
Cannabidiol Reduces Short- and Long-Term High Glutamate Release after Severe Traumatic Brain Injury and Improves Functional Recovery

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Stereotaxic Surgery
2.3. Induction of Severe TBI
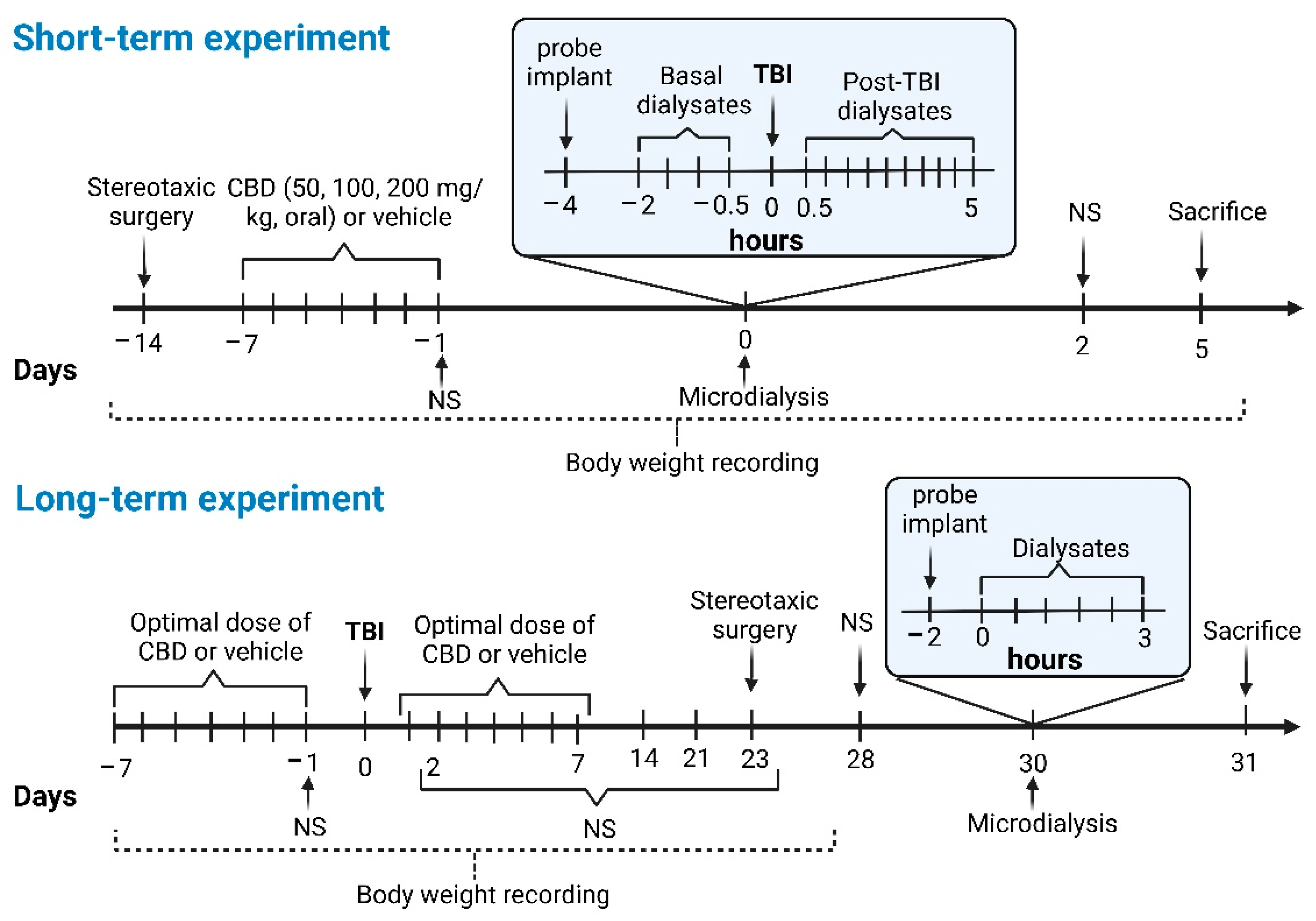
2.4. Groups for the Short-Term Experiment
2.4.1. CBD200 + TBI (n = 11)
2.4.2. CBD100 + TBI (n = 9)
2.4.3. CBD50 + TBI (n = 9)
2.4.4. TBI (n = 14)
2.4.5. SHAM (n = 7)
2.5. Groups for the Long-Term Experiment
2.5.1. CBD + TBI + CBD (n = 10)
2.5.2. CBD + TBI (n = 9)
2.5.3. TBI (n = 11)
2.5.4. CBD + SHAM (n = 7)
