
Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine

 , , and
, , and
Abstract
:1. Introduction
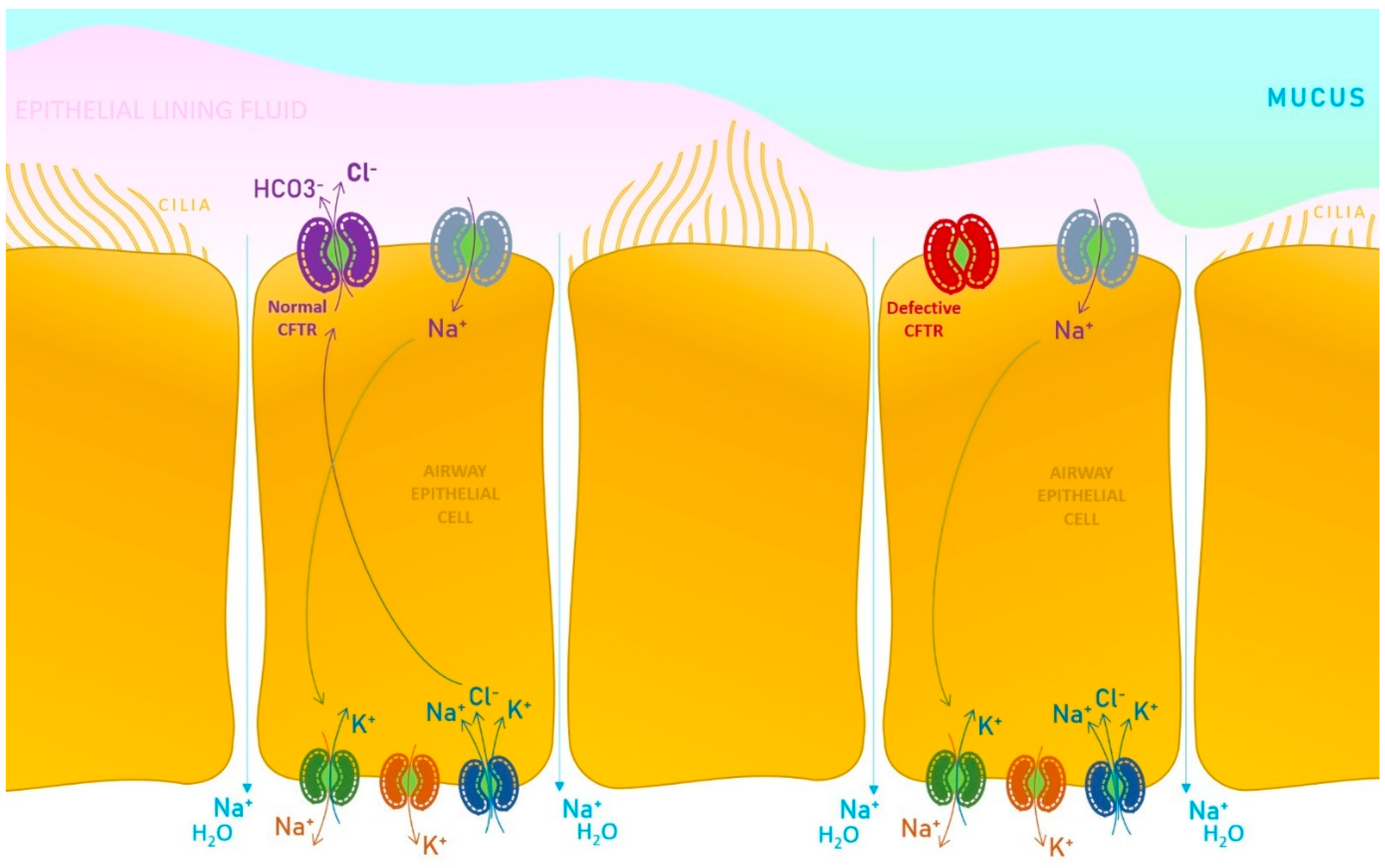
2. Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis
3. Clinical Considerations of Exocrine Pancreatic Insufficiency
3.1. Presentation
3.2. Diagnosis
3.3. Malnutrition in CF
4. Treatment
5. PERT and CFTR Modulator Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orenti, A.; Zolin, A.; Jung, A.; van Rens, J. ECFSPR Annual Report 2020. Available online: https://www.ecfs.eu/projects/ecfs-patient-registry/annual-reports (accessed on 13 November 2022).
- Erdogan, M.; Kose, M.; Pekcan, S.; Hangul, M.; Balta, B.; Kiraz, A.; Gonen, G.; Zamani, A.G.; Yildirim, M.; Ramaslı Gürsoy, T.; et al. The Genetic Analysis of Cystic Fibrosis Patients with Seven Novel Mutations in the CFTR Gene in the Central Anatolian Region of Turkey. Balk. Med. J. 2021, 38, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Qadir, A.G.; Al-Musawi, B.M.; Thejeal, R.F.; Al-Omar, S.A.-B. Molecular Analysis of CFTR Gene Mutations among Iraqi Cystic Fibrosis Patients. Egypt. J. Med. Hum. Genet. 2021, 22, 45. [Google Scholar] [CrossRef]
- CFTR2. Available online: https://cftr2.org/ (accessed on 10 October 2022).
