
Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations

 , ,
, ,  ,
, 
Abstract
:1. Introduction
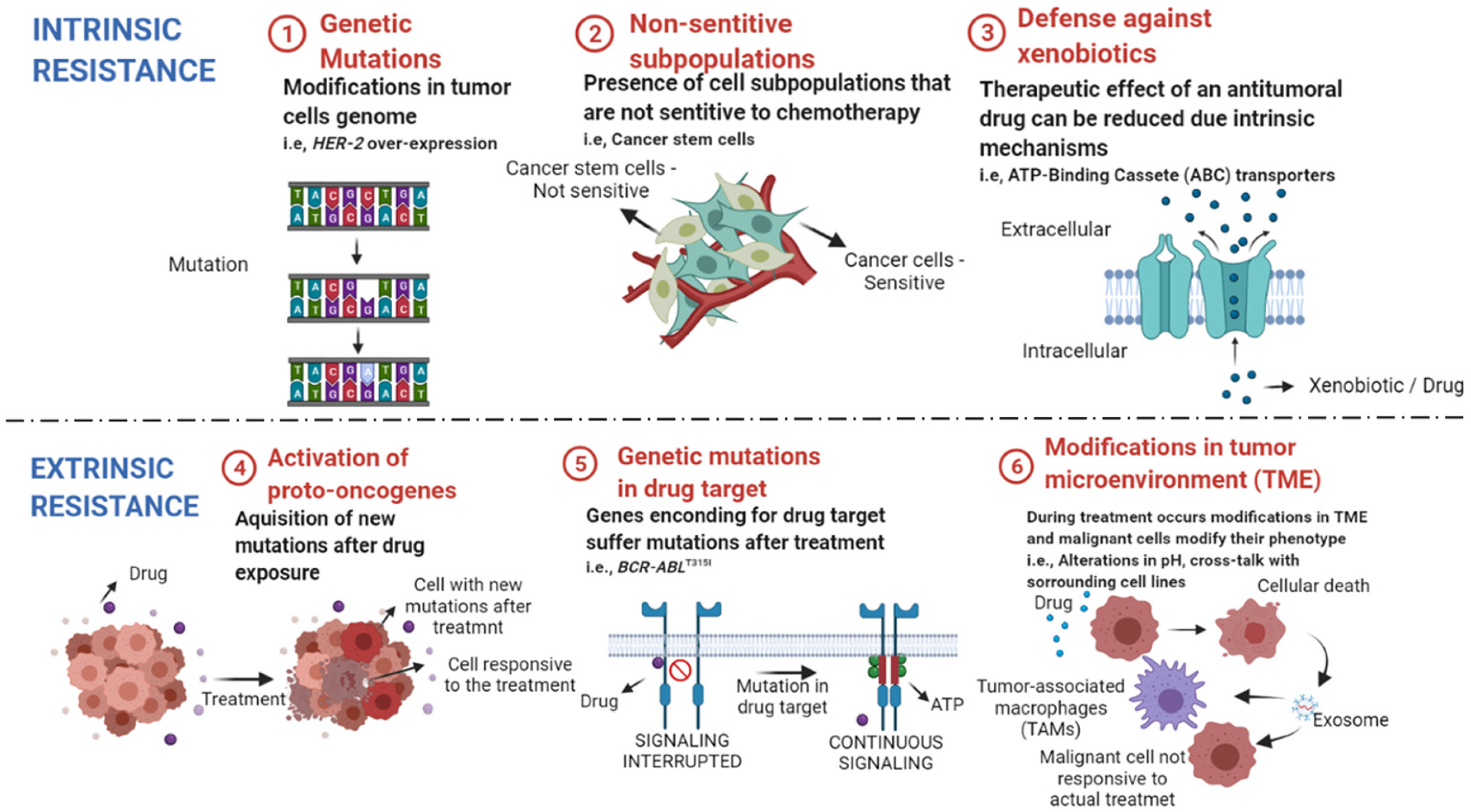
Conventional Therapies and Resistance in Hematologic Neoplasms
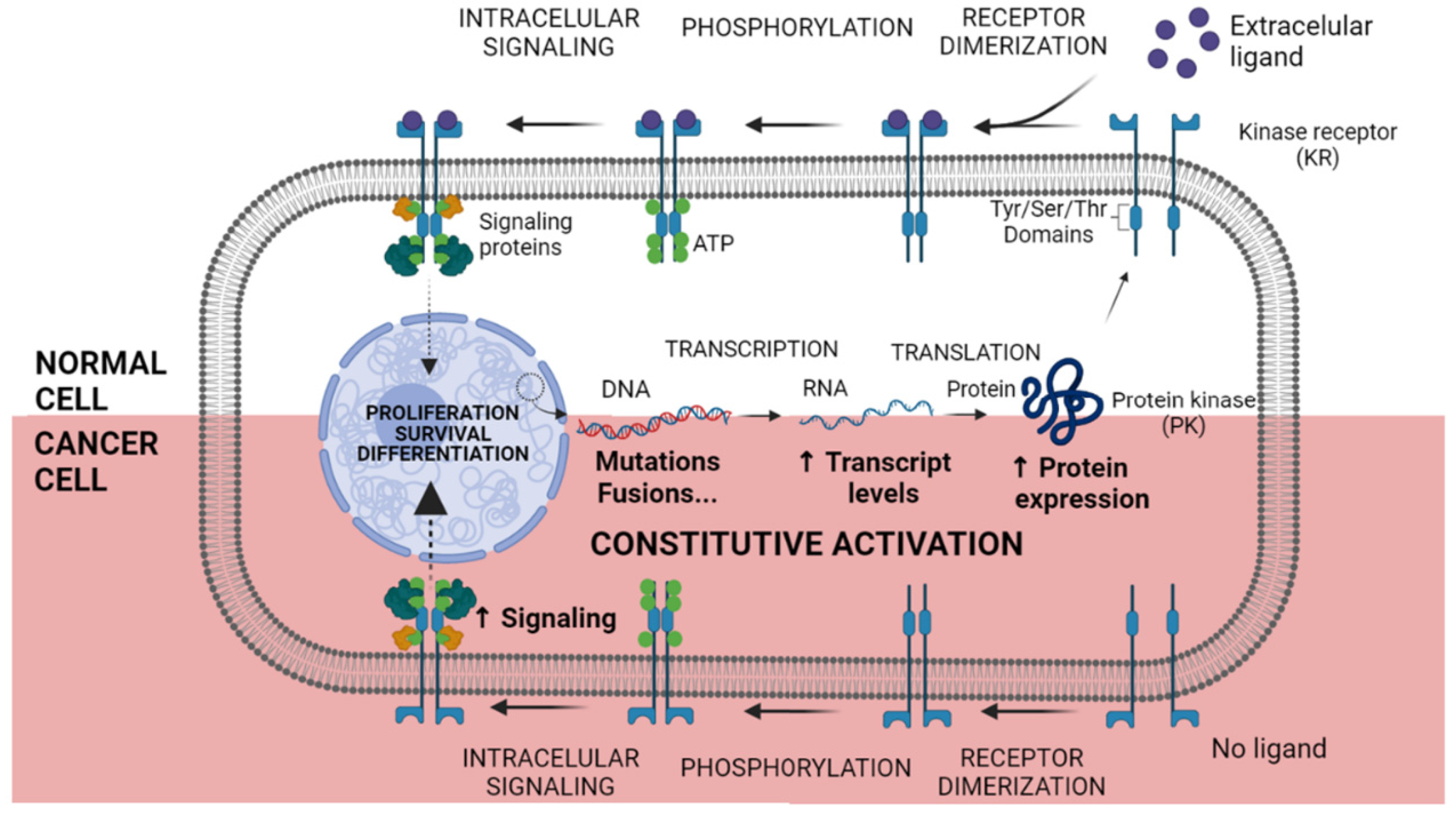
2. The Advent of Kinase Inhibition
3. Recent Prospects into Clinical Investigations
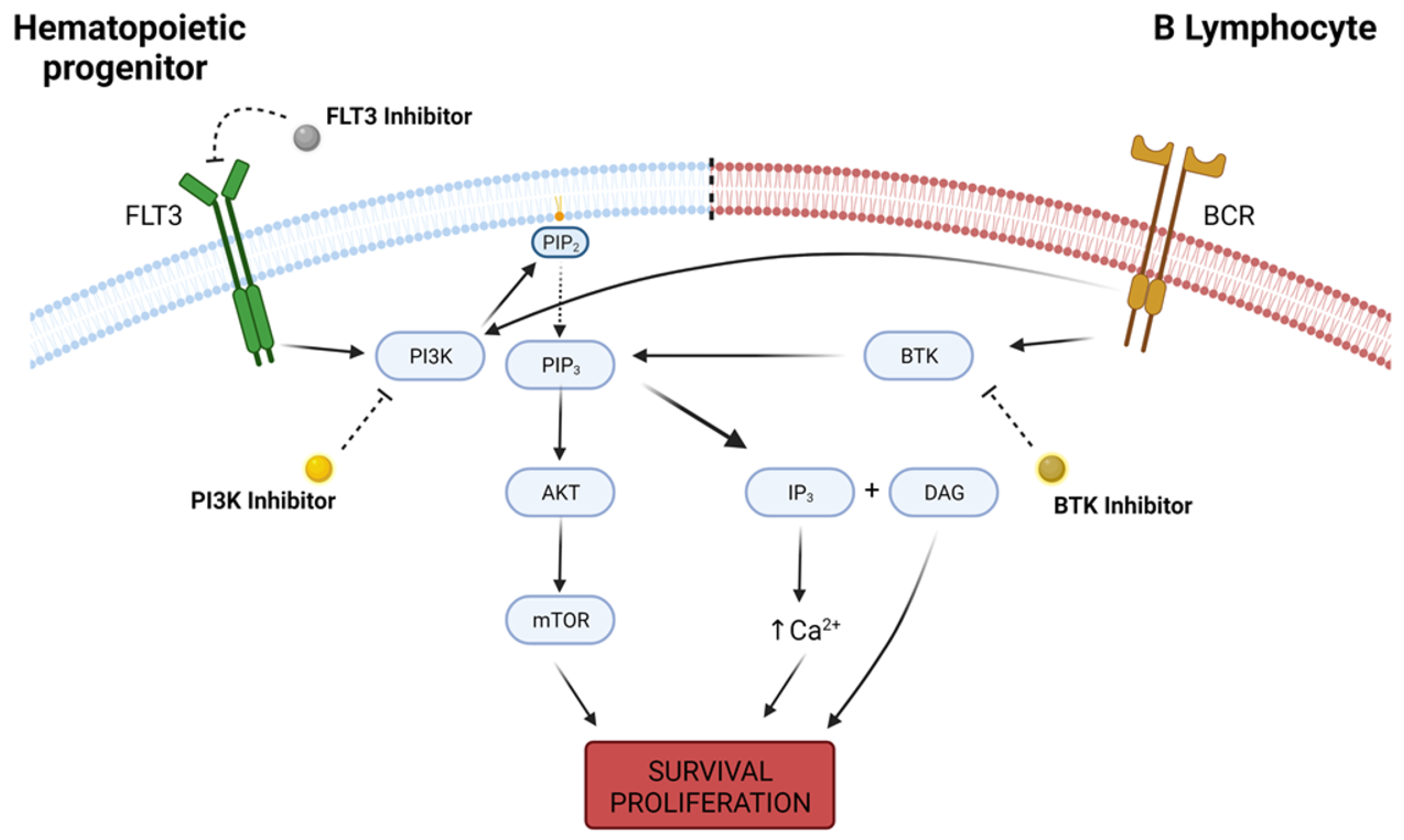
4. AML and Myeloproliferative Disorders
5. Lymphoid Malignancies
6. Toxicity Profiles
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowska, A.M.; Rudzki, M.; Rudzki, S.; Lewandowski, T.; Laskowska, B. Environmental Risk Factors for Cancer–Review Paper. Ann. Agric. Environ. Med. 2019, 26, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, J.O.; Gascoyne, R.D.; Lunning, M.A.; Cavalli, F. Non-Hodgkin Lymphoma. Lancet 2017, 390, 298–310. [Google Scholar] [CrossRef]
- Juliusson, G.; Hough, R. Leukemia. Prog. Tumor Res. 2016, 43, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov. 2021, 2021, 1–23. [Google Scholar] [CrossRef]
- Ramdass, B.; Chowdhary, A.; Koka, P.S. Hematological Malignancies: Disease Pathophysiology of Leukemic Stem Cells. J. Stem. Cells 2013, 8, 151–187. [Google Scholar]
- Nogueira, B.M.D.; Machado, C.B.; Montenegro, R.C.; Moraes, M.E.A.D.; Moreira-Nunes, C.A. Telomere Length and Hematological Disorders: A Review. In Vivo 2020, 34, 3093–3101. [Google Scholar] [CrossRef]
- Harrison, C.J. Cytogenetics of Paediatric and Adolescent Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2009, 144, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Montaño, A.; Forero-Castro, M.; Hernández-Rivas, J.-M.; García-Tuñón, I.; Benito, R. Targeted Genome Editing in Acute Lymphoblastic Leukemia: A Review. BMC Biotechnol. 2018, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.; Morgan, L.; Lin, T.T.; Hewamana, S.; Matthews, R.J.; Deaglio, S.; Rowntree, C.; Fegan, C.; Pepper, C.; Brennan, P. Genetic Modification of Primary Chronic Lymphocytic Leukemia Cells with a Lentivirus Expressing CD38. Haematologica 2010, 95, 514–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, A. The Origin and Spread of Human Leukemia. Med. Hypotheses 1992, 39, 110–118. [Google Scholar] [CrossRef]
- Beutler, E. The Treatment of Acute Leukemia: Past, Present, and Future. Leukemia 2001, 15, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, E.H.; Good, J.T.; Martin, E. Chemotherapy of malignant neoplastic diseases. J. Am. Med. Assoc. 1950, 142, 383–390. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K. Temporary Remissions in Acute Leukemia in Children Produced by Folic Acid Antagonist, 4-Aminopteroyl-Glutamic Acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef]
- Niu, C.; Yan, H.; Yu, T.; Sun, H.P.; Liu, J.X.; Li, X.S.; Wu, W.; Zhang, F.Q.; Chen, Y.; Zhou, L.; et al. Studies on Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide: Remission Induction, Follow-up, and Molecular Monitoring in 11 Newly Diagnosed and 47 Relapsed Acute Promyelocytic Leukemia Patients. Blood 1999, 94, 3315–3324. [Google Scholar] [CrossRef]
- Hajdu, S.I.; Vadmal, M. A Note from History: Landmarks in History of Cancer, Part 6. Cancer 2013, 119, 4058–4082. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.; Marie, J.; Salet, J.; Cruciani, C. Essai de Traitement Des Leucemies Aigues de l’enfance Par l’association Aminopterin-Cortison. Bull. Mem. Soc. Med. Hop. 1951, 16, 621–625. [Google Scholar]
- Gellhorn, A.; Hyman, G.A.; Ultmann, J.E. Chlorambucil in Treatment of Chronic Lymphocytic Leukemia and Certain Lymphomas. J. Am. Med. Assoc. 1956, 162, 178–183. [Google Scholar] [CrossRef]
- Burchenal, J.H.; Murphy, M.L.; Ellison, R.R.; Sykes, M.P.; Tan, T.C.; Leone, L.A.; Karnofsky, D.A.; Craver, L.F.; Dargeon, H.W.; Rhoads, C.P. Clinical Evaluation of a New Antimetabolite, 6-Mercaptopurine, in the Treatment of Leukemia and Allied Diseases. Blood 1953, 8, 965–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coggins, P.R.; Ravdin, R.G.; Eisman, S.H. Clinical Evaluation of a New Alkylating Agent: Cytoxan (Cyclophosphamide). Cancer 1960, 13, 1254–1260. [Google Scholar] [CrossRef]
- Noble, R.L.; Beer, C.T.; Cutts, J.H. Role of Chance Observations in Chemotherapy: Vinca Rosea. Ann. N. Y. Acad. Sci. 1958, 76, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Dubost, M.; Ganter, P.; Maral, R. Une Nouvel Antibiotic a Proprieties Antitumorales. C. R. Hebd. Seances Acad. Sci. 1963, 257, 1813–1818. [Google Scholar] [PubMed]
- Umezawa, H.; Ishizuka, M.; Maeda, K.; Takeuchi, T. Studies on Bleomycin. Cancer 1967, 20, 891–895. [Google Scholar] [CrossRef]
- Galton, D.A. Myleran in Chronic Myeloid Leukaemia; Results of Treatment. Lancet 1953, 264, 208–213. [Google Scholar] [CrossRef]
- Karon, M.; Freireich, E.J.; Frei, E.; Taylor, R.; Wolman, I.J.; Djerassi, I.; Lee, S.L.; Sawitsky, A.; Hananian, J.; Selawry, O.; et al. The Role of Vincristine in the Treatment of Childhood Acute Leukemia. Clin. Pharm. Ther. 1966, 7, 332–339. [Google Scholar] [CrossRef]
- Hill, J.M.; Roberts, J.; Loeb, E.; Khan, A.; MacLellan, A.; Hill, R.W. L-Asparaginase Therapy for Leukemia and Other Malignant Neoplasms. Remission in Human Leukemia. JAMA 1967, 202, 882–888. [Google Scholar] [CrossRef]
- Tan, C.; Tasaka, H.; Yu, K.-P.; Murphy, M.L.; Karnofsky, D.A. Daunomycin, an Antitumor Antibiotic, in the Treatment of Neoplastic Disease. Clinical Evaluation with Special Reference to Childhood Leukemia. Cancer 1967, 20, 333–353. [Google Scholar] [CrossRef]