2.5.5. SHAM (n = 8)
2.6. Evaluation of Sensorimotor Function with Neuroscore Test
2.7. Microdialysis and HPLC
2.8. Nissl Histology
2.9. Statistical Analysis
3. Results
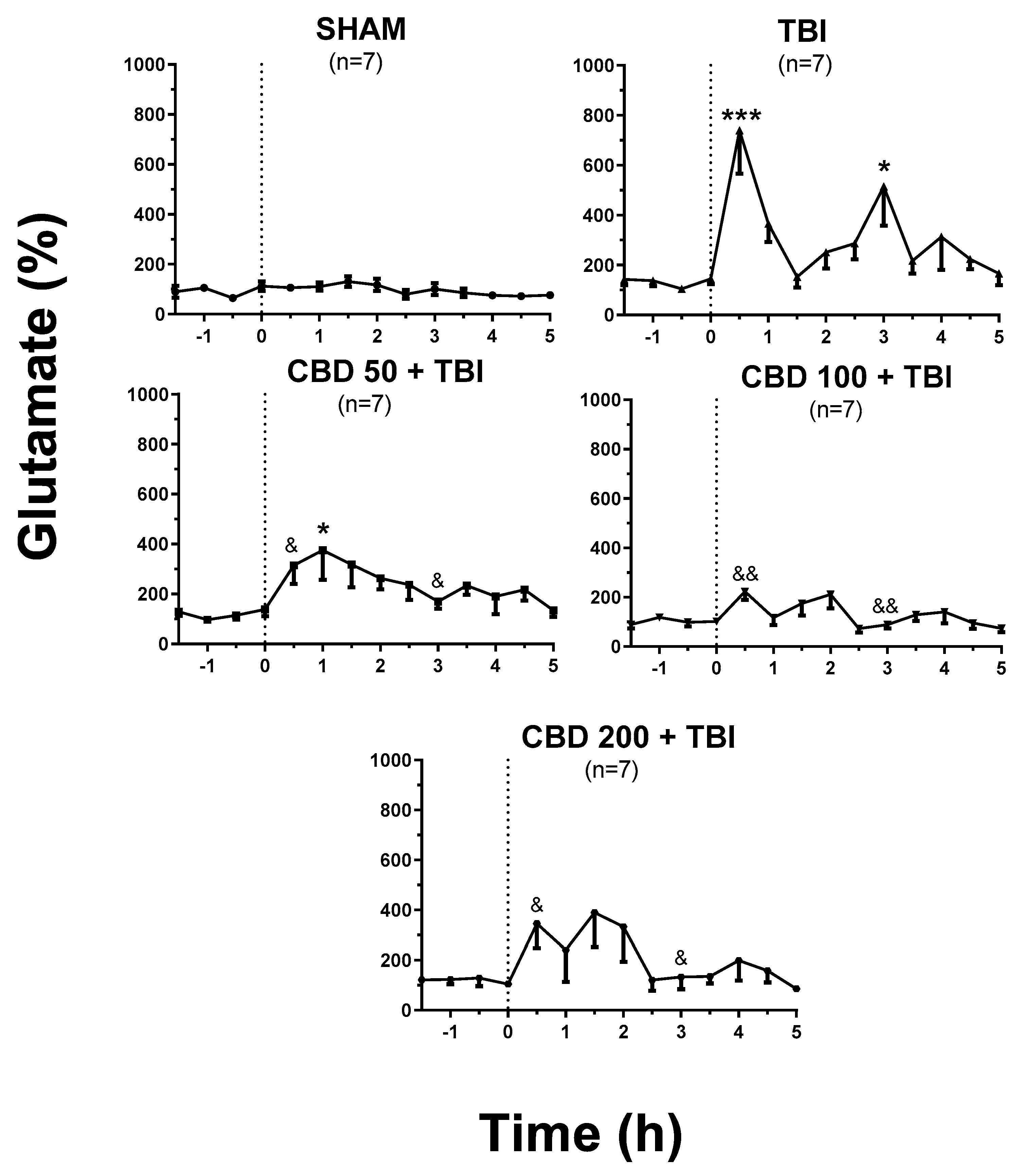
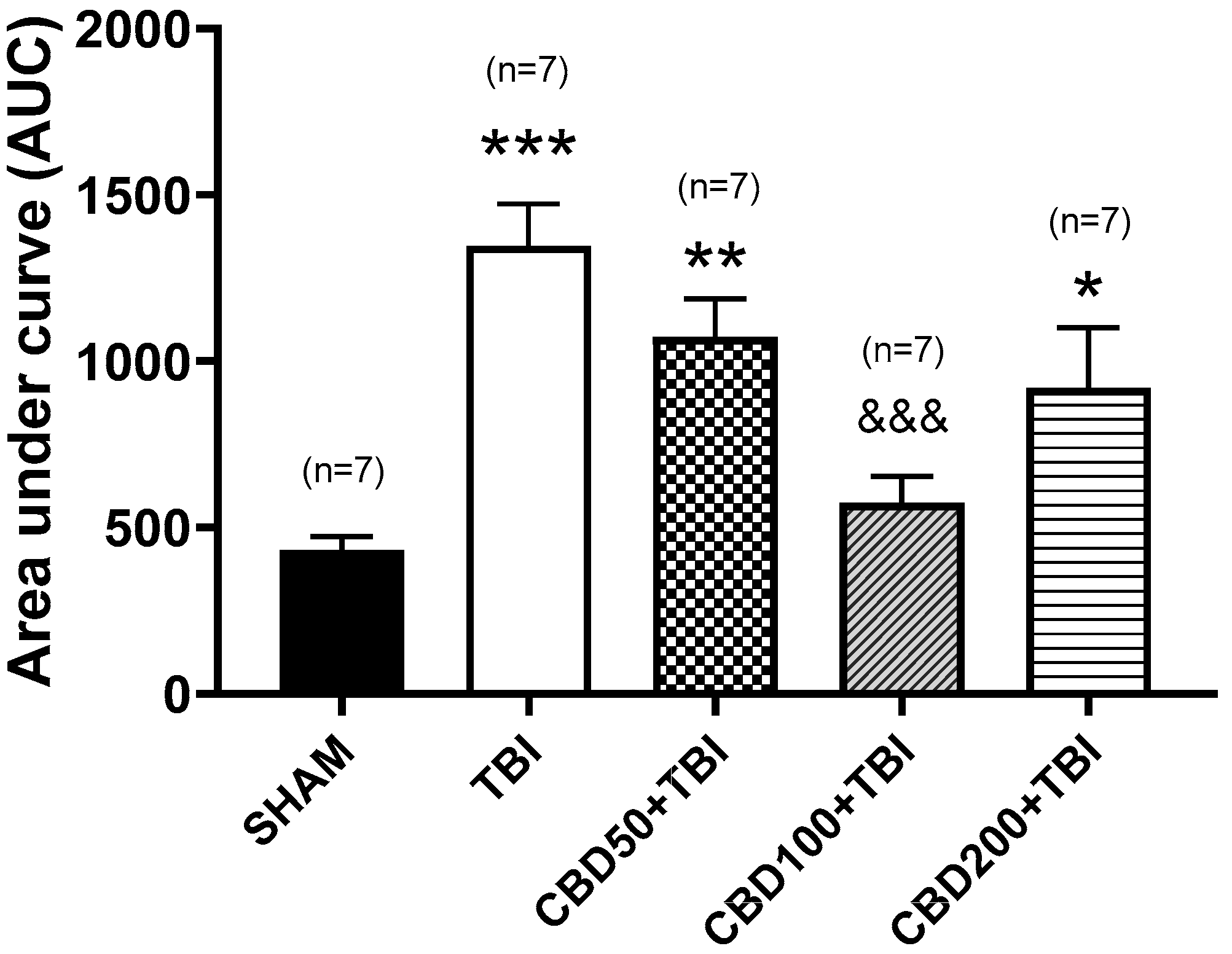
3.1. CBD Pretreatment Lessens TBI-Induced High Extracellular Glutamate Release in the Cortex Shortly after Trauma
3.2. CBD Pretreatment Does Not Modify the Short-Term TBI-Induced Changes in Body Weight and Sensorimotor Function, but Reduces Mortality
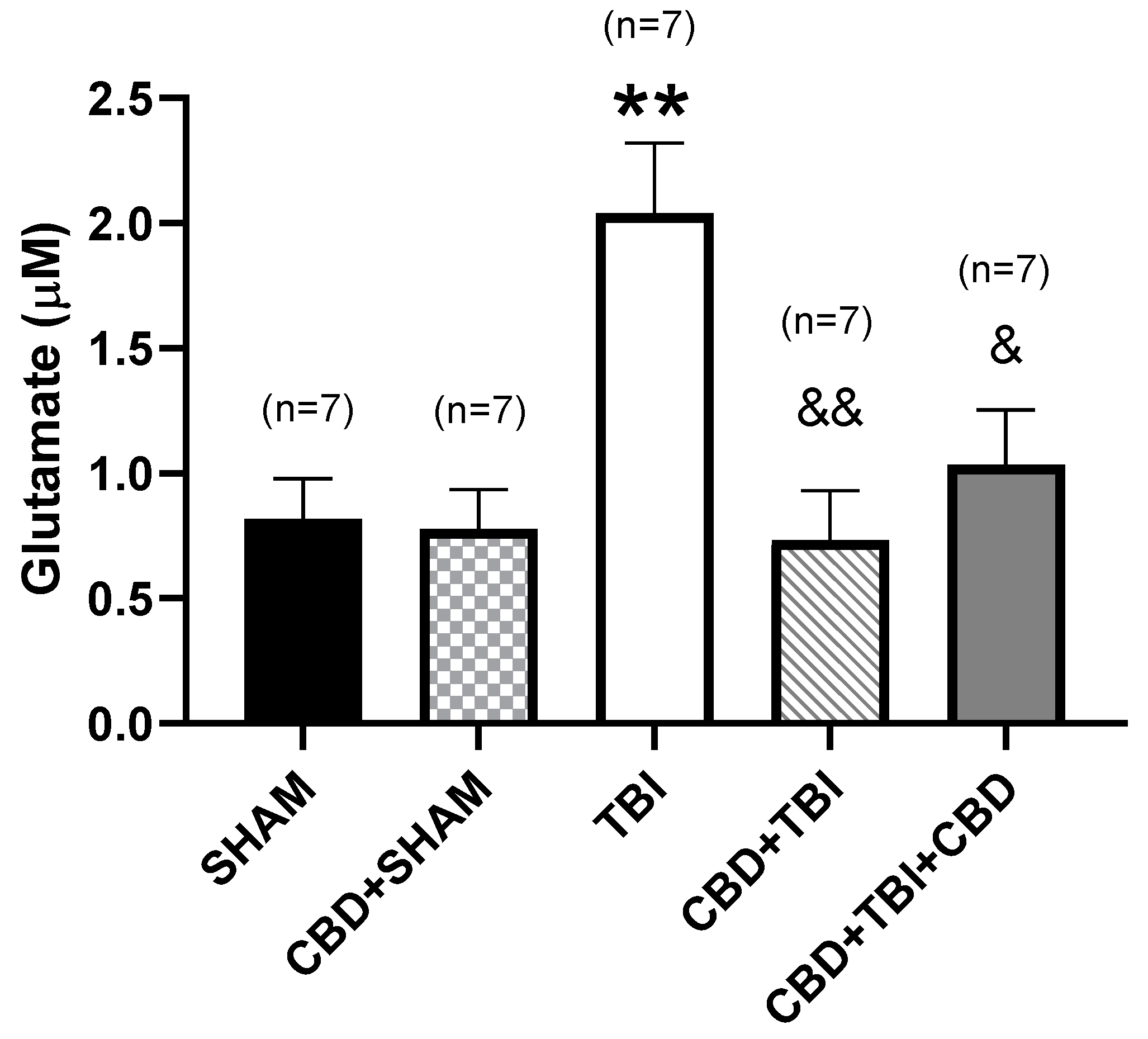
3.3. CBD Administration Attenuates Long-Term High Glutamate Release after Severe TBI
3.4. CBD Administration Diminishes Long-Term Sensorimotor Deficit, Improves Body Weight Gain, and Reduces Mortality after Severe TBI
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Centers for Disease Control and Prevention. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation; National Center for Injury Prevention and Control, Division of Unintentional Injury Prevention: Atlanta, GA, USA, 2015. Available online: https://www.cdc.gov/traumaticbraininjury/pdf/TBI_Report_to_Congress_Epi_and_Rehab-a.pdf (accessed on 15 February 2022).