- Farinha, C.M.; Callebaut, I. Molecular Mechanisms of Cystic Fibrosis—How Mutations Lead to Misfunction and Guide Therapy. Biosci. Rep. 2022, 42, BSR20212006. [Google Scholar] [CrossRef] [PubMed]
- Pranke, I.; Golec, A.; Hinzpeter, A.; Edelman, A.; Sermet-Gaudelus, I. Emerging Therapeutic Approaches for Cystic Fibrosis. From Gene Editing to Personalized Medicine. Front. Pharm. 2019, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.P.; Boeck, K.D. A New Era in the Treatment of Cystic Fibrosis: Correction of the Underlying CFTR Defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Haq, I.; Almulhem, M.; Soars, S.; Poulton, D.; Brodlie, M. Precision Medicine Based on CFTR Genotype for People with Cystic Fibrosis. Pharmgenom. Pers. Med. 2022, 15, 91–104. [Google Scholar] [CrossRef]
- Sheppard, D.N.; Davis, A.P. Pore-Forming Small Molecules Offer a Promising Way to Tackle Cystic Fibrosis. Nature 2019, 567, 315–317. [Google Scholar] [CrossRef] [Green Version]
- Rogan, M.P.; Stoltz, D.A.; Hornick, D.B. Cystic Fibrosis Transmembrane Conductance Regulator Intracellular Processing, Trafficking, and Opportunities for Mutation-Specific Treatment. Chest 2011, 139, 1480–1490. [Google Scholar] [CrossRef]
- Gentzsch, M.; Mall, M.A. Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Callebaut, I.; Chong, P.A.; Forman-Kay, J.D. CFTR Structure. J. Cyst. Fibros. 2018, 17, S5–S8. [Google Scholar] [CrossRef] [PubMed]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in Epithelial Physiology. Cell Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Atlas, A.; Rosh, J. Pediatric Gastroenterology and Liver Disease; Elsevier: Amsterdam, The Netherlands, 2005; pp. 999–1015. [Google Scholar]
- Hill, D.B.; Long, R.F.; Kissner, W.J.; Atieh, E.; Garbarine, I.C.; Markovetz, M.R.; Fontana, N.C.; Christy, M.; Habibpour, M.; Tarran, R.; et al. Pathological Mucus and Impaired Mucus Clearance in Cystic Fibrosis Patients Results from Increased Concentration, Not Altered PH. Eur. Respir. J. 2018, 52, 1801297. [Google Scholar] [CrossRef] [PubMed]
- Madácsy, T.; Pallagi, P.; Maleth, J. Cystic Fibrosis of the Pancreas: The Role of CFTR Channel in the Regulation of Intracellular Ca2+ Signaling and Mitochondrial Function in the Exocrine Pancreas. Front. Physiol. 2018, 9, 1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlett, C.; Ronan, N.J.; NiChroinin, M.; Mullane, D.; Plant, B.J. Partial Restoration of Pancreatic Function in a Child with Cystic Fibrosis. Lancet Respir. Med. 2016, 4, e21–e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, V.K.; Schwarzenberg, S.J. Pancreatic Insufficiency in Cystic Fibrosis. J. Cyst. Fibros. 2017, 16 (Suppl. S2), S70–S78. [Google Scholar] [CrossRef] [Green Version]
- Rachel, M.; Topolewicz, S.; Śliwczyński, A.; Galiniak, S. Managing Cystic Fibrosis in Polish Healthcare. Int. J. Environ. Res. Public Health 2020, 17, E7630. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.J.; Haverkort, E.B.; Tytgat, G.N.; van Leeuwen, D.J. Maldigestion Associated with Exocrine Pancreatic Insufficiency: Implications of Gastrointestinal Physiology and Properties of Enzyme Preparations for a Cause-Related and Patient-Tailored Treatment. Am. J. Gastroenterol. 1995, 90, 1383–1393. [Google Scholar]
- Fieker, A.; Philpott, J.; Armand, M. Enzyme Replacement Therapy for Pancreatic Insufficiency: Present and Future. Clin. Exp. Gastroenterol. 2011, 4, 55–73. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Corey, M.; Forstner, G.; Zielenski, J.; Tsui, L.-C.; Ellis, L.; Tullis, E.; Durie, P. Molecular Consequences of Cystic Fibrosis Transmembrane Regulator (CFTR) Gene Mutations in the Exocrine Pancreas. Gut 2003, 52, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkowiak, J.; Herzig, K.H.; Witt, M.; Pogorzelski, A.; Piotrowski, R.; Barra, E.; Sobczynska-Tomaszewska, A.; Trawinska-Bartnicka, M.; Strzykala, K.; Cichy, W.; et al. Analysis of Exocrine Pancreatic Function in Cystic Fibrosis: One Mild CFTR Mutation Does Not Exclude Pancreatic Insufficiency. Eur. J. Clin. Investig. 2001, 31, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.S.; Forsmark, C.E. Review Article: Pancreatic Function Testing. Aliment. Pharm. Ther. 2003, 17, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Niederau, C.; Grendell, J.H. Diagnosis of Chronic Pancreatitis. Gastroenterology 1985, 88, 1973–1995. [Google Scholar] [CrossRef] [PubMed]
- Ochi, K.; Mizushima, T.; Harada, H.; Matsumoto, S.; Matsumura, N.; Seno, T. Chronic Pancreatitis: Functional Testing. Pancreas 1998, 16, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Freswick, P.N.; Reid, E.K.; Mascarenhas, M.R. Pancreatic Enzyme Replacement Therapy in Cystic Fibrosis. Nutrients 2022, 14, 1341. [Google Scholar] [CrossRef] [PubMed]
- Walkowiak, J.; Herzig, K.-H.; Strzykala, K.; Przyslawski, J.; Krawczynski, M. Fecal Elastase-1 Is Superior to Fecal Chymotrypsin in the Assessment of Pancreatic Involvement in Cystic Fibrosis. Pediatrics 2002, 110, e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkowiak, J.; Lisowska, A.; Przyslawski, J.; Grzymislawski, M.; Krawczynski, M.; Herzig, K.H. Faecal Elastase-1 Test Is Superior to Faecal Lipase Test in the Assessment of Exocrine Pancreatic Function in Cystic Fibrosis. Acta Paediatr. 2004, 93, 1042–1045. [Google Scholar] [CrossRef]
- NHS. Available online: https://www.rbht.nhs.uk/childrencf (accessed on 10 October 2022).