- Whiteside, J.A.; Philips, F.S.; Dargeon, H.W.; Burchenal, J.H. Intrathecal Amethopterin in Neurological Manifestations of Leukemia. AMA Arch. Intern. Med. 1958, 101, 279–285. [Google Scholar] [CrossRef]
- Rundles, R.W.; Grizzle, J.; Bell, W.N.; Corley, C.C.; Frommeyer, W.B.; Greenberg, B.G.; Huguley, C.M.; Watson James, G.; Jones, R.; Larsen, W.E.; et al. Comparison of Chlorambucil and Myleran® in Chronic Lymphocytic and Granulocytic Leukemia. Am. J. Med. 1959, 27, 424–432. [Google Scholar] [CrossRef]
- Rundles, R.W.; Laszlo, J.; Garrison, F.E.; Hobson, J.B. The Antitumor Spectrum of Cyclophosphamide. Cancer Chemother. Rep. 1962, 16, 407–411. [Google Scholar]
- George, P.; Hernandez, K.; Hustu, O.; Borella, L.; Holton, C.; Pinkel, D. A Study of “Total Therapy” of Acute Lymphocytic Leukemia in Children. J. Pediatr. 1968, 72, 399–408. [Google Scholar] [CrossRef]
- Carbone, P.P.; Spurr, C.; Schneiderman, M.; Scotto, J.; Holland, J.F.; Shnider, B. Management of Patients with Malignant Lymphoma: A Comparative Study with Cyclophosphamide and Vinca Alkaloids. Cancer Res. 1968, 28, 811–822. [Google Scholar] [PubMed]
- Boiron, M.; Weil, M.; Jacquillat, C.; Tanzer, J.; Levy, D.; Sultan, C.; Bernard, J. Daunorubicin in the Treatment of Acute Myelocytic Leukaemia. Lancet 1969, 1, 330–333. [Google Scholar] [CrossRef]
- Kersey, J.H. Fifty Years of Studies of the Biology and Therapy of Childhood Leukemia. Blood 1997, 90, 4243–4251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devita, V.T.; Serpick, A.A.; Carbone, P.P. Combination Chemotherapy in the Treatment of Advanced Hodgkin’s Disease. Ann. Intern. Med. 1970, 73, 881–895. [Google Scholar] [CrossRef]
- Moxley, J.H.; De Vita, V.T.; Brace, K.; Frei, E. Intensive Combination Chemotherapy and X-Irradiation in Hodgkin’s Disease. Cancer Res. 1967, 27, 1258–1263. [Google Scholar] [PubMed]
- Haghbin, M.; Tan, C.C.; Clarkson, B.D.; Miké, V.; Burchenal, J.H.; Murphy, M.L. Intensive Chemotherapy in Children with Acute Lymphoblastic Leukemia (L-2 Protocol). Cancer 1974, 33, 1491–1498. [Google Scholar] [CrossRef]
- Aur, R.J.A.; Simone, J.V.; Hustu, H.O.; Verzosa, M.S. A Comparative Study of Central Nervous System Irradiation and Intensive Chemotherapy Early in Remission of Childhood Acute Lymphocytic Leukemia. Cancer 1972, 29, 381–391. [Google Scholar] [CrossRef]
- Thomas, E.D.; Buckner, C.D.; Banaji, M.; Clift, R.A.; Fefer, A.; Flournoy, N.; Goodell, B.W.; Hickman, R.O.; Lerner, K.G.; Neiman, P.E.; et al. One Hundred Patients with Acute Leukemia Treated by Chemotherapy, Total Body Irradiation, and Allogeneic Marrow Transplantation. Blood 1977, 49, 511–533. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.-S.; Carter, B.Z.; Andreeff, M. Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of Drug Resistance in Acute Myeloid Leukemia. Onco. Targets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and Impact of Subclonal Mutations in Chronic Lymphocytic Leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef] [Green Version]
- Tadesse, F.; Asres, G.; Abubeker, A.; Gebremedhin, A.; Radich, J. Spectrum of BCR-ABL Mutations and Treatment Outcomes in Ethiopian Imatinib-Resistant Patients With Chronic Myeloid Leukemia. JCO Glob. Oncol. 2021, 1187–1193. [Google Scholar] [CrossRef]
- Huang, R.; Kang, Q.; Liu, H.; Li, Y. New Insights into the Molecular Resistance Mechanisms of Chronic Myeloid Leukemia. Curr. Cancer Drug Targets 2016, 16, 323–345. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Kantarjian, H.M.; Cortes, J.E. Mechanisms of Primary and Secondary Resistance to Imatinib in Chronic Myeloid Leukemia. Cancer Control 2009, 16, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Viale, A.; De Franco, F.; Orleth, A.; Cambiaghi, V.; Giuliani, V.; Bossi, D.; Ronchini, C.; Ronzoni, S.; Muradore, I.; Monestiroli, S.; et al. Cell-Cycle Restriction Limits DNA Damage and Maintains Self-Renewal of Leukaemia Stem Cells. Nature 2009, 457, 51–56. [Google Scholar] [CrossRef]
- Michael, M.; Doherty, M.M. Tumoral Drug Metabolism: Overview and Its Implications for Cancer Therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef]
- Wolach, O.; Itchaki, G.; Bar-Natan, M.; Yeshurun, M.; Ram, R.; Herscovici, C.; Shpilberg, O.; Douer, D.; Tallman, M.S.; Raanani, P. High-Dose Cytarabine as Salvage Therapy for Relapsed or Refractory Acute Myeloid Leukemia--Is More Better or More of the Same? Hematol. Oncol. 2016, 34, 28–35. [Google Scholar] [CrossRef]
- Pinto, C.A.; DE Sousa Portilho, A.J.; Barbosa, M.C.; de Moraes, M.E.A.; de Lemos, J.A.R.; Burbano, R.M.R.; Moreira-Nunes, C.A. Combined Therapy of ATRA and Imatinib Mesylate Decreases BCR-ABL and ABCB1/MDR1 Expression Through Cellular Differentiation in a Chronic Myeloid Leukemia Model. In Vivo 2021, 35, 2661–2667. [Google Scholar] [CrossRef]
- Baudis, M.; Prima, V.; Tung, Y.H.; Hunger, S.P. ABCB1 Over-Expression and Drug-Efflux in Acute Lymphoblastic Leukemia Cell Lines with t(17;19) and E2A-HLF Expression. Pediatr. Blood Cancer 2006, 47, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug Resistance: Still a Daunting Challenge to the Successful Treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eadie, L.N.; Hughes, T.P.; White, D.L. ABCB1 Overexpression Is a Key Initiator of Resistance to Tyrosine Kinase Inhibitors in CML Cell Lines. PLoS ONE 2016, 11, e0161470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, C.B.; Nogueira, B.M.D.; de Sousa Portilho, A.J.; de Moraes Filho, M.O.; de Moraes, M.E.A.; de Fátima Aquino Moreira Nunes, C. Caracterização do Cromossomo Philadephia em Tumores Não-Sólidos: Uma Abordagem Citogenética Ao Câncer. In A Genética e a Construção de Novos Paradigmas Nas Ciências da Vida; Atenas: Ponta Grossa, Brazil, 2021; pp. 14–21. [Google Scholar]