- Dunne, J.; Quiñones-Ossa, G.A.; Still, E.G.; Suarez, M.N.; González-Soto, J.A.; Vera, D.S.; Rubiano, A.M. The epidemiology of traumatic brain injury due to traffic accidents in Latin America: A narrative review. J. Neurosci. Rural Pract. 2020, 11, 287–290. [Google Scholar] [CrossRef]
- Iaccarino, C.; Carretta, A.; Nicolosi, F.; Morselli, C. Epidemiology of severe traumatic brain injury. J. Neurosurg. Sci. 2018, 62, 535–541. [Google Scholar] [CrossRef]
- Agimi, Y.; Marion, D.; Schwab, K.; Stout, K. Estimates of long-term disability among US service members with traumatic brain injuries. J. Head Trauma Rehabil. 2021, 36, 1–9. [Google Scholar] [CrossRef]
- Brazinova, A.; Rehorcikova, V.; Taylor, M.S.; Buckova, V.; Majdan, M.; Psota, M.; Peeters, W.; Feigin, V.; Theadom, A.; Holkovic, L.; et al. Epidemiology of traumatic brain injury in Europe: A living systematic review. J. Neurotrauma 2021, 38, 1411–1440. [Google Scholar] [CrossRef] [Green Version]
- Dismuke, C.E.; Walker, R.J.; Egede, L.E. Utilization and cost of health services in individuals with traumatic brain injury. Glob. J. Health Sci. 2015, 7, 156–169. [Google Scholar] [CrossRef] [PubMed]
- van Dijck, J.T.J.M.; Mostert, C.Q.B.; Greeven, A.P.A.; Kompanje, E.J.O.; Peul, W.C.; de Ruiter, G.C.W.; Polinder, S. Functional outcome, in-hospital healthcare consumption and in-hospital costs for hospitalised traumatic brain injury patients: A Dutch prospective multicentre study. Acta Neurochir. 2020, 162, 1607–1618. [Google Scholar] [CrossRef]
- Giza, C.C.; Hovda, D.A. The pathophysiology of traumatic brain injury. In Traumatic Brain Injury in Sports, 1st ed.; Lovell, M., Barth, J., Collins, M., Echemendia, R., Eds.; Taylor & Francis: London, UK, 2004; pp. 45–70. [Google Scholar]
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, S.V.; Faden, A.I. Neuroprotective strategies for traumatic brain injury: Improving clinical translation. Int. J. Mol. Sci. 2014, 15, 1216–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef]
- Chamoun, R.; Suki, D.; Gopinath, S.P.; Goodman, J.C.; Robertson, C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury. J. Neurosurg. 2010, 113, 564–570. [Google Scholar] [CrossRef]
- Dorsett, C.R.; McGuire, J.L.; DePasquale, E.A.; Gardner, A.E.; Floyd, C.L.; McCullumsmith, R.E. Glutamate neurotransmission in rodent models of traumatic brain injury. J. Neurotrauma 2017, 34, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Murasugi, T.; Oda, T. Acute neuroinflammation exacerbates excitotoxicity in rat hippocampus in vivo. Exp. Neurol. 2002, 177, 95–104. [Google Scholar] [CrossRef]
- Chaparro-Huerta, V.; Rivera-Cervantes, M.C.; Flores-Soto, M.E.; Gómez-Pinedo, U.; Beas-Zárate, C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. J. Neuroimmunol. 2005, 165, 53–62. [Google Scholar] [CrossRef]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Abdul-Muneer, P.M. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Mandali, A.; Chakravarthy, V.S.; Ramaswamy, S. A computational model of loss of dopaminergic cells in Parkinson’s disease due to glutamate-induced excitotoxicity. Front. Neural Circuits 2019, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Olajide, O.J.; Gbadamosi, I.T.; Yawson, E.O.; Arogundade, T.; Lewu, F.S.; Ogunrinola, K.Y.; Adigun, O.O.; Bamisi, O.; Lambe, E.; Arietarhire, L.O.; et al. Hippocampal degeneration and behavioral impairment during Alzheimer-like pathogenesis involves glutamate excitotoxicity. J. Mol. Neurosci. 2021, 71, 1205–1220. [Google Scholar] [CrossRef]
- Green, J.L.; Dos Santos, W.F.; Fontana, A.C. Role of glutamate excitotoxicity and glutamate transporter EAAT2 in epilepsy: Opportunities for novel therapeutics development. Biochem. Pharmacol. 2021, 193, 114786. [Google Scholar] [CrossRef]
- Malvestio, R.B.; Medeiros, P.; Negrini-Ferrari, S.E.; Oliveira-Silva, M.; Medeiros, A.C.; Padovan, C.M.; Luongo, L.; Maione, S.; Coimbra, N.C.; de Freitas, R.L. Cannabidiol in the prelimbic cortex modulates the comorbid condition between the chronic neuropathic pain and depression-like behaviour in rats: The role of medial prefrontal cortex 5-HT1A and CB1 receptors. Brain Res. Bull. 2021, 174, 323–338. [Google Scholar] [CrossRef]
- Devinsky, O.; Cross, J.H.; Wright, S. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 2017, 377, 699–700. [Google Scholar] [CrossRef] [Green Version]
- Shbiro, L.; Hen-Shoval, D.; Hazut, N.; Rapps, K.; Dar, S.; Zalsman, G.; Mechoulam, R.; Weller, A.; Shoval, G. Effects of cannabidiol in males and females in two different rat models of depression. Physiol. Behav. 2019, 201, 59–63. [Google Scholar] [CrossRef]
- Pazos, M.R.; Cinquina, V.; Gómez, A.; Layunta, R.; Santos, M.; Fernández-Ruiz, J.; Martínez-Orgado, J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. [Google Scholar] [CrossRef]
- Devinsky, O.; Patel, A.D.; Thiele, E.A.; Wong, M.H.; Appleton, R.; Harden, C.L.; Greenwood, S.; Morrison, G.; Sommerville, K. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology 2018, 90, e1204–e1211. [Google Scholar] [CrossRef] [Green Version]
- Perucca, E.; Bialer, M. Critical Aspects Affecting Cannabidiol Oral Bioavailability and Metabolic Elimination, and Related Clinical Implications. CNS Drugs 2020, 34, 795–800. [Google Scholar] [CrossRef]