- Turck, D.; Braegger, C.P.; Colombo, C.; Declercq, D.; Morton, A.; Pancheva, R.; Robberecht, E.; Stern, M.; Strandvik, B.; Wolfe, S.; et al. ESPEN-ESPGHAN-ECFS Guidelines on Nutrition Care for Infants, Children, and Adults with Cystic Fibrosis. Clin. Nutr. 2016, 35, 557–577. [Google Scholar] [CrossRef] [Green Version]
- Ng, J.; Friedmacher, F.; Pao, C.; Charlesworth, P. Gastroesophageal Reflux Disease and Need for Antireflux Surgery in Children with Cystic Fibrosis: A Systematic Review on Inciden.nce, Surgical Complications, and Postoperative Outcomes. Eur. J. Pediatr. Surg. 2020, 31, 106–114. [Google Scholar] [CrossRef]
- Stefano, M.A.; Sandy, N.S.; Zagoya, C.; Duckstein, F.; Ribeiro, A.F.; Mainz, J.G.; Lomazi, E.A. Diagnosing Constipation in Patients with Cystic Fibrosis Applying ESPGHAN Criteria. J. Cyst. Fibros. 2022, 21, 497–501. [Google Scholar] [CrossRef]
- Patel, D.; Shan, A.; Mathews, S.; Sathe, M. Understanding Cystic Fibrosis Comorbidities and Their Impact on Nutritional Management. Nutrients 2022, 14, 1028. [Google Scholar] [CrossRef]
- Ng, C.; Prayle, A.P. Gastrointestinal Complications of Cystic Fibrosis. Paediatr. Child. Health 2020, 30, 345–349. [Google Scholar] [CrossRef]
- Valamparampil, J.J.; Gupte, G.L. Cystic Fibrosis Associated Liver Disease in Children. World J. Hepatol. 2021, 13, 1727–1742. [Google Scholar] [CrossRef]
- Mouzaki, M.; Bronsky, J.; Gupte, G.; Hojsak, I.; Jahnel, J.; Pai, N.; Quiros-Tejeira, R.E.; Wieman, R.; Sundaram, S. Nutrition Support of Children With Chronic Liver Diseases: A Joint Position Paper of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Kilinc, A.A.; Beser, O.F.; Ugur, E.P.; Cokugras, F.C.; Cokugras, H. The Effects of Nutritional Status and Intervention on Pulmonary Functions in Pediatric Cystic Fibrosis Patients. Pediatr. Int. 2021, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Soltman, S.; Hicks, R.A.; Naz Khan, F.; Kelly, A. Body Composition in Individuals with Cystic Fibrosis. J. Clin. Transl. Endocrinol. 2021, 26, 100272. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Gede, N.; Ocskay, K.; Dobai, B.-M.; Abada, A.; Vereczkei, Z.; Pázmány, P.; Kató, D.; Hegyi, P.; Párniczky, A. Association of Body Mass Index With Clinical Outcomes in Patients With Cystic Fibrosis: A Systematic Review and Meta-Analysis. JAMA Netw. Open 2022, 5, e220740. [Google Scholar] [CrossRef]
- Nicolson, W.B.; Bailey, J.; Alotaibi, N.Z.; Krick, S.; Lowman, J.D. Effects of Exercise on Nutritional Status in People with Cystic Fibrosis: A Systematic Review. Nutrients 2022, 14, 933. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Hutcheon, D.; Ziegler, J. Association Between Fat-Free Mass and Pulmonary Function in Patients With Cystic Fibrosis: A Narrative Review. Nutr. Clin. Pr. 2019, 34, 715–727. [Google Scholar] [CrossRef]
- Bailey, J.; Rozga, M.; McDonald, C.M.; Bowser, E.K.; Farnham, K.; Mangus, M.; Padula, L.; Porco, K.; Alvarez, J.A. Effect of CFTR Modulators on Anthropometric Parameters in Individuals with Cystic Fibrosis: An Evidence Analysis Center Systematic Review. J. Acad. Nutr. Diet. 2021, 121, 1364–1378.e2. [Google Scholar] [CrossRef]
- Kutney, K.A.; Sandouk, Z.; Desimone, M.; Moheet, A. Obesity in Cystic Fibrosis. J. Clin. Transl. Endocrinol. 2021, 26, 100276. [Google Scholar] [CrossRef]
- Khare, S.; Harindhanavudhi, T.; Wang, Q.; Espinosa, R.; Griffin, T.; Downs, E.; Dunitz, J.; Moran, A.; Moheet, A. Factors Associated with Weight Gain and Improvement in FEV1 in People with Cystic Fibrosis on Elexacaftor-Tezacaftor-Ivacaftor. In Pediatric Pulmonology; Wiley: Hoboken, NJ, USA, 2020; Volume 55, p. S81. [Google Scholar]
- Beswick, D.M.; Humphries, S.M.; Balkissoon, C.D.; Strand, M.; Vladar, E.K.; Lynch, D.A.; Taylor-Cousar, J.L. Impact of Cystic Fibrosis Transmembrane Conductance Regulator Therapy on Chronic Rhinosinusitis and Health Status: Deep Learning CT Analysis and Patient-Reported Outcomes. Ann. Am. Thorac. Soc. 2022, 19, 12–19. [Google Scholar] [CrossRef]
- Bonhoure, A.; Boudreau, V.; Litvin, M.; Colomba, J.; Bergeron, C.; Mailhot, M.; Tremblay, F.; Lavoie, A.; Rabasa-Lhoret, R. Overweight, Obesity and Significant Weight Gain in Adult Patients with Cystic Fibrosis Association with Lung Function and Cardiometabolic Risk Factors. Clin. Nutr. 2020, 39, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- 2021 Annual Data Report. Available online: https://www.cff.org (accessed on 13 November 2022).