- Manouchehri, A.; Kanu, E.; Mauro, M.J.; Aday, A.W.; Lindner, J.R.; Moslehi, J. Tyrosine Kinase Inhibitors (TKI) in Leukemia and Cardiovascular Events-From Mechanism to Patient Care. Arter. Thromb. Vasc. Biol. 2020, 40, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yaghmaie, M.; Yeung, C.C. Molecular Mechanisms of Resistance to Tyrosine Kinase Inhibitors. Curr. Hematol. Malig. Rep. 2019, 14, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Chaar, M.; Kamta, J.; Ait-Oudhia, S. Mechanisms, Monitoring, and Management of Tyrosine Kinase Inhibitors–Associated Cardiovascular Toxicities. OncoTargets Ther. 2018, 11, 6227. [Google Scholar] [CrossRef] [Green Version]
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Baccarani, M. Tyrosine Kinase Inhibitors in Chronic Myeloid Leukaemia: Which, When, for Whom? Nat. Rev. Clin. Oncol. 2017, 14, 141–154. [Google Scholar] [CrossRef]
- Pophali, P.A.; Patnaik, M.M. The Role of New Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Cancer J. (Sudbury Mass.) 2016, 22, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waller, C. Imatinib Mesylate. Recent Results Cancer Res. 2018, 212, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Ame, S.; Berger, M.; Cayuela, J.-M.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Dubruille, V.; Dulucq, S.; Etienne, G.; et al. Discontinuation of Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia: Recommendations for Clinical Practice from the French Chronic Myeloid Leukemia Study Group. Cancer 2018, 124, 2956–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Azevedo, L.D.; Bastos, M.M.; de Oliveira, A.P.; Boechat, N. Syntheses and properties of drugs inhibitors of bcr-abl tyrosine kinase, used in the treatment of chronic myeloid leukemia. Química Nova 2017, 40, 791–809. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Martinez, R., III.; Defnet, A.; Shapiro, P. Avoiding or Co-Opting ATP Inhibition: Overview of Type III, IV, V, and VI Kinase Inhibitors. Next Gener. Kinase Inhib. 2020, 29. [Google Scholar] [CrossRef]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, F.M.; Gray, N.S. Kinase Inhibitors: The Road Ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Antar, A.; Otrock, Z.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 Inhibitors in Acute Myeloid Leukemia: Ten Frequently Asked Questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Lee, H.; Basso, I.; Kim, D. Target Spectrum of the BCR-ABL Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Int. J. Hematol. 2021, 113, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Deuker, M.; Chun, F.; Karakiewicz, P. Second-Line Tyrosine Kinase Inhibitor-Therapy after Immunotherapy-Failure. Curr. Opin. Support. Palliat. Care 2020, 14, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Capponi, M.; Ambrosetti, A.; Lucia, E.; Antolino, A.; Pulsoni, A.; Ferrari, S.; Zinzani, P.L.; et al. Vemurafenib plus Rituximab in Refractory or Relapsed Hairy-Cell Leukemia. N. Engl. J. Med. 2021, 384, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Sunami, K.; Minami, H.; Hatake, K.; Sekiguchi, R.; Natsume, K.; Ishikawa, N.; Rinne, M.; Taniwaki, M. A Phase I, Dose-Escalation Study of Oral PIM447 in Japanese Patients with Relapsed and/or Refractory Multiple Myeloma. Int. J. Hematol. 2021, 113, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Quach, H.; Nicol, A.; Badoux, X.; Rose, H.; Miles Prince, H.; Leahy, M.F.; Eek, R.; Wickham, N.; Patil, S.S.; et al. Zanubrutinib (BGB-3111) plus Obinutuzumab in Patients with Chronic Lymphocytic Leukemia and Follicular Lymphoma. Blood Adv. 2020, 4, 4802–4811. [Google Scholar] [CrossRef]
- Borthakur, G.; Zeng, Z.; Cortes, J.E.; Chen, H.C.; Huang, X.; Konopleva, M.; Ravandi, F.; Kadia, T.; Patel, K.P.; Daver, N.; et al. Phase 1 Study of Combinatorial Sorafenib, G-CSF, and Plerixafor Treatment in Relapsed/Refractory, FLT3-ITD-Mutated Acute Myelogenous Leukemia Patients. Am. J. Hematol. 2020, 95, 1296–1303. [Google Scholar] [CrossRef]
- Navada, S.C.; Garcia-Manero, G.; OdchimarReissig, R.; Pemmaraju, N.; Alvarado, Y.; Ohanian, M.N.; John, R.B.; Demakos, E.P.; Zbyszewski, P.S.; Maniar, M.; et al. Rigosertib in Combination with Azacitidine in Patients with Myelodysplastic Syndromes or Acute Myeloid Leukemia: Results of a Phase 1 Study. Leuk. Res. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, S.; Zhou, K.; Pan, L.; Li, Z.; Zhou, J.; Gao, S.; Zhou, D.; Hu, J.; Feng, R.; et al. Treatment of Relapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma with the BTK Inhibitor Zanubrutinib: Phase 2, Single-Arm, Multicenter Study. J. Hematol. Oncol. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Levis, M.J.; Frankfurt, O.; Pagel, J.M.; Roboz, G.J.; Stone, R.M.; Wang, E.S.; Severson, P.L.; West, B.L.; Le, M.H.; et al. A Phase 1/2 Study of the Oral FLT3 Inhibitor Pexidartinib in Relapsed/ Refractory FLT3-ITD–Mutant Acute Myeloid Leukemia. Blood Adv. 2020, 4, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Nierman, P.; Kendall, E.K.; Cheung, J.; Gulrajani, M.; Herman, S.E.M.; Pleyer, C.; Ahn, I.E.