
Potent Antiplasmodial Derivatives of Dextromethorphan Reveal the Ent-Morphinan Pharmacophore of Tazopsine-Type Alkaloids

 ,
,  , , , , and
, , , , and
Abstract
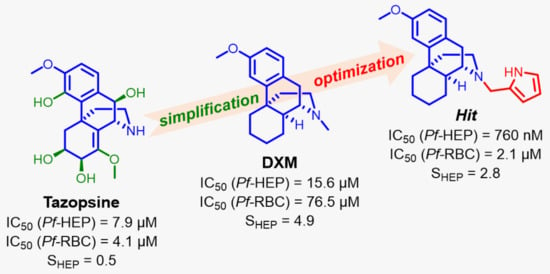
:
1. Introduction
2. Experimental Section
2.1. Reagents, Solvents, and Equipment
2.2. Synthetic Chemistry
2.2.1. Chemical Derivations of Tazopsine 1 (4,6,7,10-Tetrahydroxy-8,14-didehydro-3,8- dimethoxymorphinan)
2.2.2. Chemical Derivations of DXM [(9,13,14)-17-Methyl-3-methoxymorphinan] 3
2.3. Parasite Maintenance and Inhibition Assays (By Order of Appearance in the Manuscript)
2.3.1. P. yoelii Growth Inhibition Assays In Vitro
2.3.2. P. yoelii Growth Inhibition Assays In Vivo
2.3.3. P. falciparum and P. berghei Liver Stages Growth Inhibition Assays In Vitro
2.3.4. P. falciparum Asexual Blood Stages
2.3.5. P. falciparum Sexual Blood Stages
3. Results
3.1. Extended SAR in the Tazopsine Series
3.2. DXM Repurposing against Malaria
3.3. DXM Pharmacomodulation towards Improved Antiplasmodial Derivatives
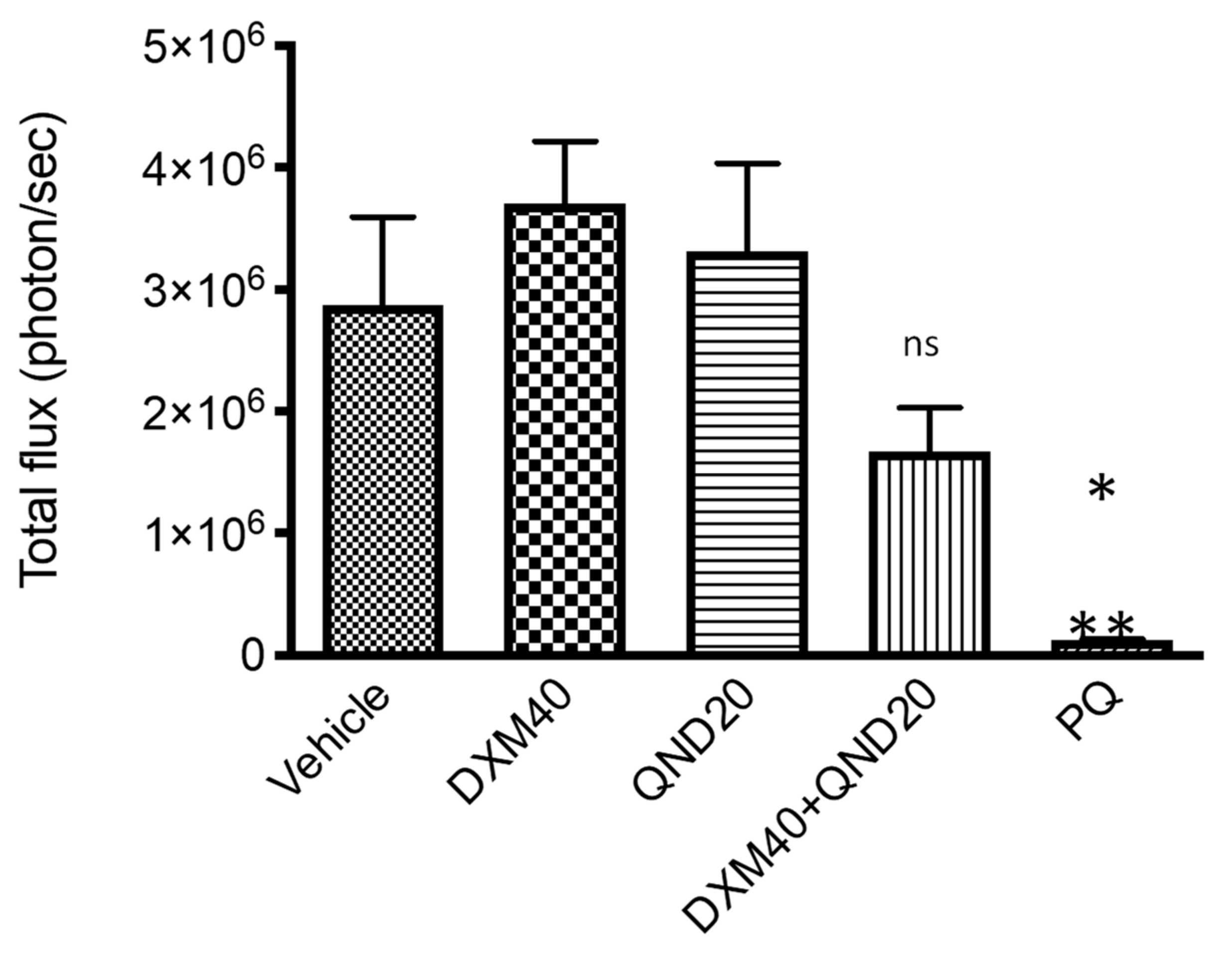
3.4. In Vitro Pre-Screening against P. berghei Liver Stages
3.5. In Vitro Screening against P. falciparum Liver and Asexual Blood Stages
3.6. In Vitro Screening against P. falciparum Sexual Blood Stages
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- WHO. World Malaria Report; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Ritchie, H.; Roser, M.; Causes of Death. Our World in Data 2018. Available online: https://ourworldindata.org/causes-of-death (accessed on 28 December 2021).
- Bhatt, S.; Weiss, D.J.; Cameron, E.; Bisanzio, D.; Mappin, B.; Dalrymple, U.; Battle, K.E.; Moyes, C.L.; Henry, A.; Eckhoff, P.A.; et al. The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature 2015, 526, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Primers 2017, 3, 17050. [Google Scholar] [CrossRef] [PubMed]
- Imwong, M.; Suwannasin, K.; Kunasol, C.; Sutawong, K.; Mayxay, M.; Rekol, H.; Smithuis, F.M.; Hlaing, T.M.; Tun, K.M.; van der Pluijm, R.W.; et al. The spread of artemisinin-resistant Plasmodium falciparum in the Greater Mekong subregion: A molecular epidemiology observational study. Lancet Infect. Dis. 2017, 17, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Kaneko, M.; Tachibana, S.I.; Balikagala, B.; Sakurai-Yatsushiro, M.; Yatsushiro, S.; Takahashi, N.; Yamauchi, M.; Sekihara, M.; Hashimoto, M.; et al. Artemisinin-Resistant Plasmodium falciparum with High Survival Rates, Uganda, 2014-2016. Emerg. Infect. Dis. 2018, 24, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Uwimana, A.; Umulisa, N.; Venkatesan, M.; Svigel, S.S.; Zhou, Z.; Munyaneza, T.; Habimana, R.M.; Rucogoza, A.; Moriarty, L.F.; Sandford, R.; et al. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: An open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect. Dis. 2021, 21, 1120–1128. [Google Scholar] [CrossRef]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef]