- Smith, M.A.; McGarry, M.E.; Ly, N.P.; Zinter, M.S. Outcomes of Children With Cystic Fibrosis Admitted to PICUs. Pediatr Crit Care Med. 2020, 21, e879–e887. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, B.A.; Goldsweig, B.K.; Sidhaye, A.; Blackman, S.M.; Schindler, T.; Moran, A. Cystic Fibrosis Related Diabetes: Nutrition and Growth Considerations. J. Cyst. Fibros. 2019, 18 (Suppl. S2), S32–S37. [Google Scholar] [CrossRef] [Green Version]
- Anthony, H.; Collins, C.E.; Davidson, G.; Mews, C.; Robinson, P.; Shepherd, R.; Stapleton, D. Pancreatic Enzyme Replacement Therapy in Cystic Fibrosis: Australian Guidelines. Pediatric Gastroenterological Society and the Dietitians Association of Australia. J. Paediatr. Child Health 1999, 35, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, D.; Baker, R.D.; Stallings, V. Consensus Report on Nutrition for Pediatric Patients with Cystic Fibrosis. J Pediatr Gastroenterol. Nutr. 2002, 35, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinaasappel, M.; Stern, M.; Littlewood, J.; Wolfe, S.; Steinkamp, G.; Heijerman, H.G.M.; Robberecht, E.; Döring, G. Nutrition in Patients with Cystic Fibrosis: A European Consensus. J. Cyst. Fibros. 2002, 1, 51–75. [Google Scholar] [CrossRef] [Green Version]
- Olsen, M.F.; Kjøller-Svarre, M.S.; Møller, G.; Katzenstein, T.L.; Nielsen, B.U.; Pressler, T.; Lewis, J.I.; Mathiesen, I.H.; Mølgaard, C.; Faurholt-Jepsen, D. Correlates of Pancreatic Enzyme Replacement Therapy Intake in Adults with Cystic Fibrosis: Results of a Cross-Sectional Study. Nutrients 2022, 14, 1330. [Google Scholar] [CrossRef]
- Rajindrajith, S.; Zeevenhooven, J.; Devanarayana, N.M.; Perera, B.J.C.; Benninga, M.A. Functional Abdominal Pain Disorders in Children. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 369–390. [Google Scholar] [CrossRef]
- Calvo-Lerma, J.; Boon, M.; Colombo, C.; de Koning, B.; Asseiceira, I.; Garriga, M.; Roca, M.; Claes, I.; Bulfamante, A.; Walet, S.; et al. Clinical Evaluation of an Evidence-Based Method Based on Food Characteristics to Adjust Pancreatic Enzyme Supplements Dose in Cystic Fibrosis. J. Cyst. Fibros. 2021, 20, e33–e39. [Google Scholar] [CrossRef]
- Kraisinger, M.; Hochhaus, G.; Stecenko, A.; Bowser, E.; Hendeles, L. Clinical Pharmacology of Pancreatic Enzymes in Patients with Cystic Fibrosis and in Vitro Performance of Microencapsulated Formulations. J. Clin. Pharm. 1994, 34, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, D.S.; Grand, R.J.; Durie, P.R. Use of Pancreatic Enzyme Supplements for Patients with Cystic Fibrosis in the Context of Fibrosing Colonopathy. J. Pediatr. 1995, 127, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation; Borowitz, D.; Robinson, K.A.; Rosenfeld, M.; Davis, S.D.; Sabadosa, K.A.; Spear, S.L.; Michel, S.H.; Parad, R.B.; White, T.B.; et al. Cystic Fibrosis Foundation Evidence-Based Guidelines for Management of Infants with Cystic Fibrosis. J. Pediatr. 2009, 155, S73–S93. [Google Scholar] [CrossRef] [PubMed]
- Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 17 December 2022).
- Baker, S.S.; Borowitz, D.; Duffy, L.; Fitzpatrick, L.; Gyamfi, J.; Baker, R.D. Pancreatic Enzyme Therapy and Clinical Outcomes in Patients with Cystic Fibrosis. J. Pediatr. 2005, 146, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.S. Delayed Release Pancrelipase for the Treatment of Pancreatic Exocrine Insufficiency Associated with Cystic Fibrosis. Ther. Clin. Risk Manag. 2008, 4, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Gifford, A.H.; Sanville, J.L.; Sathe, M.; Heltshe, S.L.; Goss, C.H. Use of Proton Pump Inhibitors Is Associated with Lower Hemoglobin Levels in People with Cystic Fibrosis. Pediatr. Pulmonol. 2021, 56, 2048–2056. [Google Scholar] [CrossRef]
- O’Keefe, S.J.; Cariem, A.K.; Levy, M. The Exacerbation of Pancreatic Endocrine Dysfunction by Potent Pancreatic Exocrine Supplements in Patients with Chronic Pancreatitis. J. Clin. Gastroenterol. 2001, 32, 319–323. [Google Scholar] [CrossRef]
- Safdi, M.; Bekal, P.K.; Martin, S.; Saeed, Z.A.; Burton, F.; Toskes, P.P. The Effects of Oral Pancreatic Enzymes (Creon 10 Capsule) on Steatorrhea: A Multicenter, Placebo-Controlled, Parallel Group Trial in Subjects with Chronic Pancreatitis. Pancreas 2006, 33, 156–162. [Google Scholar] [CrossRef]
- Stern, R.C.; Eisenberg, J.D.; Wagener, J.S.; Ahrens, R.; Rock, M.; doPico, G.; Orenstein, D.M. A Comparison of the Efficacy and Tolerance of Pancrelipase and Placebo in the Treatment of Steatorrhea in Cystic Fibrosis Patients with Clinical Exocrine Pancreatic Insufficiency. Am. J. Gastroenterol. 2000, 95, 1932–1938. [Google Scholar] [CrossRef]
- FitzSimmons, S.C.; Burkhart, G.A.; Borowitz, D.; Grand, R.J.; Hammerstrom, T.; Durie, P.R.; Lloyd-Still, J.D.; Lowenfels, A.B. High-Dose Pancreatic-Enzyme Supplements and Fibrosing Colonopathy in Children with Cystic Fibrosis. N. Engl. J. Med. 1997, 336, 1283–1289. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Questions and Answers: Delayed-Release Pancreatic Enzyme Product Receives FDA Approval. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/postmarket-drug-safety-information-patients-and-providers (accessed on 10 October 2022).