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Clinical and Biological Implications of Target Occupancy in CLL Treated with the BTK Inhibitor Acalabrutinib. Blood 2020, 136, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Hawkes, E.; Verhoef, G.; Haioun, C.; Thye Lim, S.; Seog Heo, D.; Ardeshna, K.; Chong, G.; Haaber, J.; Shi, W.; et al. Single-Agent Activity of Phosphatidylinositol 3-Kinase Inhibition with Copanlisib in Patients with Molecularly Defined Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Leukemia 2020, 34, 2184–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Wierda, W.G.; Schuh, A.; Devereux, S.; Chaves, J.M.; Brown, J.R.; Hillmen, P.; Martin, P.; Awan, F.T.; Stephens, D.M.; et al. Acalabrutinib Monotherapy in Patients with Relapsed/ Refractory Chronic Lymphocytic Leukemia: Updated Phase 2 Results. Blood 2020, 135, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lam, S.S.Y.; Leung, G.M.K.; Tsui, S.P.; Yang, N.; Ng, N.K.L.; Ip, H.W.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; et al. Sorafenib and Omacetaxine Mepesuccinate as a Safe and Effective Treatment for Acute Myeloid Leukemia Carrying Internal Tandem Duplication of Fms-like Tyrosine Kinase 3. Cancer 2020, 126, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and Umbralisib in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma and Chronic Lymphocytic Leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final Analysis from RESONATE: Up to Six Years of Follow-up on Ibrutinib in Patients with Previously Treated Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Flinn, I.W.; Cherry, M.A.; Maris, M.B.; Matous, J.V.; Berdeja, J.G.; Patel, M. Combination Trial of Duvelisib (IPI-145) with Rituximab or Bendamustine/Rituximab in Patients with Non-Hodgkin Lymphoma or Chronic Lymphocytic Leukemia. Am. J. Hematol. 2019, 94, 1325–1334. [Google Scholar] [CrossRef]
- Takahashi, T.; Usuki, K.; Matsue, K.; Ohno, H.; Sakura, T.; Imanaka, R.; Murakami, M.; Ohwada, S.; Takagi, T.; Sakajiri, S. Efficacy and Safety of Quizartinib in Japanese Patients with FLT3-ITD Positive Relapsed or Refractory Acute Myeloid Leukemia in an Open-Label, Phase 2 Study. Int. J. Hematol. 2019, 110, 665–674. [Google Scholar] [CrossRef]
- Kessler, T.; Koschmieder, S.; Schliemann, C.; Crysandt, M.; Mikesch, J.H.; von Stillfried, S.; Stelljes, M.; Pohlen, M.; Lenz, G.; Kirsch, A.; et al. Phase II Clinical Trial of Pazopanib in Patients with Acute Myeloid Leukemia (AML), Relapsed or Refractory or at Initial Diagnosis without an Intensive Treatment Option (PazoAML). Ann. Hematol. 2019, 98, 1393–1401. [Google Scholar] [CrossRef]
- Munakata, W.; Ando, K.; Hatake, K.; Fukuhara, N.; Kinoshita, T.; Fukuhara, S.; Shirasugi, Y.; Yokoyama, M.; Ichikawa, S.; Ohmachi, K.; et al. Phase I Study of Tirabrutinib (ONO-4059/GS-4059) in Patients with Relapsed or Refractory B-Cell Malignancies in Japan. Cancer Sci. 2019, 110, 1686–1694. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Ramchandren, R.; Yacoub, A.; Wertheim, M.S.; Edenfield, W.J.; Caimi, P.; Gutierrez, M.; Akard, L.; Escobar, C.; Call, J.; et al. Parsaclisib, a Potent and Highly Selective PI3Kd Inhibitor, in Patients with Relapsed or Refractory B-Cell Malignancies. Blood 2019, 133, 1742–1752. [Google Scholar] [CrossRef] [Green Version]
- Maiti, A.; Naqvi, K.; Kadia, T.M.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.G.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-Mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Nicotra, A.; Savell, A.; Francoeur, K.; Hellman, J.M.; Bazemore, J.; Miskin, H.P.; Sportelli, P.; Stampleman, L.; et al. Umbralisib in Combination with Ibrutinib in Patients with Relapsed or Refractory Chronic Lymphocytic Leukaemia or Mantle Cell Lymphoma: A Multicentre Phase 1-1b Study. Lancet Haematol. 2019, 6, e38–e47. [Google Scholar] [CrossRef]
- Flinn, I.W.; Hillmen, P.; Montillo, M.; Nagy, Z.; Illés, Á.; Etienne, G.; Delgado, J.; Kuss, B.J.; Tam, C.S.; Gasztonyi, Z.; et al. The Phase 3 DUO Trial: Duvelisib vs Ofatumumab in Relapsed and Refractory CLL/SLL. Blood 2018, 132, 2446–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaza, Y.; Hidalgo-Lopez, J.E.; Verstovsek, S.; Jabbour, E.; Ravandi, F.; Borthakur, G.; Estrov, Z.; Alvarado, Y.; Burger, J.; Schneider, H.; et al. Phase I Study of Ruxolitinib in Previously Treated Patients with Low or Intermediate-1 Risk Myelodysplastic Syndrome with Evidence of NF-KB Activation. Leuk. Res. 2018, 73, 78–85. [Google Scholar] [CrossRef]
- Dalle, I.A.; Cortes, J.E.; Pinnamaneni, P.; Lamothe, B.; Duque, A.D.; Randhawa, J.; Pemmaraju, N.; Jabbour, E.; Ferrajoli, A.; Wierda, W.G.; et al. A Pilot Phase II Study of Erlotinib for the Treatment of Patients with Relapsed/Refractory Acute Myeloid Leukemia. Acta Haematol. 2018, 140, 30–39. [Google Scholar] [CrossRef]
- Usuki, K.; Sakura, T.; Kobayashi, Y.; Miyamoto, T.; Iida, H.; Morita, S.; Bahceci, E.; Kaneko, M.; Kusano, M.; Yamada, S.; et al. Clinical Profile of Gilteritinib in Japanese Patients with Relapsed/Refractory Acute Myeloid Leukemia: An Open-Label Phase 1 Study. Cancer Sci. 2018, 109, 3235–3244. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b Study of 2 Dosing Regimens of Quizartinib Monotherapy in FLT3-ITD–Mutated, Relapsed or Refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Burris, H.A.; Flinn, I.W.; Patel, M.R.; Fenske, T.S.; Deng, C.; Brander, D.M.; Gutierrez, M.; Essell, J.H.; Kuhn, J.G.; Miskin, H.P.; et al. Umbralisib, a Novel PI3Kδ and Casein Kinase-1ε Inhibitor, in Relapsed or Refractory Chronic Lymphocytic Leukaemia and Lymphoma: An Open-Label, Phase 1, Dose-Escalation, First-in-Human Study. Lancet Oncol. 2018, 19, 486–496. [Google Scholar] [CrossRef]