- Witkowski, B.; Amaratunga, C.; Khim, N.; Sreng, S.; Chim, P.; Kim, S.; Lim, P.; Mao, S.; Sopha, C.; Sam, B.; et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: In-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 2013, 13, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Bloland, P.B.; Ettling, M. Making malaria-treatment policy in the face of drug resistance. Ann. Trop. Med. Parasitol. 1999, 93, 5–23. [Google Scholar]
- Derbyshire, E.R.; Prudencio, M.; Mota, M.M.; Clardy, J. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc. Natl. Acad. Sci. USA 2012, 109, 8511–8516. [Google Scholar] [CrossRef] [Green Version]
- Kato, N.; Comer, E.; Sakata-Kato, T.; Sharma, A.; Sharma, M.; Maetani, M.; Bastien, J.; Brancucci, N.M.; Bittker, J.A.; Corey, V.; et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538, 344–349. [Google Scholar] [CrossRef]
- Le Manach, C.; Dam, J.; Woodland, J.G.; Kaur, G.; Khonde, L.P.; Brunschwig, C.; Njoroge, M.; Wicht, K.J.; Horatscheck, A.; Paquet, T.; et al. Identification and Profiling of a Novel Diazaspiro[3.4]octane Chemical Series Active against Multiple Stages of the Human Malaria Parasite Plasmodium falciparum and Optimization Efforts. J. Med. Chem. 2021, 64, 2291–2309. [Google Scholar] [CrossRef] [PubMed]
- Carraz, M.; Jossang, A.; Franetich, J.F.; Siau, A.; Ciceron, L.; Hannoun, L.; Sauerwein, R.; Frappier, F.; Rasoanaivo, P.; Snounou, G.; et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS Med. 2006, 3, e513. [Google Scholar] [CrossRef] [PubMed]
- Carraz, M.; Jossang, A.; Rasoanaivo, P.; Mazier, D.; Frappier, F. Isolation and antimalarial activity of new morphinan alkaloids on Plasmodium yoelii liver stage. Bioorg. Med. Chem. 2008, 16, 6186–6192. [Google Scholar] [CrossRef] [PubMed]
- Zahari, A.; Cheah, F.K.; Mohamad, J.; Sulaiman, S.N.; Litaudon, M.; Leong, K.H.; Awang, K. Antiplasmodial and antioxidant isoquinoline alkaloids from Dehaasia longipedicellata. Planta Med. 2014, 80, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, J.E.; Riss, P.J. Transition metal free, late-stage, regiospecific, aromatic fluorination on a preparative scale using a KF/crypt-222 complex. RSC Adv. 2018, 8, 21288–21291. [Google Scholar] [CrossRef] [Green Version]
- Jozwiak, K.; Targowska-Duda, K.M.; Kaczor, A.A.; Kozak, J.; Ligeza, A.; Szacon, E.; Wrobel, T.M.; Budzynska, B.; Biala, G.; Fornal, E.; et al. Synthesis, in vitro and in vivo studies, and molecular modeling of N-alkylated dextromethorphan derivatives as non-competitive inhibitors of alpha3beta4 nicotinic acetylcholine receptor. Bioorg. Med. Chem. 2014, 22, 6846–6856. [Google Scholar] [CrossRef] [PubMed]