- Trapnell, B.C.; Maguiness, K.; Graff, G.R.; Boyd, D.; Beckmann, K.; Caras, S. Efficacy and Safety of Creon 24,000 in Subjects with Exocrine Pancreatic Insufficiency Due to Cystic Fibrosis. J. Cyst. Fibros. 2009, 8, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Wooldridge, J.L.; Heubi, J.E.; Amaro-Galvez, R.; Boas, S.R.; Blake, K.V.; Nasr, S.Z.; Chatfield, B.; McColley, S.A.; Woo, M.S.; Hardy, K.A.; et al. EUR-1008 Pancreatic Enzyme Replacement Is Safe and Effective in Patients with Cystic Fibrosis and Pancreatic Insufficiency. J. Cyst. Fibros. 2009, 8, 405–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, G.R.; Maguiness, K.; McNamara, J.; Morton, R.; Boyd, D.; Beckmann, K.; Bennett, D. Efficacy and Tolerability of a New Formulation of Pancrelipase Delayed-Release Capsules in Children Aged 7 to 11 Years with Exocrine Pancreatic Insufficiency and Cystic Fibrosis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled, Two-Period Crossover, Superiority Study. Clin. Ther. 2010, 32, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.; Major, G.; Smyth, A.R. Dosing Regimens for Pancreatic Enzyme Replacement Therapy (PERT) in Cystic Fibrosis. Cochrane Database Syst. Rev. 2019, 2019, CD013488. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Chen, S.; Khurmi, R.; Bodhani, A.; Kapoor, M.; Haupt, M. Hospitalization Rates among Patients with Cystic Fibrosis Using Pancreatic Enzyme Replacement Therapy. Chronic Respir. Dis. 2020, 17, 1479973119900612. [Google Scholar] [CrossRef]
- Phong, R.Y.; Taylor, S.L.; Robinson, B.A.; Jhawar, S.; Nandalike, K. Utility of Mid-Upper Arm Circumference in Diagnosing Malnutrition in Children With Cystic Fibrosis. Nutr. Clin. Pr. 2020, 35, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS Best Practice Guidelines: The 2018 Revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapnadak, S.G.; Ramos, K.J.; Lopriore, A.M.; Goss, C.H.; Aitken, M.L. A Survey Identifying Nutritional Needs in a Contemporary Adult Cystic Fibrosis Cohort. BMC Nutr. 2019, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Poulimeneas, D.; Grammatikopoulou, M.G.; Petrocheilou, A.; Kaditis, A.G.; Vassilakou, T. Triage for Malnutrition Risk among Pediatric and Adolescent Outpatients with Cystic Fibrosis, Using a Disease-Specific Tool. Children 2020, 7, 269. [Google Scholar] [CrossRef]
- McDonald, C.M.; Alvarez, J.A.; Bailey, J.; Bowser, E.K.K.; Farnham, K.; Mangus, M.; Padula, L.; Porco, K.; Rozga, M. Academy of Nutrition and Dietetics: 2020 Cystic Fibrosis Evidence Analysis Center Evidence-Based Nutrition Practice Guideline. J. Acad. Nutr. Diet. 2021, 121, 1591–1636.e3. [Google Scholar] [CrossRef]
- Ashkenazi, M.; Nathan, N.; Sarouk, I.; Aluma, B.E.B.; Dagan, A.; Bezalel, Y.; Keler, S.; Vilozni, D.; Efrati, O. Nutritional Status in Childhood as a Prognostic Factor in Patients with Cystic Fibrosis. Lung 2019, 197, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Niedermayr, K.; Gasser, V.; Rueckes-Nilges, C.; Appelt, D.; Eder, J.; Fuchs, T.; Naehrlich, L.; Ellemunter, H. Personalized Medicine with Drugs Targeting the Underlying Protein Defect in Cystic Fibrosis: Is Monitoring of Treatment Response Necessary? Ther. Adv. Chronic Dis. 2022, 13, 20406223221108628. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Durie, P.R. Pathology of Pancreatic and Intestinal Disorders in Cystic Fibrosis. J. R. Soc. Med. 1998, 91 (Suppl. S34), 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallings, V.A.; Stark, L.J.; Robinson, K.A.; Feranchak, A.P.; Quinton, H. Clinical Practice Guidelines on Growth and Nutrition Subcommittee; Ad Hoc Working Group Evidence-Based Practice Recommendations for Nutrition-Related Management of Children and Adults with Cystic Fibrosis and Pancreatic Insufficiency: Results of a Systematic Review. J. Am. Diet. Assoc. 2008, 108, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Konrad, J.; Eber, E.; Stadlbauer, V. Changing Paradigms in the Treatment of Gastrointestinal Complications of Cystic Fibrosis in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulators. Paediatr. Respir. Rev. 2022, 42, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Comanici, V.; Codreanu, I.; Balanescu, A.; Belivacă, A.; Stan, I.; Craiu, M.; Ritivoiu, M.; Matei, D. Acute Pancreatitis Prevalence in Children with Cystic Fibrosis and PIP Score Prediction Applicability. Rom. J. Med. Pract. 2021, 16, 248–253. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, F.; Chen, J. Molecular Structure of the ATP-Bound, Phosphorylated Human CFTR. Proc. Natl. Acad. Sci. USA 2018, 115, 12757–12762. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, B.P.; Freedman, S.D. Cystic Fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- McKay, I.R.; Ooi, C.Y. The Exocrine Pancreas in Cystic Fibrosis in the Era of CFTR Modulation: A Mini Review. Front. Pediatr. 2022, 10, 914790. [Google Scholar] [CrossRef]
- Skov, M.; Hansen, C.R.; Pressler, T. Cystic Fibrosis—An Example of Personalized and Precision Medicine. APMIS 2019, 127, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: Shedding Light on Precision Medicine for Cystic Fibrosis. Front. Pharm. 2016, 7, 275. [Google Scholar] [CrossRef] [Green Version]
- CFF. Available online: https://www.cff.org/ (accessed on 22 November 2022).
