [11]).

Figure 3.
Conjugation chemistries applied in the synthesis of antiviral PDCs. Labels (i, ii, iii-A, iii-B and iv) as described in the text above. In amide bond formation (i), conjugation is performed while the CPP is anchored on the solid support. In thioether bond formation (iii-B), a thiol-maleimide reaction is depicted. In (iv), copper (I)-catalyzed alkyne-azide cycloaddition is shown as a representative click reaction.
Figure 3.
Conjugation chemistries applied in the synthesis of antiviral PDCs. Labels (i, ii, iii-A, iii-B and iv) as described in the text above. In amide bond formation (i), conjugation is performed while the CPP is anchored on the solid support. In thioether bond formation (iii-B), a thiol-maleimide reaction is depicted. In (iv), copper (I)-catalyzed alkyne-azide cycloaddition is shown as a representative click reaction.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Reported CPPs and categories (from CPPsite 2.0, [14]).
Table 1.
Reported CPPs and categories (from CPPsite 2.0, [14]).
| Total Numbers | Percentage (%) a | ||
|---|---|---|---|
| Sequence type | Linear | 1753 | 94.5 |
| Cyclic | 102 | 5.5 | |
| Peptide class | Cationic | 714 | 38.5 |
| Amphipathic | 391 | 21.1 | |
| Origin | Protein | 774 | 41.7 |
| Synthetic | 1017 | 54.8 | |
| Chimeric | 64 | 3.5 | |
| Chirality | L | 1564 | 84.3 |
| D | 63 | 3.4 | |
| Mixed | 32 | 1.7 | |
| Modified | 110 | 5.9 | |
| Length | Up to 5 AA | 60 | 3.2 |
| 6–10 AA | 384 | 20.7 | |
| 11–15 AA | 550 | 29.6 | |
| 16–20 AA | 446 | 24.1 | |
| 21–30 AA | 320 | 17.3 | |
| >30 AA | 95 | 5.1 | |
a Relative to the total number of peptides (1855) in the database.
Table 2.
Antiviral PDCs a reported.
| Entry | Antiviral Cargo | CPP | Conjugation Chemistry | Targeted Virus | Experimental System | Literature |
|---|---|---|---|---|---|---|
| 1 | Indole | βAla-EYAARIEALIRAAQEQQEKNEAALRE | Click chemistry | HIV-1 | Cell culture (HL2/3 and MT-2 cells) | [28] |
| 2 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 3 | Indole | βAla-EYAARIEALIRAAQEQQKKNEE | ||||
| 4 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 5 | Indole | βAla-EYAARIEALIRAAQEQQKK | ||||
| 6 | N-carboxyphenylpyrrole derivative (Gls) | |||||
| 7 | Carboxymethyl derivative of N-(3-carboxy-4-hydroxyphenyl)-2,5-dimethylpyrrole (Aoc) | βAla-NNYTSLIHSLIEESQNQQEKNEQELL | Amide bond formation | HIV-1 | Cell culture (HL2/3 and MT-2 cells) | [29] |
| 8 | Carboxymethyl derivative of N-(4-carboxy-3-hydroxyphenyl)-2,5-dimethylpyrrole (Noc) | |||||
| 9 | 2-5A 2′ 5′-phosphodiester linker oligoadenylate | GGRRKKRRQRRR (HIV-Tat) | Click chemistry | HIV | Cell culture (HeLa M cells) | [30] |
| 10 | 2-5A 2′ 5′-phosphodiester linker oligoadenylate | CGGRKKRRQRRR (HIV-Tat) | Thiol-chloroacetyl ligation | |||
| 11 | N-3 aminopropyl TSAO-T | VAVP | Amide bond formation | HIV-1 | Cell culture (Human T lymphocytic CEM and MT-4 cells) | [31] |
| 12 | VAVA | |||||
| 13 | KPDP | |||||