- Navada, S.C.; Fruchtman, S.M.; Odchimar-Reissig, R.; Demakos, E.P.; Petrone, M.E.; Zbyszewski, P.S.; Holland, J.F.; Silverman, L.R. A Phase 1/2 Study of Rigosertib in Patients with Myelodysplastic Syndromes (MDS) and MDS Progressed to Acute Myeloid Leukemia. Leuk. Res. 2018, 64, 10–16. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective Inhibition of FLT3 by Gilteritinib in Relapsed or Refractory Acute Myeloid Leukaemia: A Multicentre, First-in-Human, Open-Label, Phase 1–2 Study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Schuster, M.W.; Jain, N.; Advani, A.; Jabbour, E.; Gamelin, E.; Rasmussen, E.; Juan, G.; Anderson, A.; Chow, V.F.; et al. A Phase 1 Study of AMG 900, an Orally Administered Pan-Aurora Kinase Inhibitor, in Adult Patients with Acute Myeloid Leukemia. Am. J. Hematol. 2017, 92, 660–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.A.; Robak, T.; Brown, J.R.; Awan, F.T.; Badoux, X.; Coutre, S.; Loscertales, J.; Taylor, K.; Vandenberghe, E.; Wach, M.; et al. Efficacy and Safety of Idelalisib in Combination with Ofatumumab for Previously Treated Chronic Lymphocytic Leukaemia: An Open-Label, Randomised Phase 3 Trial. Lancet Haematol. 2017, 4, e114–e126. [Google Scholar] [CrossRef]
- Schliemann, C.; Gerss, J.; Wiebe, S.; Mikesch, J.H.; Knoblauch, N.; Sauer, T.; Angenendt, L.; Kewitz, T.; Urban, M.; Butterfass-Bahloul, T.; et al. A Phase I Dose Escalation Study of the Triple Angiokinase Inhibitor Nintedanib Combined with Low-Dose Cytarabine in Elderly Patients with Acute Myeloid Leukemia. PLoS ONE 2016, 11, e0164499. [Google Scholar] [CrossRef] [Green Version]
- Yee, K.W.L.; Chen, H.W.T.; Hedley, D.W.; Chow, S.; Brandwein, J.; Schuh, A.C.; Schimmer, A.D.; Gupta, V.; Sanfelice, D.; Johnson, T.; et al. A Phase I Trial of the Aurora Kinase Inhibitor, ENMD-2076, in Patients with Relapsed or Refractory Acute Myeloid Leukemia or Chronic Myelomonocytic Leukemia. Investig. New Drugs 2016, 34, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Kantarjian, H. Chronic Myeloid Leukemia: 2018 Update on Diagnosis, Therapy and Monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Manabe, A. Treatment and Biology of Pediatric Acute Lymphoblastic Leukemia. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2018, 60, 4–12. [Google Scholar] [CrossRef]
- Aldoss, I.; Stein, A. Advances in Adult Acute Lymphoblastic Leukemia Therapy. Leuk. Lymphoma 2018, 59, 1033–1050. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.; Chávez-Valencia, V. FLT3-ITD and Its Current Role in Acute Myeloid Leukaemia. Med. Oncol. (Northwood Lond. Engl.) 2017, 34. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 Mutations in Acute Myeloid Leukemia: Therapeutic Paradigm beyond Inhibitor Development. Cancer Sci. 2020, 111, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Garten, A.; Grohmann, T.; Kluckova, K.; Lavery, G.G.; Kiess, W.; Penke, M. Sorafenib-Induced Apoptosis in Hepatocellular Carcinoma Is Reversed by SIRT1. Int. J. Mol. Sci. 2019, 20, 4048. [Google Scholar] [CrossRef] [Green Version]
- Abdelgalil, A.A.; Alkahtani, H.M.; Al-Jenoobi, F.I. Sorafenib. Profiles Drug Subst. Excip. Relat. Methodol. 2019, 44, 239–266. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Konopleva, M.; Shi, Y.; McQueen, T.; Harris, D.; Ling, X.; Estrov, Z.; Quintás-Cardama, A.; Small, D.; Cortes, J.; et al. Mutant FLT3: A Direct Target of Sorafenib in Acute Myelogenous Leukemia. JNCI J. Natl. Cancer Inst. 2008, 100, 184–198. [Google Scholar] [CrossRef]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I Study of Sorafenib in Patients with Refractory or Relapsed Acute Leukemias. Haematologica 2011, 96, 62. [Google Scholar] [CrossRef] [Green Version]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3–Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef]
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib Maintenance in Patients with FLT3-ITD Acute Myeloid Leukaemia Undergoing Allogeneic Haematopoietic Stem-Cell Transplantation: An Open-Label, Multicentre, Randomised Phase 3 Trial. Lancet Oncol. 2020, 21, 1201–1212. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Garcia-Horton, A.; Yee, K.W. Quizartinib for the Treatment of Acute Myeloid Leukemia. Expert Opin. Pharmacother. 2020, 21, 2077–2090. [Google Scholar] [CrossRef]
- Tarver, T.C.; Hill, J.E.; Rahmat, L.; Perl, A.E.; Bahceci, E.; Mori, K.; Smith, C.C. Gilteritinib Is a Clinically Active FLT3 Inhibitor with Broad Activity against FLT3 Kinase Domain Mutations. Blood Adv. 2020, 4, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Li, C.; Zhu, X. FLT3 Inhibitors in Acute Myeloid Leukemia. J. Hematol. Oncol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K Signaling in Cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Fujishita, T. Oncogenic Roles of the PI3K/AKT/MTOR Axis. Curr. Top. Microbiol. Immunol. 2017, 407, 153–189. [Google Scholar] [CrossRef] [PubMed]
- Arafeh, R.; Samuels, Y. PIK3CA in Cancer: The Past 30 Years. Semin. Cancer Biol. 2019, 59, 36–49. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Gilead Sciences. ZYDELIG® (Idelalisib) Tablets, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205858lbl.pdf (accessed on 21 August 2021).