- Millar, S.A.; Stone, N.L.; Yates, A.S.; O’Sullivan, S.E. A Systematic Review on the Pharmacokinetics of Cannabidiol in Humans. Front. Pharmacol. 2018, 9, 1365. [Google Scholar] [CrossRef]
- Cherniakov, I.; Izgelov, D.; Domb, A.J.; Hoffman, A. The effect of Pro NanoLipospheres (PNL) formulation containing natural absorption enhancers on the oral bioavailability of delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) in a rat model. Eur. J. Pharm. Sci. 2017, 109, 21–30. [Google Scholar] [CrossRef]
- Urquhart, L. Regulatory watch: FDA new drug approvals in Q2 2018. Nat. Rev. Drug Discov. 2018, 17, 536–537. [Google Scholar] [CrossRef]
- Khaksar, S.; Bigdeli, M.R. Intra-cerebral cannabidiol infusion-induced neuroprotection is partly associated with the TNF-α/TNFR1/NF-κB pathway in transient focal cerebral ischaemia. Brain Inj. 2017, 31, 1932–1943. [Google Scholar] [CrossRef]
- Gobira, P.H.; Vilela, L.R.; Gonçalves, B.D.; Santos, R.P.; de Oliveira, A.C.; Vieira, L.B.; Aguiar, D.C.; Crippa, J.A.; Moreira, F.A. Cannabidiol, a Cannabis sativa constituent, inhibits cocaine-induced seizures in mice: Possible role of the mTOR pathway and reduction in glutamate release. Neurotoxicology 2015, 50, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kong, W.; Chambers, C.R.; Yu, D.; Ganea, D.; Tuma, R.F.; Ward, S.J. The non-psychoactive phytocannabinoid cannabidiol (CBD) attenuates pro-inflammatory mediators, T cell infiltration, and thermal sensitivity following spinal cord injury in mice. Cell Immunol. 2018, 329, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Patel, V.; Saito, K.; Matsumoto, S.; Kashiwaya, Y.; Horváth, B.; Mukhopadhyay, B.; Becker, L.; et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 2115–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do Val-da Silva, R.A.; Peixoto-Santos, J.E.; Kandratavicius, L.; De Ross, J.B.; Esteves, I.; De Martinis, B.S.; Alves, M.N.; Scandiuzzi, R.C.; Hallak, J.E.; Zuardi, A.W.; et al. Protective effects of cannabidiol against seizures and neuronal death in a rat model of mesial temporal lobe epilepsy. Front. Pharmacol. 2017, 8, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, L.K.; Peng, H.; Zeman, R.J. Cannabidiol reduces lesion volume and restores vestibulomotor and cognitive function following moderately severe traumatic brain injury. Exp. Neurol. 2021, 346, 113844. [Google Scholar] [CrossRef] [PubMed]
- Belardo, C.; Iannotta, M.; Boccella, S.; Rubino, R.C.; Ricciardi, F.; Infantino, R.; Pieretti, G.; Stella, L.; Paino, S.; Marabese, I.; et al. Oral cannabidiol prevents allodynia and neurological dysfunctions in a mouse model of mild traumatic brain injury. Front. Pharmacol. 2019, 10, 352. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: New York, NY, USA, 1998. [Google Scholar]
- McIntosh, T.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef]
- Segovia-Oropeza, M.; Santiago-Castañeda, C.; Orozco-Suárez, S.A.; Concha, L.; Rocha, L. Sodium cromoglycate decreases sensorimotor impairment and hippocampal alterations induced by severe traumatic brain injury in rats. J. Neurotrauma 2020, 37, 2595–2603. [Google Scholar] [CrossRef] [PubMed]
- Frías-Soria, C.L.; Pérez-Pérez, D.; Orozco-Suárez, S.; Rocha, L. Cannabidiol modifies the seizure expression and effects of antiseizure drugs in a rat model of recurrent severe seizures. Seizure 2021, 90, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Maidment, N.T.; Brumbaugh, D.R.; Rudolph, V.D.; Erdelyi, E.; Evans, C.J. Microdialysis of extracellular endogenous opioid peptides from rat brain in vivo. Neuroscience 1989, 33, 549–557. [Google Scholar] [CrossRef]
- Santana-Gómez, C.E.; Orozco-Suárez, S.A.; Talevi, A.; Bruno-Blanch, L.; Magdaleno-Madrigal, V.M.; Fernández-Mas, R.; Rocha, L. Propylparaben applied after pilocarpine-induced status epilepticus modifies hippocampal excitability and glutamate release in rats. Neurotoxicology 2017, 59, 110–120. [Google Scholar] [CrossRef]
- Pruessner, J.C.; Kirschbaum, C.; Meinlschmid, G.; Hellhammer, D.H. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003, 28, 916–931. [Google Scholar] [CrossRef]
- Fleischmann, C.; Shohami, E.; Trembovler, V.; Heled, Y.; Horowitz, M. Cognitive effects of astaxanthin pretreatment on recovery from traumatic brain injury. Front. Neurol. 2020, 11, 999. [Google Scholar] [CrossRef]
- Samini, F.; Samarghandian, S.; Borji, A.; Mohammadi, G.; Bakaian, M. Curcumin pretreatment attenuates brain lesion size and improves neurological function following traumatic brain injury in the rat. Pharmacol. Biochem. Behav. 2013, 110, 238–244. [Google Scholar] [CrossRef]
- Stokum, J.A.; Keledjian, K.; Hayman, E.; Karimy, J.K.; Pampori, A.; Imran, Z.; Woo, S.K.; Gerzanich, V.; Simard, J.M. Glibenclamide pretreatment protects against chronic memory dysfunction and glial activation in rat cranial blast traumatic brain injury. Behav. Brain Res. 2017, 333, 43–53. [Google Scholar] [CrossRef]
- Panter, S.S.; Faden, A.I. Pretreatment with NMDA antagonists limits release of excitatory amino acids following traumatic brain injury. Neurosci. Lett. 1992, 136, 165–168. [Google Scholar] [CrossRef]
- Pérez-Pinzón, M.A.; Alonso, O.; Kraydieh, S.; Dietrich, W.D. Induction of tolerance against traumatic brain injury by ischemic preconditioning. Neuroreport 1999, 10, 2951–2954. [Google Scholar] [CrossRef]