- Bell, S.C.; De Boeck, K.; Amaral, M.D. New Pharmacological Approaches for Cystic Fibrosis: Promises, Progress, Pitfalls. Pharm. Ther. 2015, 145, 19–34. [Google Scholar] [CrossRef]
- Slae, M.; Wilschanski, M. Prevention of Malnutrition in Cystic Fibrosis. Curr. Opin. Pulm. Med. 2019, 25, 674–679. [Google Scholar] [CrossRef]
- Nick, J.A.; St. Clair, C.; Jones, M.C.; Lan, L.; Higgins, M. VX12-770-113 Study Team Ivacaftor in Cystic Fibrosis with Residual Function: Lung Function Results from an N-of-1 Study. J. Cyst. Fibros. 2020, 19, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.C.; Wainwright, C.E.; Sawicki, G.S.; Higgins, M.N.; Campbell, D.; Harris, C.; Panorchan, P.; Haseltine, E.; Tian, S.; Rosenfeld, M. Ivacaftor in Infants Aged 4 to <12 Months with Cystic Fibrosis and a Gating Mutation. Results of a Two-Part Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 585–593. [Google Scholar] [CrossRef]
- Gramegna, A.; Contarini, M.; Aliberti, S.; Casciaro, R.; Blasi, F.; Castellani, C. From Ivacaftor to Triple Combination: A Systematic Review of Efficacy and Safety of CFTR Modulators in People with Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, E5882. [Google Scholar] [CrossRef] [PubMed]
- CFTR Modulator Therapies|Cystic Fibrosis Foundation. Available online: https://www.cff.org/managing-cf/cftr-modulator-therapies (accessed on 13 November 2022).
- Volkova, N.; Moy, K.; Evans, J.; Campbell, D.; Tian, S.; Simard, C.; Higgins, M.; Konstan, M.W.; Sawicki, G.S.; Elbert, A.; et al. Disease Progression in Patients with Cystic Fibrosis Treated with Ivacaftor: Data from National US and UK Registries. J. Cyst. Fibros. 2020, 19, 68–79. [Google Scholar] [CrossRef]
- Hubert, D.; Dehillotte, C.; Munck, A.; David, V.; Baek, J.; Mely, L.; Dominique, S.; Ramel, S.; Danner Boucher, I.; Lefeuvre, S.; et al. Retrospective Observational Study of French Patients with Cystic Fibrosis and a Gly551Asp-CFTR Mutation after 1 and 2 years of Treatment with Ivacaftor in a Real-World Setting. J. Cyst. Fibros. 2018, 17, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Hug, C.; Marigowda, G.; Tian, S.; Huang, X.; Stanojevic, S.; Milla, C.E.; Robinson, P.D.; Waltz, D.; Davies, J.C.; et al. Efficacy and Safety of Lumacaftor and Ivacaftor in Patients Aged 6-11 Years with Cystic Fibrosis Homozygous for F508del-CFTR: A Randomised, Placebo-Controlled Phase 3 Trial. Lancet Respir. Med. 2017, 5, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, J.E.; Chilvers, M.; Ratjen, F.; McNamara, J.J.; Owen, C.A.; Tian, S.; Zahigian, R.; Cornell, A.G.; McColley, S.A. Long-Term Safety of Lumacaftor-Ivacaftor in Children Aged 2–5 Years with Cystic Fibrosis Homozygous for the F508del-CFTR Mutation: A Multicentre, Phase 3, Open-Label, Extension Study. Lancet Respir. Med. 2021, 9, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric Folding Correction of F508del and Rare CFTR Mutants by Elexacaftor-Tezacaftor-Ivacaftor (Trikafta) Combination. JCI Insight 2020, 5, 139983. [Google Scholar] [CrossRef]
- Sondo, E.; Cresta, F.; Pastorino, C.; Tomati, V.; Capurro, V.; Pesce, E.; Lena, M.; Iacomino, M.; Baffico, A.M.; Coviello, D.; et al. The L467F-F508del Complex Allele Hampers Pharmacological Rescue of Mutant CFTR by Elexacaftor/Tezacaftor/Ivacaftor in Cystic Fibrosis Patients: The Value of the Ex Vivo Nasal Epithelial Model to Address Non-Responders to CFTR-Modulating Drugs. Int. J. Mol. Sci. 2022, 23, 3175. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and Safety of the Elexacaftor plus Tezacaftor plus Ivacaftor Combination Regimen in People with Cystic Fibrosis Homozygous for the F508del Mutation: A Double-Blind, Randomised, Phase 3 Trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Bass, R.; Brownell, J.N.; Stallings, V.A. The Impact of Highly Effective CFTR Modulators on Growth and Nutrition Status. Nutrients 2021, 13, 2907. [Google Scholar] [CrossRef] [PubMed]