- Bouyer, G.; Barbieri, D.; Dupuy, F.; Marteau, A.; Sissoko, A.; N’Dri, M.E.; Neveu, G.; Bedault, L.; Khodabux, N.; Roman, D.; et al. Plasmodium falciparum sexual parasites regulate infected erythrocyte permeability. Commun. Biol. 2020, 3, 726. [Google Scholar] [CrossRef]
- Peet, N.P. N-Demethylation of dextromethorphan. J. Pharm. Sci. 1980, 69, 1447–1448. [Google Scholar] [CrossRef]
- Cevenini, L.; Camarda, G.; Michelini, E.; Siciliano, G.; Calabretta, M.M.; Bona, R.; Kumar, T.R.; Cara, A.; Branchini, B.R.; Fidock, D.A.; et al. Multicolor bioluminescence boosts malaria research: Quantitative dual-color assay and single-cell imaging in Plasmodium falciparum parasites. Anal. Chem. 2014, 86, 8814–8821. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, C.; van Loon, W.; Habarugira, F.; Tacoli, C.; Jager, J.C.; Savelsberg, D.; Nshimiyimana, F.; Rwamugema, E.; Mbarushimana, D.; Ndoli, J.; et al. Increase in Kelch 13 Polymorphisms in Plasmodium falciparum, Southern Rwanda. Emerg. Infect. Dis. 2021, 27, 294–296. [Google Scholar] [CrossRef]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.M.; Warsame, M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.B.; Munguti, K.; et al. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Cui, L. In Vitro Activities of Primaquine-Schizonticide Combinations on Asexual Blood Stages and Gametocytes of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015, 59, 7650–7656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borch, R.F.; Bernstein, M.D.; Dupont Durst, H. Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Renia, L.; Mattei, D.; Goma, J.; Pied, S.; Dubois, P.; Miltgen, F.; Nussler, A.; Matile, H.; Menegaux, F.; Gentilini, M.; et al. A Malaria Heat-Shock-Like Determinant Expressed on the Infected Hepatocyte Surface Is the Target of Antibody-Dependent Cell-Mediated Cytotoxic Mechanisms by Nonparenchymal Liver-Cells. Eur. J. Immunol. 1990, 20, 1445–1449. [Google Scholar] [CrossRef]
- Bosson-Vanga, H.; Franetich, J.F.; Soulard, V.; Sossau, D.; Tefit, M.; Kane, B.; Vaillant, J.C.; Borrmann, S.; Muller, O.; Dereuddre-Bosquet, N.; et al. Differential activity of methylene blue against erythrocytic and hepatic stages of Plasmodium. Malar. J. 2018, 17, 143. [Google Scholar] [CrossRef]