- Ward, R.J.; Colivicchi, M.A.; Allen, R.; Schol, F.; Lallemand, F.; de Witte, P.; Ballini, C.; Corte, L.D.; Dexter, D. Neuroinflammation induced in the hippocampus of ‘binge drinking’ rats may be mediated by elevated extracellular glutamate content. J. Neurochem. 2009, 111, 1119–1128. [Google Scholar] [CrossRef]
- Parfenova, H.; Basuroy, S.; Bhattacharya, S.; Tcheranova, D.; Qu, Y.; Regan, R.F.; Leffler, C.W. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: Contributions of HO-1 and HO-2 to cytoprotection. Am. J. Physiol. Cell Physiol. 2006, 290, C1399–C1410. [Google Scholar] [CrossRef]
- Binvignat, O.; Olloquequi, J. Excitotoxicity as a target against neurodegenerative processes. Curr. Pharm. Des. 2020, 26, 1251–1262. [Google Scholar] [CrossRef]
- Blaylock, R.L.; Maroon, J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy—A unifying hypothesis. Surg. Neurol. Int. 2011, 2, 107. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.M.; Marion, D.W.; Botscheller, M.L.; Swedlow, P.E.; Styren, S.D.; DeKosky, S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993, 61, 2015–2024. [Google Scholar] [CrossRef]
- Shutter, L.; Tong, K.A.; Holshouser, B.A. Proton MRS in acute traumatic brain injury: Role for glutamate/glutamine and choline for outcome prediction. J. Neurotrauma 2004, 21, 1693–1705. [Google Scholar] [CrossRef]
- Globus, M.Y.; Alonso, O.; Dietrich, W.D.; Busto, R.; Ginsberg, M.D. Glutamate release and free radical production following brain injury: Effects of posttraumatic hypothermia. J. Neurochem. 1995, 65, 1704–1711. [Google Scholar] [CrossRef]
- Vespa, P.; Prins, M.; Ronne-Engstrom, E.; Caron, M.; Shalmon, E.; Hovda, D.A.; Martin, N.A.; Becker, D.P. Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: A microdialysis study. J. Neurosurg. 1998, 89, 971–982. [Google Scholar] [CrossRef]
- Krishna, G.; Bromberg, C.; Connell, E.C.; Mian, E.; Hu, C.; Lifshitz, J.; Adelson, P.D.; Thomas, T.C. Traumatic brain injury-induced sex-dependent changes in late-onset sensory hypersensitivity and glutamate neurotransmission. Front. Neurol. 2020, 11, 749. [Google Scholar] [CrossRef]
- Rovegno, M.; Soto, P.A.; Sáez, J.C.; von Bernhardi, R. Mecanismos biológicos involucrados en la propagación del daño en el traumatismo encéfalo craneano [Biological mechanisms involved in the spread of traumatic brain damage]. Med. Intensiva 2012, 36, 37–44. [Google Scholar] [CrossRef]
- Sulhan, S.; Lyon, K.A.; Shapiro, L.A.; Huang, J.H. Neuroinflammation and blood-brain barrier disruption following traumatic brain injury: Pathophysiology and potential therapeutic targets. J. Neurosci. Res. 2020, 98, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Wang, J.; Sun, X.; Shao, L.; Guo, Z.; Li, Y. Morphological and functional alterations of astrocytes responding to traumatic brain injury. J. Integr. Neurosci. 2019, 18, 203–215. [Google Scholar] [CrossRef]
- Cantu, D.; Walker, K.; Andresen, L.; Taylor-Weiner, A.; Hampton, D.; Tesco, G.; Dulla, C.G. Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb. Cortex 2015, 25, 2306–2320. [Google Scholar] [CrossRef] [Green Version]
- Piao, C.S.; Holloway, A.L.; Hong-Routson, S.; Wainwright, M.S. Depression following traumatic brain injury in mice is associated with down-regulation of hippocampal astrocyte glutamate transporters by thrombin. J. Cereb. Blood Flow Metab. 2019, 39, 58–73. [Google Scholar] [CrossRef]
- Mkrtchyan, G.V.; Üçal, M.; Müllebner, A.; Dumitrescu, S.; Kames, M.; Moldzio, R.; Molcanyi, M.; Schaefer, S.; Weidinger, A.; Schaefer, U.; et al. Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex. Biochim. Biophys. Acta 2018, 1859, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590. [Google Scholar] [CrossRef]
- Walker, W.C.; Pickett, T.C. Motor impairment after severe traumatic brain injury: A longitudinal multicenter study. J. Rehabil. Res. Dev. 2007, 44, 975–982. [Google Scholar] [CrossRef]
- Santiago-Castañeda, C.; Segovia-Oropeza, M.; Concha, L.; Orozco-Suárez, S.A.; Rocha, L. Propylparaben reduces the long-term consequences in hippocampus induced by traumatic brain injury in rats: Its implications as therapeutic strategy to prevent neurodegenerative diseases. J. Alzheimers Dis. 2021, 82, S215–S226. [Google Scholar] [CrossRef]
- Frankenfield, D.C.; Ashcraft, C.M. Description and prediction of resting metabolic rate after stroke and traumatic brain injury. Nutrition 2012, 28, 906–911. [Google Scholar] [CrossRef]
- Kousi, C.; Lampri, E.; Voulgaris, S.; Vougiouklakis, T.; Galani, V.; Mitselou, A. Expression of orexin-A (hypocretin-A) in the hypothalamus after traumatic brain injury: A postmortem evaluation. Forensic Sci. Int. 2021, 327, 110961. [Google Scholar] [CrossRef]
- Mediavilla, C. Bidirectional gut-brain communication: A role for orexin-A. Neurochem. Int. 2020, 141, 104882. [Google Scholar] [CrossRef]
- Schröder, N.; da Silva, V.K.; Hallak, J.E.C.; Zuardi, A.W.; de Souza Crippa, J.A. Cannabidiol and neuroprotection: Evidence from preclinical studies. In Handbook of Cannabis and Related Pathologies, 1st ed.; Preedy, V.R., Ed.; Academic Press: London, UK, 2017; pp. 802–812. [Google Scholar]
- Levin, R.; Peres, F.F.; Almeida, V.; Calzavara, M.B.; Zuardi, A.W.; Hallak, J.E.; Crippa, J.A.; Abílio, V.C. Effects of cannabinoid drugs on the deficit of prepulse inhibition of startle in an animal model of schizophrenia: The SHR strain. Front. Pharmacol. 2014, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Martínez-Aguirre, C.; Carmona-Cruz, F.; Velasco, A.L.; Velasco, F.; Aguado-Carrillo, G.; Cuéllar-Herrera, M.; Rocha, L. Cannabidiol acts at 5-HT1A receptors in the human brain: Relevance for treating temporal lobe epilepsy. Front. Behav. Neurosci. 2020, 14, 611278. [Google Scholar] [CrossRef]