- Zirlik, K.; Veelken, H. Idelalisib. Recent Results Cancer Res. 2018, 212, 243–264. [Google Scholar] [CrossRef]
- Bayer. ALIQOPATM (Copanlisib) for Injection, for Intravenous Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209936s000lbl.pdf (accessed on 21 August 2021).
- Verastem, Inc. COPIKTRA (Duvelisib), Capsules for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/211155s000lbl.pdf (accessed on 21 August 2021).
- TG Therapeutics. UKONIQTM (Umbralisib) Tablets, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213176s000lbl.pdf (accessed on 21 August 2021).
- Ghia, P.; Ljungström, V.; Tausch, E.; Agathangelidis, A.; Scheffold, A.; Scarfo, L.; Jebaraj, B.M.C.; Owen, C.J.; Barrientos, J.C.; Zapatka, M.; et al. Whole-Exome Sequencing Revealed No Recurrent Mutations within the PI3K Pathway in Relapsed Chronic Lymphocytic Leukemia Patients Progressing Under Idelalisib Treatment. Blood 2016, 128, 2770. [Google Scholar] [CrossRef]
- Ondrisova, L.; Mraz, M. Genetic and Non-Genetic Mechanisms of Resistance to BCR Signaling Inhibitors in B Cell Malignancies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Murali, I.; Kasar, S.; McWilliams, E.M.; Itchaki, G.; Tyekucheva, S.; Livitz, D.; Leshchiner, I.; Dong, S.; Fernandes, S.M.; Getz, G.; et al. Activating MAPK Pathway Mutations Mediate Primary Resistance to PI3K Inhibitors in Chronic Lymphocytic Leukemia (CLL). Blood 2018, 132, 587. [Google Scholar] [CrossRef]
- Scheffold, A.; Jebaraj, B.M.C.; Tausch, E.; Bloehdorn, J.; Ghia, P.; Yahiaoui, A.; Dolnik, A.; Blätte, T.J.; Bullinger, L.; Dheenadayalan, R.P.; et al. IGF1R as Druggable Target Mediating PI3K-δ Inhibitor Resistance in a Murine Model of Chronic Lymphocytic Leukemia. Blood 2019, 134, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kokabee, L.; Kokabee, M.; Conklin, D.S. Bruton’s Tyrosine Kinase and Its Isoforms in Cancer. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s Tyrosine Kinase (BTK) as a Promising Target in Solid Tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Pharmacyclics. IMBRUVICA® (Ibrutinib) Capsules, for Oral Use. U.S. Food and Drug Administration Website. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210563s000lbl.pdf (accessed on 21 August 2021).
- Deeks, E. Ibrutinib: A Review in Chronic Lymphocytic Leukaemia. Drugs 2017, 77, 225–236. [Google Scholar] [CrossRef]
- Zi, F.; Yu, L.; Shi, Q.; Tang, A.; Cheng, J. Ibrutinib in CLL/SLL: From Bench to Bedside (Review). Oncol. Rep. 2019, 42, 2213–2227. [Google Scholar] [CrossRef]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286. [Google Scholar] [CrossRef] [Green Version]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 2352. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib Resistance Mechanisms and Treatment Strategies for B-Cell Lymphomas. Cancers 2020, 12, 1328. [Google Scholar] [CrossRef]
- Wu, J.; Liu, C.; Tsui, S.T.; Liu, D. Second-Generation Inhibitors of Bruton Tyrosine Kinase. J. Hematol. Oncol. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.A.A.; Furman, R.R.; O’Brien, S.M.; Yenerel, M.N.; Illés, Á.; Kay, N.E.; et al. First Results of a Head-to-Head Trial of Acalabrutinib versus Ibrutinib in Previously Treated Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2021, 39, 7500. [Google Scholar] [CrossRef]
- Tam, C.S.; Opat, S.; D’Sa, S.; Jurczak, W.; Lee, H.-P.; Cull, G.; Owen, R.G.; Marlton, P.; Wahlin, B.E.; Sanz, R.G.; et al. A Randomized Phase 3 Trial of Zanubrutinib vs Ibrutinib in Symptomatic Waldenström Macroglobulinemia: The ASPEN Study. Blood 2020, 136, 2038. [Google Scholar] [CrossRef]
- Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A Medicinal Chemistry and Drug Delivery Perspective. Int. J. Mol. Sci. 2021, 22, 7641. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Haap, M.; Kopp, H.; Lipp, H. Tyrosine Kinase Inhibitors-A Review on Pharmacology, Metabolism and Side Effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. Kinetic and Catalytic Mechanisms of Protein Kinases. Chem. Rev. 2001, 101, 2271–2290. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Xu, Z.; Yang, B.; He, Q.; Luo, P. Sorafenib-Associated Hand-Foot Skin Reaction: Practical Advice on Diagnosis, Mechanism, Prevention, and Management. Expert Rev. Clin. Pharmacol. 2019, 12, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, I.B.; Flowers, C.R.; Blum, K.A.; Cohen, J.B. Clinical Use of PI3K Inhibitors in B-Cell Lymphoid Malignancies: Today and Tomorrow. Expert Rev. Anticancer Ther. 2017, 17, 271–279. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Preite, S.; Gomez-Rodriguez, J.; Cannons, J.; Schwartzberg, P. T and B-Cell Signaling in Activated PI3K Delta Syndrome: From Immunodeficiency to Autoimmunity. Immunol. Rev. 2019, 291, 154–173. [Google Scholar] [CrossRef] [PubMed]
- Paydas, S. Management of Adverse Effects/Toxicity of Ibrutinib. Crit. Rev. Oncol. Hematol. 2019, 136, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, A.; Lamanna, N. Managing Toxicities of Bruton Tyrosine Kinase Inhibitors. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 336. [Google Scholar] [CrossRef]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia 2021, 35, 312. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Byrd, J.C.; Brown, J.R.; Coutre, S.E.; Benson, D.M.; Wagner-Johnston, N.D.; Flinn, I.W.; Kahl, B.S.; Spurgeon, S.E.; Lannutti, B.; et al. CAL-101, An Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase P110δ, Demonstrates Clinical Activity and Pharmacodynamic Effects In Patients with Relapsed or Refractory Chronic Lymphocytic Leukemia. Blood 2010, 116, 55. [Google Scholar] [CrossRef]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged Lymphocytosis during Ibrutinib Therapy Is Associated with Distinct Molecular Characteristics and Does Not Indicate a Suboptimal Response to Therapy. Blood 2014, 123, 1810. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, J.C.; Burger, J.A.; Byrd, J.C.; Hillmen, P.; Zhou, C.; Ninomoto, J.; James, D.F.; Kipps, T.J. Characterizing the Kinetics of Lymphocytosis in Patients with Chronic Lymphocytic Leukemia Treated with Single-Agent Ibrutinib. Leuk. Lymphoma 2018, 60, 1000–1005. [Google Scholar] [CrossRef] [Green Version]
- Marconi, G.; De Polo, S.; Martinelli, G.; Nanni, J.; Bertamini, L.; Talami, A.; Olivi, M.; Ragaini, S.; Abbenante, M.C.; Sartor, C.; et al. Safety Profile and Impact on Survival of Tyrosine Kinase Inhibitors versus Conventional Therapy in Relapse or Refractory FLT3 Positive Acute Myeloid Leukemia Patients. Leuk. Res. 2021, 101, 106497. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A. BTK Inhibitors: Present and Future. Cancer J. (Sudbury Mass.) 2019, 25, 386. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Study Phase | Leukemia Subtype | Targeted Kinase | Kinase Inhibitor | Associated Treatment | Clinical Outcome | Adverse Events | References |
|---|---|---|---|---|---|---|---|
| II | HCL | BRAF | Vemurafenib, 960 mg twice daily, orally administered for a total of 8 weeks | Eight intravenous rituximab doses (375 mg per square meter of body surface) over 18 weeks | Complete response achieved in 87% of the patients with PFS of 78% at a median follow-up of 37 months | Mostly grade 1 or 2 involving neutropenia, cutaneous rash, photosensitivity, fever, fatigue and others; Liver and pancreatic biochemical alterations were also detected | [77] |