- Manzoni, G.; Briquet, S.; Risco-Castillo, V.; Gaultier, C.; Topcu, S.; Ivanescu, M.L.; Franetich, J.F.; Hoareau-Coudert, B.; Mazier, D.; Silvie, O. A rapid and robust selection procedure for generating drug-selectable marker-free recombinant malaria parasites. Sci. Rep. 2014, 4, 4760. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.R.; Agrawal, S.; Sabir, S.; Taylor, A. Dextromethorphan; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Shin, E.J.; Bach, J.H.; Lee, S.Y.; Kim, J.M.; Lee, J.; Hong, J.S.; Nabeshima, T.; Kim, H.C. Neuropsychotoxic and neuroprotective potentials of dextromethorphan and its analogs. J. Pharmacol. Sci. 2011, 116, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Annoura, T.; Chevalley, S.; Janse, C.J.; Franke-Fayard, B.; Khan, S.M. Quantitative analysis of Plasmodium berghei liver stages by bioluminescence imaging. Methods Mol. Biol. 2013, 923, 429–443. [Google Scholar]
- Schadel, M.; Wu, D.; Otton, S.V.; Kalow, W.; Sellers, E.M. Pharmacokinetics of dextromethorphan and metabolites in humans: Influence of the CYP2D6 phenotype and quinidine inhibition. J. Clin. Psychopharmacol. 1995, 15, 263–269. [Google Scholar] [CrossRef]
- Capon, D.A.; Bochner, F.; Kerry, N.; Mikus, G.; Danz, C.; Somogyi, A.A. The influence of CYP2D6 polymorphism and quinidine on the disposition and antitussive effect of dextromethorphan in humans. Clin. Pharmacol. Ther. 1996, 60, 295–307. [Google Scholar] [CrossRef]
- Gorski, J.C.; Jones, D.R.; Wrighton, S.A.; Hall, S.D. Characterization of dextromethorphan N-demethylation by human liver microsomes. Contribution of the cytochrome P450 3A (CYP3A) subfamily. Biochem. Pharmacol. 1994, 48, 173–182. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. Reductive Amination of Aldehydes and Ketones with Sodium Triacetoxyborohydride. Studies on Direct and Indirect Reductive Amination Procedures(1). J. Org. Chem. 1996, 61, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Adjalley, S.H.; Johnston, G.L.; Li, T.; Eastman, R.T.; Ekland, E.H.; Eappen, A.G.; Richman, A.; Sim, B.K.; Lee, M.C.; Hoffman, S.L.; et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc. Natl. Acad. Sci. USA 2011, 108, E1214–E1223. [Google Scholar] [CrossRef] [Green Version]
- Le Nagard, H.; Vincent, C.; Mentre, F.; Le Bras, J. Online analysis of in vitro resistance to antimalarial drugs through nonlinear regression. Comput. Methods Prog. Biomed. 2011, 104, 10–18. [Google Scholar] [CrossRef]
- Kaddouri, H.; Nakache, S.; Houze, S.; Mentre, F.; Le Bras, J. Assessment of the drug susceptibility of Plasmodium falciparum clinical isolates from africa by using a Plasmodium lactate dehydrogenase immunodetection assay and an inhibitory maximum effect model for precise measurement of the 50-percent inhibitory concentration. Antimicrob. Agents Chemother. 2006, 50, 3343–3349. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | Reaction Time (h) | Yield (%) |
|---|---|---|
| 16a | 2 | 45 |
| 16b | 2 | 75 |
| 16c | 2 | 56 |
| 16d | 2 | 56 |