- Castillo, A.; Tolón, M.R.; Fernández-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D.; Giuffrida, A.; Calignano, A.; De Fonseca, F.R. The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol. Sci. 2000, 21, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; De Petrocellis, L.; Fezza, F.; Ligresti, A.; Bisogno, T. Anandamide receptors. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 377–391. [Google Scholar] [CrossRef]
- Yaguchi, T.; Nishizaki, T. Extracellular high K+ stimulates vesicular glutamate release from astrocytes by activating voltage-dependent calcium channels. J. Cell. Physiol. 2010, 225, 512–518. [Google Scholar] [CrossRef]
- Bouron, A. Phyto and endocannabinoids exert complex actions on calcium and zinc signaling in mouse cortical neurons. Biochem. Pharmacol. 2018, 152, 244–251. [Google Scholar] [CrossRef]
- Bano, D.; Ankarcrona, M. Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci. Lett. 2018, 663, 79–85. [Google Scholar] [CrossRef]
- Secondo, A.; Staiano, R.I.; Scorziello, A.; Sirabella, R.; Boscia, F.; Adornetto, A.; Valsecchi, V.; Molinaro, P.; Canzoniero, L.M.; Di Renzo, G.; et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium 2007, 42, 521–535. [Google Scholar] [CrossRef]
- Szydlowska, K.; Tymianski, M. Calcium, ischemia and excitotoxicity. Cell Calcium 2010, 47, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Khaksar, S.; Bigdeli, M.R. Anti-excitotoxic effects of cannabidiol are partly mediated by enhancement of NCX2 and NCX3 expression in animal model of cerebral ischemia. Eur. J. Pharmacol. 2017, 794, 270–279. [Google Scholar] [CrossRef]
- Ceprián, M.; Jiménez-Sánchez, L.; Vargas, C.; Barata, L.; Hind, W.; Martínez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery in a neonatal rat model of arterial ischemic stroke. Neuropharmacology 2017, 116, 151–159. [Google Scholar] [CrossRef]
- Carvalho, R.K.; Souza, M.R.; Santos, M.L.; Guimarães, F.S.; Pobbe, R.L.H.; Andersen, M.L.; Mazaro-Costa, R. Chronic cannabidiol exposure promotes functional impairment in sexual behavior and fertility of male mice. Reprod. Toxicol. 2018, 81, 34–40. [Google Scholar] [CrossRef]
- Kingston, A.E.; O’Neill, M.J.; Lam, A.; Bales, K.R.; Monn, J.A.; Schoepp, D.D. Neuroprotection by metabotropic glutamate receptor glutamate receptor agonists: LY354740, LY379268 and LY389795. Eur. J. Pharmacol. 1999, 377, 155–165. [Google Scholar] [CrossRef]
- Ali, S.A.; Zaitone, S.A.; Dessouki, A.A.; Ali, A.A. Pregabalin affords retinal neuroprotection in diabetic rats: Suppression of retinal glutamate, microglia cell expression and apoptotic cell death. Exp. Eye Res. 2019, 184, 78–90. [Google Scholar] [CrossRef]
- Jiang, H.; Li, H.; Cao, Y.; Zhang, R.; Zhou, L.; Zhou, Y.; Zeng, X.; Wu, J.; Wu, D.; Wu, D.; et al. Effects of cannabinoid (CBD) on blood-brain barrier permeability after brain injury in rats. Brain Res. 2021, 1768, 147586. [Google Scholar] [CrossRef]
- Khaksar, S.; Bigdeli, M.; Samiee, A.; Shirazi-Zand, Z. Antioxidant and anti-apoptotic effects of cannabidiol in model of ischemic stroke in rats. Brain Res. Bull. 2022, 180, 118–130. [Google Scholar] [CrossRef]
- Majdi, F.; Taheri, F.; Salehi, P.; Motaghinejad, M.; Safari, S. Cannabinoids Δ9-tetrahydrocannabinol and cannabidiol may be effective against methamphetamine induced mitochondrial dysfunction and inflammation by modulation of Toll-like type-4(Toll-like 4) receptors and NF-κB signaling. Med. Hypotheses 2019, 133, 109371. [Google Scholar] [CrossRef] [PubMed]
- Gáll, Z.; Farkas, S.; Albert, Á.; Ferencz, E.; Vancea, S.; Urkon, M.; Kolcsár, M. Effects of chronic cannabidiol treatment in the rat chronic unpredictable mild stress model of depression. Biomolecules 2020, 10, 801. [Google Scholar] [CrossRef]
- Chaves, Y.C.; Genaro, K.; Stern, C.A.; de Oliveira Guaita, G.; de Souza Crippa, J.A.; da Cunha, J.M.; Zanoveli, J.M. Two-weeks treatment with cannabidiol improves biophysical and behavioral deficits associated with experimental type-1 diabetes. Neurosci. Lett. 2020, 729, 135020. [Google Scholar] [CrossRef]
- Victor, T.R.; Hage, Z.; Tsirka, S.E. Prophylactic administration of cannabidiol reduces microglial inflammatory response to kainate-induced seizures and neurogenesis. Neuroscience 2022, in press. [Google Scholar] [CrossRef]
- Cassol, O.J., Jr.; Comim, C.M.; Silva, B.R.; Hermani, F.V.; Constantino, L.S.; Felisberto, F.; Petronilho, F.; Hallak, J.E.; De Martinis, B.S.; Zuardi, A.W.; et al. Treatment with cannabidiol reverses oxidative stress parameters, cognitive impairment and mortality in rats submitted to sepsis by cecal ligation and puncture. Brain Res. 2010, 1348, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.A.; Glyn, S.E.; Akiyama, S.; Hill, T.D.; Hill, A.J.; Weston, S.E.; Burnett, M.D.; Yamasaki, Y.; Stephens, G.J.; Whalley, B.J.; et al. Cannabidiol exerts anti-convulsant effects in animal models of temporal lobe and partial seizures. Seizure 2012, 21, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusardi, T.A.; Lytle, N.K.; Szybala, C.; Boison, D. Caffeine prevents acute mortality after TBI in rats without increased morbidity. Exp. Neurol. 2012, 234, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, F.S.; Naik, B.; Simpson, J. Hypoxia-ischemia stimulates hippocampal glutamate efflux in perinatal rat brain: An in vivo microdialysis study. Pediatr. Res. 1991, 30, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.H.; Nonaka, M.; Miller, R.; Leoni, M.; Chen, X.H.; Alsop, D.; Meaney, D.F. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. J. Neurosurg. 2000, 93, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Sellmann, T.; Miersch, D.; Kienbaum, P. The impact of arterial hypertension on polytrauma and traumatic brain injury. Dtsch. Arztebl. Int. 2012, 109, 849–856. [Google Scholar] [CrossRef]