| 14 | Acyclovir | VPVP | Amide bond formation | HSV-1, HSV-2 | Cell culture (HEL cells) | [32] |
| 15 | VPV | Ester bond formation | ||||
| 16 | Zidovudine (AZT) | Boc-FP; Boc-NFP; Boc-FPI; Boc-NFPI; Fmoc-FP; Fmoc-NFP; Fmoc-FPI; Fmoc-NFPI; Z-FP; Z-NFP; Z-FPI; Z-NFPI; Qnc-FP; Qnc-NFP; Qnc-FPI; Qnc-NFPI | Ester bond formation | HIV-1 | Cell culture (CEM-SS TK+, CEM-SS TK- and MT-4 cells) | [33] |
| 17 | Zidovudine monophosphate (AZT-MT) | FP-OMe; FPI-OMe; NFP-OMe; NFPI-OMe; AFP-OMe; AFPI-OMe; ANFP-OMe; ANFPI-OMe | Phosphoramidate bond formation | |||
| 18 | Rhodanine b | Arg-Lys-Nle | Amide bond formation | Dengue virus, West Nile fever virus | Cell culture (Huh-7 cells) | [34] |
| 19 | Thiazolidinedione a | |||||
| 20 | GRL0617 (C20H20N2O) | ECLRGM (cyclic) | Amide bond formation | SARS-CoV-2 | Cell culture (Human kidney cells 293T; Human lung adenocarcinoma A549 cells; HCT116 cells) | [35] |
| EMLRGC (cyclic) | ||||||
| 21 | 25-Hydroxycholesterol (25-HC) | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKELGSGSG | Amide bond formation through linker | SARS-CoV-2 | Human kidney 293T cells; Huh-7 cells; RD cells; Caco2 cells | [36,37] |
| Palmitic acid (C16) | ||||||
| 22 | PMO | (RAhx c R)4 | Amide bond formation through linker | SARS-CoV-2 | Vero-E6 cells | [38] |
| 23 | PMO | (RAhxR)4-Ahx-βAla | Amide bond formation through linker; thioether bond formation through linker; | West Nile fever virus, Japanese encephalitis virus, St. Louis encephalitis virus, Coxsackievirus B2, Coxsackievirus B3, poliovirus 1, human rhinovirus 14, mouse hepatitis virus, Venezuelan equine encephalitis virus, respiratory syncytial virus, measles virus, influenza A virus, Kaposi’s sarcoma-associated herpesvirus, herpesvirus type 1 | Cell culture (KSHV-infected BC-1 and BCBL-1 cells; MDCK cells; Vero or Vero/hSLAM cells; HeLa and HL-1 cells; BHK-21 cells), in vivo mouse infection model | [39,40,41,42,43,44,45,46,47,48,49,50,51] |
| 24 | PMO | RRRRRFFRRRRC; RRRRRRRRRFFC; (RAhxR)4-Ahx-βAla | Amide bond formation through linker; thioether bond formation through linker | Dengue virus | Cell culture (Vero and BHK-21 cells), in vivo mouse infection model | [52,53,54] |
| 25 | PMO | RRRRRFFRRRRC; RRRRRRRRRFFC | Thioether bond formation through linker; | SARS-CoV1 | Cell culture (Vero-E6 cells) | [55] |
| 26 | PMO | RRRRRRRRRFFC | Thioether bond formation through linker | Equine arteritis virus, foot-and-mouth disease virus, poliovirus 1, human rhinovirus 14, coxsackievirus B2, Mouse hepatitis virus, Sindbis virus | Cell culture (BHK-21 and Vero cells; DBT cells; HeLa cells; Vero-E6 cells) | [41,44,56,57,58] |
| 27 | PMO | RRRRRFFRRRRC | Thioether bond formation through linker | Influenza A virus, porcine reproductive and respiratory syndrome virus, Kaposis sarcoma-associated herpesvirus | Cell culture (ATCC CRL11171 cell line; BC-1 and BCBL-1 cells; MDCK cells) | [47,50,59,60] |
| 28 | PMO | RRRRRRRRRFFC; (RAhxR)4-Ahx-βAla; (RβAla)8βAla; (RAhx)n=2-8βAla | Amide bond formation through linker; thioether bond formation through linker | Ebola virus | Cell culture (Vero and Vero-E6 cells) in vivo mouse infection model | [61,62] |