| I | MM | PIM | PIM447, 250 mg or 300 mg, orally administered daily in a 28 continuous days cycle | NR | ORR was 15.4%, DCR was 69.2%, and CBR was 23.1%; All patients discontinued treatment due to physician decision or progressive disease | At least one grade 3 or 4 AE in all of the studied patients; The most common AEs associated with the treatment were thrombocytopenia, anemia, leukopenia and lymphopenia | [78] |
| Ib | CLL/SLL; FL | BTK | Zanubrutinib, 160 mg twice daily or 320 mg once daily in a 28 continuous days cycle | Obinutuzumab intravenously administered in 6 28-day cycles at different doses dependent on the cycle day | R/R CLL/SLL patients had a 92% ORR with 28% CR and median PFS was not reached; R/R FL patients had a 72% ORR with 39% CR and PFS of 25 months | The most common reported AE was upper respiratory tract infections in both patient cohorts; Neutropenia was the most common grade ≥3 AE; Only 6.17% of patients went through treatment discontinuation due to AEs | [79] |
| I | AML | FLT3 | Sorafenib orally administered in 400 mg, 600 mg and 800 mg doses twice daily in a 28 continuous days cycle | G-CSF and plerixafor at 10 mcg/kg and 240 mcg/kg subcutaneous doses, respectively; Administration occurred every other day for seven doses starting on day 1 | 36% of patients responded to treatment with a median duration of response of 5.3 months; Most patients that responded remained FLT3-ITD positive during therapy | 71.4% of patients presented grade ≥3 nonhematologic AE beyond cycle 1 including skin associated toxicities, cardiac arrhythmias, liver enzyme elevations, bone pain and others | [80] |
| I | MDS; CMML; AML | RAS | Rigosertib in 140 mg or 280 mg doses twice daily or in a total 840 mg daily dose in a 4-week cycle consisting in 3 weeks of treatment and 1 week of rest | Azacitidine in subcutaneous or intravenous doses of 75 mg/m2/kg for seven days starting one week after rigosertib initial treatment | Responses to treatment were achieved in 56% of patients with a median duration of response of 5.8 months; | 89% of the patients experienced grade ≥3 AE including pneumonia, neutropenia and thrombocytopenia; 72% reported serious AEs, but none were considered related to study treatment; 33% reported genitourinary AEs of mostly grade 1 or 2 | [81] |
| II | CLL/SLL | BTK | Zanubrutinib,160 mg twice daily in 28-day cycles | NR | 84.6% of patients achieved a response, but only 3.3% achieved CR; 92.9% of responders did not have disease progression at 12 months follow-up | 63.7% of patients reported grade 3 AEs, 8.8% reported grade 4 AEs and 3.3% reported grade 5 AEs. The most common AEs were neutropenia, upper respiratory tract infections, petechiae, anemia and hematuria | [82] |
| I/II | AML | FLT3 | Pexidartinib orally administered twice daily in a dose expanding protocol of 800 mg to 5000 mg in a 28-day cycle; Pexidartinib in 3000 mg doses in phase II | NR | ORR for all treated patients was 21%; Patients in phase II had a median duration of response of 76 days, median PFS of 48 days and median OS of 112 days; Parameters were highly enhanced in responders | Most common grade ≥3 AEs were febrile neutropenia and anemia while the most common general AEs included diarrhea, fatigue and nausea; Only 1 patient experienced a fatal AE that was considered related to treatment; MTD was not reached | [83] |
| II | CLL/SLL | BTK | Acalabrutinib, 100 mg doses twice daily or 200 mg doses once daily each in 28-day cycles | NR | Median time for initial response was 5.5 months; ORR for all patients was 87.5%; Estimated PFS at 24 months was 84.3% for R/R patients | Most common AEs were grade 1 or 2 and included headache, contusion, diarrhea and upper respiratory tract infection; Grade ≥3 AEs occurred in 43.8% of patients and manifested primarily as neutropenia; One patient experienced grade 5 liver failure considered related to treatment | [84] |
| II | DLBCL | PI3K | Copanlisib in a 60 mg dose as an IV infusion in days 1, 8 and 15 of a 28-day cycle | NR | ORR was 19.4% with CR corresponding to 7.5% of patients; Median PFS in the overall cohort was 1.8 months and median OS was 7.4 months | 97% of patients experienced some kind of AE; 86.6% of AEs were grade ≥3, the main ones being hypertension and hyperglycemia, and 65.7% of patients experienced serious AEs; Drug-related AEs leading to treatment discontinuation occurred in 11.9% of patients | [85] |
| I/II | CLL/SLL | BTK | Acalabrutinib in dose escalation of 100 mg to 400 mg once daily or 200 mg twice daily; Acalabrutinib in doses of 100 mg twice daily or 200 mg once daily in phase II | NR | ORR was 94% and CR was 4%; Median time to initial response of PR or better was 4.7 months; Estimated PFS at 45 months was 62% | AEs occurring in ≥10% of patients were generally mild to moderate and included diarrhea, headache, upper respiratory tract infection and fatigue; Grade ≥3 AEs occurred in 66% of patients and were mainly neutropenia and pneumonia; 10 patients (7.5%) had AE-related deaths | [86] |
| II | AML | FLT3 | Sorafenib administered in 400 mg doses twice daily in a 21-day cycle | OME administered IV from day 1 to 7 in 2 mg/d doses; After CR/CRi, OME was administered from days 1 to 5 in new cycles | 71.8% of R/R patients achieved CR/CRi after 1 or 2 cycles; Patients who achieved CR/CRi and did not undergo hematopoietic stem cell transplant relapsed despite continuous treatment; OS in nonresponders was shorter than 11 months while median OS in those who achieved CR/CRi was 10.9 months | Most AEs were grade 2 or lower and mainly included fever, rash and anemia; Grade ≥3 AEs included neutropenia, thrombocytopenia and hematuria | [87] |
| I/Ib | B-NHL; CLL/SLL | PI3K-δ | Umbralisib orally administered once daily at doses of 800 mg or 1200 mg (initial formulation) or escalating doses of 400 mg to 1200 mg (micronized formulation) | Ublituximab doses of 900 mg to B-NHL patients and 600 mg or 900 mg to CLL patients administered IV on different days according to cycle progression | ORR was 46% and CR was 17% with median time to first response of 8 weeks; Virtually all patients who responded (29 of 32) received therapeutic-dose levels of umbralisib | The majority of AEs were grade 1 or 2 and the most common were diarrhea, nausea and fatigue; Neutropenia was the most common grade ≥3 AE | [88] |
| III | CLL/SLL | BTK | Ibrutinib orally administered in doses of 420 mg daily | NR | ORR was 91% and CR/CRi was 11%; Median OS was 67.7 months and PFS was 44.1 months | Commonly reported grade ≥3 hematologic AEs were neutropenia, thrombocytopenia and anemia while nonhematologic AEs included pneumonia and hypertension; 10% of patients experienced major hemorrhage; Prevalence of AEs decreased over time for patients on continuous therapy | [89] |
| I | NHL; CLL | PI3K-δ; PI3K-γ | Duvelisib orally administered twice a day in 25 mg daily doses in a 28-day cycle | Rituximab (375 mg/m2 IV) on cycle day one (Arm 1); Rituximab (375 mg/m2 IV) with addition of variable doses of Bendamustine (Arm 2) | ORR in Arm 1 was 78.3% compared to 62.5% in Arm 2; Overall median PFS was 13.7 months and median OS of 9.1 months was only reached for NHL patients in Arm 2 | 95.7% of patients were afflicted by an AE considered related to treatment; 87% of patients experienced grade ≥3, the most common being neutropenia; Serious AEs related to treatment happened on 25.9% of patients in Arm 1 and 10.5% of patients in Arm 2 | [90] |