- Alves, F.H.; Crestani, C.C.; Gomes, F.V.; Guimarães, F.S.; Correa, F.M.; Resstel, L.B. Cannabidiol injected into the bed nucleus of the stria terminalis modulates baroreflex activity through 5-HT1A receptors. Pharmacol. Res. 2010, 62, 228–236. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Lafuente, H.; Rey-Santano, M.C.; Mielgo, V.E.; Gastiasoro, E.; Rueda, M.; Martinez-Orgado, J. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr. Res. 2008, 64, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Hanscom, M.; Loane, D.J.; Shea-Donohue, T. Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury. J. Clin. Investig. 2021, 131, e143777. [Google Scholar] [CrossRef]
- Ferrara, M.; Bertozzi, G.; Zanza, C.; Longhitano, Y.; Piccolella, F.; Lauritano, C.E.; Volonnino, G.; Manetti, A.C.; Maiese, A.; Russa, R. Traumatic Brain Injury and Gut Brain Axis: The Disruption of an Alliance. Rev. Recent Clin. Trials 2022, in press. [Google Scholar] [CrossRef]
- Medel-Matus, J.S.; Lagishetty, V.; Santana-Gomez, C.; Shin, D.; Mowrey, W.; Staba, R.J.; Galanopoulou, A.S.; Sankar, R.; Jacobs, J.P.; Mazarati, A.M. Susceptibility to epilepsy after traumatic brain injury is associated with preexistent gut microbiome profile. Epilepsia 2022, 63, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N.; Miranda, K.; Wilson, K.; Holloman, B.L.; Nagarkatti, P.; Nagarkatti, M. Effects of orally administered cannabidiol on neuroinflammation and intestinal inflammation in the attenuation of experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2021, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Citti, C.; Pacchetti, B.; Vandelli, M.A.; Forni, F.; Cannazza, G. Analysis of cannabinoids in commercial hemp seed oil and decarboxylation kinetics studies of cannabidiolic acid (CBDA). J. Pharm. Biomed. Anal. 2018, 149, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Mercier, M.S.; Hill, T.D.; Glyn, S.E.; Jones, N.A.; Yamasaki, Y.; Futamura, T.; Duncan, M.; Stott, C.G.; Stephens, G.J.; et al. Cannabidivarin is anticonvulsant in mouse and rat. Br. J. Pharmacol. 2012, 167, 1629–1642. [Google Scholar] [CrossRef] [PubMed]
- Amada, N.; Yamasaki, Y.; Williams, C.M.; Whalley, B.J. Cannabidivarin (CBDV) suppresses pentylenetetrazole (PTZ)-induced increases in epilepsy-related gene expression. PeerJ 2013, 1, e214. [Google Scholar] [CrossRef]
- Zamberletti, E.; Rubino, T.; Parolaro, D. Therapeutic potential of cannabidivarin for epilepsy and autism spectrum disorder. Pharmacol. Ther. 2021, 226, 107878. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups (at the Beginning of the Experiment) | Body Weight in g (% vs. Basal) (n = 7) | Neuroscore (n = 7) | Percentage Mortality (after TBI) |
|---|---|---|---|
| SHAM (n = 7) | 294 ± 6.7 (+1.3%) | 27.0 ± 0.3 | 0% |
| TBI (n = 14) | 263 ± 10.5 *** (−9.7%) @@@ | 13.5 ± 1.6 ### | 50% |
| CBD50 + TBI (n = 9) | 269 ± 11.8 ** (−6.1%) @ | 16.4 ± 1.4 ## | 22.2% &&& |
| CBD100 + TBI (n = 9) | 253 ± 11.5 *** (−7.2%) @@ | 14.8 ± 2.8 ### | 22.2% &&& |
| CBD200 + TBI (n = 11) | 268 ± 23.4 *** (−9.6%) @@@ | 15.1 ± 2.3 ### | 36.3% & |
| Groups (at the Beginning of the Experiment) | Body Weight in g (% vs. Basal) (n = 7) | Neuroscore (n = 7) | Percentage Mortality (after TBI) |
|---|---|---|---|
| SHAM (n = 8) | 401 ± 13.5 (+40.9%) @@@ | 26.8 ± 0.4 | 0% |
| CBD + SHAM (n = 7) | 409 ± 18 (+35.8%) @@@ | 26.7 ± 0.4 | 0% |
| TBI (n = 11) | 329 ± 15 ## (+21.6%) @@@ | 17.5 ± 1.1 *** | 36.3% |
| CBD + TBI (n = 9) | 375 ± 15 && (+39.3%) @@@ | 22.0 ± 0.6 ***/ə | 22.2% ●● |
| CBD + TBI + CBD (n = 10) | 398 ± 24 & (+27.3%) @@@ | 22.4 ± 0.7 **/ə | 30% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago-Castañeda, C.; Huerta de la Cruz, S.; Martínez-Aguirre, C.; Orozco-Suárez, S.A.; Rocha, L. Cannabidiol Reduces Short- and Long-Term High Glutamate Release after Severe Traumatic Brain Injury and Improves Functional Recovery. Pharmaceutics 2022, 14, 1609. https://doi.org/10.3390/pharmaceutics14081609
Santiago-Castañeda C, Huerta de la Cruz S, Martínez-Aguirre C, Orozco-Suárez SA, Rocha L. Cannabidiol Reduces Short- and Long-Term High Glutamate Release after Severe Traumatic Brain Injury and Improves Functional Recovery. Pharmaceutics. 2022; 14(8):1609. https://doi.org/10.3390/pharmaceutics14081609
Chicago/Turabian StyleSantiago-Castañeda, Cindy, Saúl Huerta de la Cruz, Christopher Martínez-Aguirre, Sandra Adela Orozco-Suárez, and Luisa Rocha. 2022. "Cannabidiol Reduces Short- and Long-Term High Glutamate Release after Severe Traumatic Brain Injury and Improves Functional Recovery" Pharmaceutics 14, no. 8: 1609. https://doi.org/10.3390/pharmaceutics14081609