| 29 | PNA | CGWTLNSAGYLLGKINLKALAALAKKIL; (Npys) d GWTLNSAGYLLGKINLKALAALAKKIL; RQIKIWFQNRRMKWKK; GRKKRRQRRRPPQ; GWYLNSAGYLLGK(Cys)INLKALAALAKKIL; AGYLLGK(Cys)INLKALAALAKKIL; GWYLNSAGYLLGK(Cys)INLKALAAL; GRKKRRQRRRP; GWTLNSAGYLLGKINLKALAALAKKIL; GWYLNSAGYLLGKINLKALAALAKKIL; PKKKRKV; GRKKRRQRRRPC; RQIKIWFQNRRMKWKKGGC; RRRRRRRRRFFC; RRRRRRRQIKIWFQNRRMKWKKGGC | Disulfide bridge formation; amide bond formation through linker | HIV-1 | Cell culture (HeLa cells; 293T cells; CEM CD4+ cells; Jurkat T-cell lymphocites; Vero and Vero E6 cells). in vivo mouse infection model | [63,64,65,66,67,68,69,70] |
| 30 | PNA | GRKKRRQRRRPPQ; GRKKRRQRRRPPC; YGRRRRRRRRR; RKKRRQRRR | Amide bond formation through linker; thiol-maleimide bond formation | Japanese encephalitis virus, hepatitis B virus, hepatitis C virus, SARS-CoV | Cell culture (Huh7 cells; Vero and BHK-21 cells; HepG2.2.1.5, HepG2 and L-02 cells), in vivo mouse infection model | [71,72,73,74] |
| 31 | siRNA | CYGRKKRRQRRR; RRRRRRRRR; KETWWETWWTEWSQPGRKKRRQRRR; GWTLNSAGYLLGKINLKALAALAKKILrrrrrrrrr e | Disulfide bridge formation; thiol-maleimide bond formation; non-covalent complex formation | Hepatitis C virus, HIV-1 influenza virus | Cell culture (Huh7 cells; MDCK and A549 cells; MDM cells), in vivo mouse infection model | [75,76,77,78] |
| 32 | Protein | RRRRRRRRR; YGRKKRRQRRR | Cell expression | Human papilloma virus type 18, hepatitis B virus, mucosal influenza | Cell culture (MDCK cells; Huh7 and HepG2.2.1.5; human cell line 293H), in vivo mouse infection model | [79,80,81] |
| 33 | Porphyrin | AGILKRW AGILKRWK VQQLTKRFSL VQQLTKRFSLK SGTQEEY SGTQEEYK | Amide bond formation | HIV-1 Zika virus | Cell culture (Vero and TZM-bl cells) | [82,83] |
a We have pondered whether the acronym CPPDC would describe the conjugates more accurately than PDC, since to our best knowledge all reported PDCs contain a cell-penetrating peptide (CPP) motif, but have finally decided for the more widely accepted PDC acronym. b Twenty-six different rhodamine and thiazolidinedione substituents were evaluated. c Ahx stands for 6-aminohexanoic acid. d Npys stands for 3-nitro-2-pyridinesulfenyl. e Lower case denotes D-amino acid residues.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Todorovski, T.; Kalafatovic, D.; Andreu, D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics 2023, 15, 357. https://doi.org/10.3390/pharmaceutics15020357
AMA Style
Todorovski T, Kalafatovic D, Andreu D. Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives. Pharmaceutics. 2023; 15(2):357. https://doi.org/10.3390/pharmaceutics15020357
Chicago/Turabian StyleTodorovski, Toni, Daniela Kalafatovic, and David Andreu. 2023. "Antiviral Peptide-Based Conjugates: State of the Art and Future Perspectives" Pharmaceutics 15, no. 2: 357. https://doi.org/10.3390/pharmaceutics15020357
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.