- Guimbellot, J.S.; Baines, A.; Paynter, A.; Heltshe, S.L.; VanDalfsen, J.; Jain, M.; Rowe, S.M.; Sagel, S.D. GOAL-e2 Investigators Long Term Clinical Effectiveness of Ivacaftor in People with the G551D CFTR Mutation. J. Cyst. Fibros. 2021, 20, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Begnel, L.; Wallendorf, M.; Litvin, M. Effect of Elexacaftor-Tezacaftor-Ivacaftor on Body Weight and Metabolic Parameters in Adults with Cystic Fibrosis. J. Cyst. Fibros. 2022, 21, 265–271. [Google Scholar] [CrossRef]
- Rosenfeld, M.; Wainwright, C.E.; Higgins, M.; Wang, L.T.; McKee, C.; Campbell, D.; Tian, S.; Schneider, J.; Cunningham, S.; Davies, J.C.; et al. Ivacaftor Treatment of Cystic Fibrosis in Children Aged 12 to <24 Months and with a CFTR Gating Mutation (ARRIVAL): A Phase 3 Single-Arm Study. Lancet Respir. Med. 2018, 6, 545–553. [Google Scholar] [CrossRef]
- Sun, X.; Yi, Y.; Yan, Z.; Rosen, B.H.; Liang, B.; Winter, M.C.; Evans, T.I.A.; Rotti, P.G.; Yang, Y.; Gray, J.S.; et al. In Utero and Postnatal VX-770 Administration Rescues Multiorgan Disease in a Ferret Model of Cystic Fibrosis. Sci. Transl. Med. 2019, 11, eaau7531. [Google Scholar] [CrossRef] [Green Version]
- Mennella, J.A.; Inamdar, L.; Pressman, N.; Schall, J.I.; Papas, M.A.; Schoeller, D.; Stallings, V.A.; Trabulsi, J.C. Type of Infant Formula Increases Early Weight Gain and Impacts Energy Balance: A Randomized Controlled Trial. Am. J. Clin. Nutr. 2018, 108, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Stallings, V.A.; Tindall, A.M.; Mascarenhas, M.R.; Maqbool, A.; Schall, J.I. Improved Residual Fat Malabsorption and Growth in Children with Cystic Fibrosis Treated with a Novel Oral Structured Lipid Supplement: A Randomized Controlled Trial. PLoS ONE 2020, 15, e0232685. [Google Scholar] [CrossRef]
- Gould, M.J.; Smith, H.; Rayment, J.H.; Machida, H.; Gonska, T.; Galante, G.J. CFTR Modulators Increase Risk of Acute Pancreatitis in Pancreatic Insufficient Patients with Cystic Fibrosis. J. Cyst. Fibros. 2022, 21, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Megalaa, R.; Gopalareddy, V.; Champion, E.; Goralski, J.L. Time for a Gut Check: Pancreatic Sufficiency Resulting from CFTR Modulator Use. Pediatr. Pulmonol. 2019, 54, E16–E18. [Google Scholar] [CrossRef]
- Ramsey, M.L.; Gokun, Y.; Sobotka, L.A.; Wellner, M.R.; Porter, K.; Kirkby, S.E.; Li, S.S.; Papachristou, G.I.; Krishna, S.G.; Stanich, P.P.; et al. Cystic Fibrosis Transmembrane Conductance Regulator Modulator Use Is Associated With Reduced Pancreatitis Hospitalizations in Patients With Cystic Fibrosis. Am. J. Gastroenterol. 2021, 116, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Taylor-Cousar, J.L. Triple Combination Cystic Fibrosis Transmembrane Conductance Regulator Modulator Therapy in the Real World—Opportunities and Challenges. Curr. Opin. Pulm. Med. 2021, 27, 554–566. [Google Scholar] [CrossRef]
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, E589. [Google Scholar] [CrossRef] [PubMed]
- Barry, P.J.; Plant, B.J. Editorial: The Changing Landscape of Cystic Fibrosis: New Therapies, Challenges and a Global Pandemic. Curr. Opin. Pulm. Med. 2020, 26, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Havermans, T.; Duff, A.J.A. Changing Landscape: Psychological Care in the Era of Cystic Fibrosis Transmembrane Conductance Regulator Modulators. Curr. Opin. Pulm. Med. 2020, 26, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Wilschanski, M.; Peckham, D. Nutritional and Metabolic Management for Cystic Fibrosis in a Post-Cystic Fibrosis Transmembrane Conductance Modulator Era. Curr. Opin. Pulm. Med. 2022, 28, 577–583. [Google Scholar] [CrossRef]