| 16e | 2 | 52 |
| 16f | 2 | 68 |
| 16g | 2 | 35 |
| 16h | 2 | 68 |
| 16i | 2 | 53 |
| 16j | 21 | 48 |
| 16k | 15 | 33 |
| 16l | 2 | 82 |
| 16m | 2 | 97 |
| Cpd. | Substitutions | IC50 (Py265BY-PMH, µM) |
|---|---|---|
| Tazopsine 1 | R1 = OH, R2 = H | 3.1 ± 0.2 |
| 10-epi-tazopsine 4 | R1 = H, R2 = OH | 16.1 ± 1.9 |
| Sinococuline 5 | R1 = R2 = H | 4.5 ± 0.4 |
| 4-O-Me-tazopsine 6 | NA | >100 |
| N-methyl-tazopsine 7a | R = Me | 5.8 ± 0.4 |
| N-n-propyl-tazopsine 7b | R = n-Pro | 12.6 ± 1.7 |
| N-4′-hydroxybenzyl-tazopsine 7c | R = 4-OH-Bn | 14.2 ± 2.2 |
| N-4′-methoxybenzyl-tazopsine 7d | R = 4-OMe-Bn | 24.2 ± 0.7 |
| N-3′,4′-methylenedioxybenzyl-tazopsine 7e | R = 3,4-methylenedioxy-Bn | >100 |
| N-4′-chlorobenzyl-tazopsine 7f | R = 4-Cl-Bn | 5.8 ± 1.1 |
| N-4′-bromobenzyl-tazopsine 7g | R = 4-Br-Bn | 4.2 ± 0.3 |
| N-acetyl-tazopsine 8 | R = Ac | >100 |
| Sinoacutine 9 | NA | >100 |
| Sinomenine 10 | NA | >100 |
| PQ | NA | 0.62 ± 0.03 |
| Cpds. | IC50 (PHH, µM) |
|---|---|
| Tazopsine 1 | 7.88 ± 3.05 |
| DXM 3 | 15.59 ± 1.19 |
| PQ | 0.75 ± 0.15 |
| Cpds. | R | IC50 (µM) | S | |
|---|---|---|---|---|
| PfNF54-PHH | Pf3D7-HE | |||
| Tazopsine 1 | NA | 7.88 ± 3.05 | 4.07 ± 0.87 | 0.5 |
| DXM 3 | Me | 15.59 ± 1.19 | 76.5 ± 0.9 | 4.9 |
| 11 | NA | 4.10 ± 2.77 | 61.7 ± 5.3 | 15.0 |
| 16a | n-propyl | 2.25 ± 3.03 | 43.3 ± 2.3 | 19.2 |
| 16g | 2′-furanylmethyl | 1.56 ± 0.59 | 56.2 ± 2.7 | 36.0 |
| 16i | 2′-pyrrolylmethyl | 0.76 ± 0.13 | 2.1 ± 0.4 | 2.8 |
| 16l | 2′-indolylmethyl | 1.98 ± 0.34 | 6.5 ± 0.4 | 3.3 |
| PQ | NA | 0.75 ± 0.15 | NT | NA |
| CQ | NA | NT | 0.033 ± 0.016 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keita, A.; Franetich, J.-F.; Carraz, M.; Valentin, L.; Bordessoules, M.; Baron, L.; Bigeard, P.; Dupuy, F.; Geay, V.; Tefit, M.; et al. Potent Antiplasmodial Derivatives of Dextromethorphan Reveal the Ent-Morphinan Pharmacophore of Tazopsine-Type Alkaloids. Pharmaceutics 2022, 14, 372. https://doi.org/10.3390/pharmaceutics14020372
Keita A, Franetich J-F, Carraz M, Valentin L, Bordessoules M, Baron L, Bigeard P, Dupuy F, Geay V, Tefit M, et al. Potent Antiplasmodial Derivatives of Dextromethorphan Reveal the Ent-Morphinan Pharmacophore of Tazopsine-Type Alkaloids. Pharmaceutics. 2022; 14(2):372. https://doi.org/10.3390/pharmaceutics14020372
Chicago/Turabian StyleKeita, Antoinette, Jean-François Franetich, Maëlle Carraz, Loïse Valentin, Mallaury Bordessoules, Ludivine Baron, Pierre Bigeard, Florian Dupuy, Valentine Geay, Maurel Tefit, and et al. 2022. "Potent Antiplasmodial Derivatives of Dextromethorphan Reveal the Ent-Morphinan Pharmacophore of Tazopsine-Type Alkaloids" Pharmaceutics 14, no. 2: 372. https://doi.org/10.3390/pharmaceutics14020372