- Higgins, M.; Volkova, N.; Moy, K.; Marshall, B.C.; Bilton, D. Real-World Outcomes Among Patients with Cystic Fibrosis Treated with Ivacaftor: 2012–2016 Experience. Pulm. Ther. 2020, 6, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Allan, K.M.; Farrow, N.; Donnelley, M.; Jaffe, A.; Waters, S.A. Treatment of Cystic Fibrosis: From Gene- to Cell-Based Therapies. Front. Pharm. 2021, 12, 639475. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR Modulator Theratyping: Current Status, Gaps and Future Directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Stan, I. 25(OH) vitamin D deficiency in cystic fibrosis children—A prospective study on prevalence and treatment outcome. Farmacia 2019, 67, 423–429. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| PERT Pharmaceutical Product | Units of Lipase Contents | Enteric-Coated or Non-Enteric Coated Formulations | Posology/Dose and Administration Recommendations |
|---|---|---|---|
| Kreon/Creon® | 3000; 6000; 10,000; 12,000; 24,000; 28,000; 36,000 | Enteric-coated microspheres | 500–2500 U/kg body weight/meal 500–4000 U/g of fat/day 2000–5000 U/breastfeed or 100–120 mL infant formula (Not to be mixed directly with the formula or breast milk) Not to exceed 10,000 U/kg/day |
| Pankreal® | 35,000 | Non-enteric-coated capsules | |
| Zenpep® | 3000; 5000; 10,000; 15,000; 20,000; 25,000; 40,000 | Enteric-coated beads | |
| Pancreaze/Pancrease® | 2600; 4200; 10,500; 16,800; 21,000; 37,000 | Enteric-coated microtablets | |
| Ultresa® | 4000; 13,800; 20,700; 23,000 | Enteric-coated beads | |
| Viokase/Viokace® | 10,440; 20,880 (requires acid suppression: proton pump inhibitor) 8000; 16,000; 16,800 per each 0.7 g powder (¼ teaspoonful) | Non-enteric-coated tablets or powder | |
| Pertzye® | 4000; 8000; 16,000; 24,000 | Enteric-coated microspheres | |
| Nutrizym 22® | 22,000 | Enteric-coated tablets | |
| Pancrex V® | 2950; 8000 | Non-enteric-coated hard capsules | Pancrex V Powder may be administered via a nasogastric or a gastrostomy tube. |
| 5600 25,000/1 g powder | Enteric-coated tablets or powder |
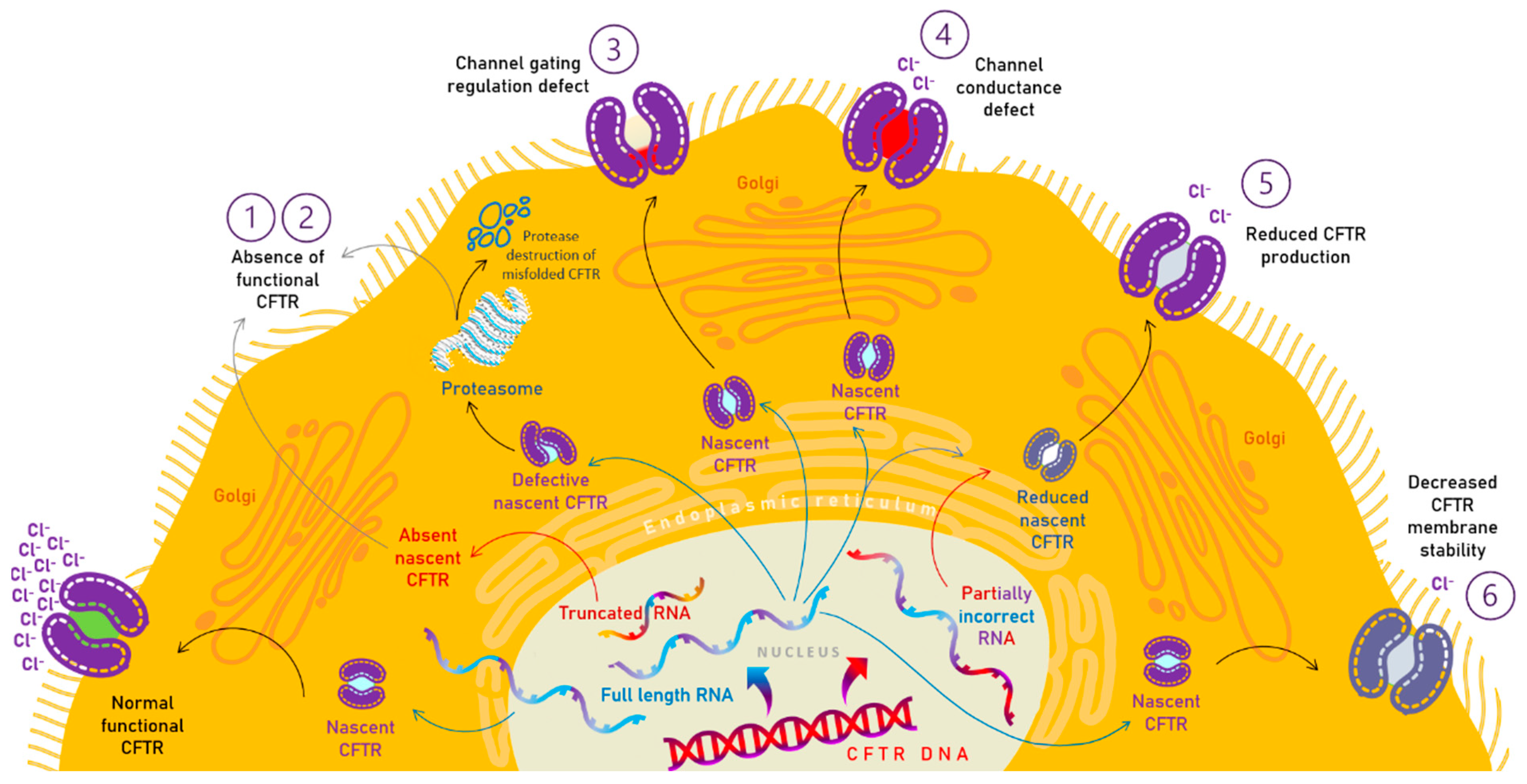
| Class | Type of Defect | Risk of EPI | Examples of CFTR Mutations | Therapeutic Approach | Available Drugs |
|---|---|---|---|---|---|
| I | protein synthesis defect | 98% 99% 97% | G542x W1282x R553x | Genetic therapies Read-through agents | — |
| II | protein trafficking | 98% 98% | F508del N1303K | Corrector (and potentiator) | Elexacaftor/tezacaftor/ivacaftor Lumacaftor/ivacaftor Tezacaftor/ivacaftor |
| III | gatting defect | 100% 96% 33% | G970R G551D G551S | Potentiator | Ivacaftor |
| IV | conductance defect | 0% 40% 68% | G314E R334W R347P | Potentiator | Ivacaftor |
| V | reduced protein synthesis | 43% 33% 29% | 2789 + 5G → A 3849 + 10KbC → T 3272-26A → G | Amplifier | — |
| VI | decreased stability | — | c.120del123 rPhe580del | Stabiliser | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritivoiu, M.-E.; Drăgoi, C.M.; Matei, D.; Stan, I.V.; Nicolae, A.C.; Craiu, M.; Dumitrescu, I.-B.; Ciolpan, A.A. Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics 2023, 15, 162. https://doi.org/10.3390/pharmaceutics15010162
Ritivoiu M-E, Drăgoi CM, Matei D, Stan IV, Nicolae AC, Craiu M, Dumitrescu I-B, Ciolpan AA. Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine. Pharmaceutics. 2023; 15(1):162. https://doi.org/10.3390/pharmaceutics15010162
Chicago/Turabian StyleRitivoiu, Mirela-Elena, Cristina Manuela Drăgoi, Dumitru Matei, Iustina Violeta Stan, Alina Crenguţa Nicolae, Mihai Craiu, Ion-Bogdan Dumitrescu, and Alina Angelica Ciolpan. 2023. "Current and Future Therapeutic Approaches of Exocrine Pancreatic Insufficiency in Children with Cystic Fibrosis in the Era of Personalized Medicine" Pharmaceutics 15, no. 1: 162. https://doi.org/10.3390/pharmaceutics15010162