| II | AML | FLT3 | Quizartinib in daily doses of 20, 30 or 60 mg that increased by one dose level at a time in patients who did not achieve CRc | NR | CRc rate was 53.8%; Median duration of CRc was 16.1 weeks and median OS was 34.1 weeks | The most common AEs, as well as most common grade ≥3, were febrile neutropenia and platelet count decrease; Serious AEs were reported in 45.9% of patients | [91] |
| II | AML | VEGFR | Pazopanib in doses of 800 mg once daily | NR | PR was achieved in 10% of patients and was the best reported response; SD was reported in 70% of patients; Median PFS was 65 days and median OS was 191 days | Majority of AEs were gastrointestinal and included nausea, diarrhea and decreased appetite; The most common grade 3 AE was nausea and no grade ≥4 was reported; No serious AEs were considered related to treatment | [92] |
| I | B-NHL; CLL/SLL | BTK | Tirabrutinib administered in 160 mg, 320 mg or 480 mg once daily or 300 mg twice daily | NR | ORR for all cohorts was 76.5%, with variations when analyzing specific treatment cohorts and malignant subtypes; 12 out of 16 patients showed ≥50% reduction in tumor diameter | One DLT was observed in the 300 mg twice daily cohort; Most common AEs were rash, vomiting and neutropenia; Grade ≥3 AEs mainly included hematologic toxicities; 4 serious AEs related to treatment were reported | [93] |
| I/II | B-NHL | PI3K-δ; JAK 1 | Parsaclisib in dose escalation that was later amended to doses of 20 mg once daily for the first 9 weeks, followed by parsaclisib 20 mg once weekly | Itacitinib 300 mg once daily or chemotherapy (rituximab, ifosfamide, carboplatin and etoposide) | As a monotherapy, ORR varied by disease type, with worse response of DLBCL patients (30%) and best response of MZL patients (78%); Durable responses were observed in patients following the once-weekly dosing schedule; In combination therapies, response rates varied highly and were inconsistent due to small amount of patients per group | As a monotherapy, the most common nonhematologic AE were diarrhea, nausea, fatigue and rash, while any-grade neutropenia was experienced by 44% of patients; In treatment with itacitinib, 45% of patients experienced grade 3/4 AEs and in chemotherapy combination, patients reported grade 4 thrombocytopenia and neutropenia | [94] |
| II | AML | MEK 1/2 | Binimetinib administered in doses of 30 mg or 45 mg twice daily | NR | Only one patient (8%) achieved a CRi; Median OS was 1.8 months | The most common AEs were diarrhea, hypokalemia, hypotension, hypoalbuminemia and hypocalcemia; Only one serious AE related to treatment was identified; No treatment-related deaths occured | [95] |
| I/Ib | CLL; MCL | PI3K-δ; BTK | Umbralisib orally at doses of 400 mg, 600 mg or 800 mg; Ibrutinib at doses of 420 mg for CLL patients and 560 mg for MCL patients | NR | ORR for CLL patients was 90%; CLL 2-years PFS and OS were 90% and 95%, respectively; ORR for MCL patients was 67%; MCL 2-years PFS and OS were 49% and 58%, respectively; Median time to best response was 2 months in both cohorts | Common AEs included diarrhea, infection and nausea; Grade 3/4 hematologic AEs were not common and included neutropenia, thrombocytopenia and anemia, but were not considered related to study drugs; Serious AEs were experienced by 29% of patients | [96] |
| III | CLL/SLL | PI3K-δ; PI3K-γ | Duvelisib twice daily in doses of 25 mg in 28-day cycles | NR | Median PFS was 13.3 months and estimated 12-month PFS was 60%; ORR was 73.8% with PR being 72.5% of cases; Estimated 12-month OS was 86% | Most common hematologic AEs included neutropenia, anemia and thrombocytopenia; Grade ≥3 AE occurred in 87% of patients and 4 serious AEs experienced by patients were considered related to study drug | [97] |
| I | MDS; CMML | JAK 1/2 | Ruxolitinib in doses of 5, 10, 15 or 20 mg twice a day in 28-day cycles | NR | Responses were achieved in 4 out of 18 patients, but 2 patients relapsed after 1 and 4 months; Median OS was 14.5 months with different outcomes depending on disease cohort | Most common nonhematologic AEs were hyperglycemia, fatigue and elevated liver function; Thrombocytopenia and anemia were the most common hematologic AEs and were also observed as grade ≥3 | [98] |
| II | AML | SYK | Erlotinib in daily doses of 150 mg | NR | ORR was 10% with only one patient (3%) achieving CR; All patients discontinued therapy due to PD and median OS was 3.5 months | AEs considered related to study drug included fatigue, diarrhea, nausea and rash; Only 7% of patients had AEs that required dose reduction and treatment discontinuation | [99] |
| I | AML | FLT3; AXL | Gilteritinib in escalating doses of 20 to 300 mg daily | NR | ORR was 47.4% with CRc being 36.8% of overall cases; ORR and CRc rates were enhanced in patients who were FLT3 mutation-postive | DLTs were reported at 120 mg and 300 mg cohorts and MTD was established at 200 mg/day; Most drug-related AEs were elevated liver enzyme levels, elevated blood creatine phosphokinase and elevated blood lactate dehydrogenase; Serious AEs were experienced by 29.2% of patients | [100] |
| IIb | AML | FLT3 | Quizartinib in daily doses of 30 mg or 60 mg in 28-day cycles | NR | Overall CRc was 47.4% and ORR was 61% and 71% for patients in the 30 mg and 60 mg groups, respectively; Duration of CRc and OS was longer on the 60 mg group | AEs considered related to treatment were reported evenly among both treatment groups and the most common included hematologic events, diarrhea and fatigue; Serious AEs considered related to treatment occurred in 26% of patients in 30 mg group and 22% of patients in 60 mg group | [101] |
| I | CLL/SLL; B-NHL; T-NHL; HL | PI3K-δ | Umbralisib daily in 28-day cycles following a dose escalation protocol to a maximum of 1800 mg a day; Later amendments transitioned all doses to 800 mg a day | NR | Of all patients treated, 62% reported reduction in disease burden, 33% had an objective response and 30% had a partial response; Efficacy varied highly among disease subtypes and best parameters of ORR, DOR and PFS were achieved in the CLL patients subgroup | DLTs were observed in cohorts of 800 mg and 1800 mg and the MTD was determined to be 1200 mg; Most common AEs were diarrhea, nausea and fatigue and were generally grade 1 or 2; Grade ≥3 AE included neutropenia, anemia and thrombocytopenia; Discontinuation of treatment due to AEs occurred in 7% of patients | [102] |
| I/II | MDS; AML | RAS | Rigosertib in continuous IV infusion at initial doses of 650 mg/m2 for 3 days on 14-day cycles; Dose escalation was possible depending on toxicity and effectiveness | NR | 26.5% of patients achieved bone marrow/peripheral blood response and the same amount of patients achieved SD; Median OS for responding patients was 15.7 months in contrast to OS of 2 months for nonresponders | MTD was determined as 1700 mg/m2 and recommended phase 2 dosage was 1350 mg/m2; Most common AEs were fatigue, diarrhea and pyrexia; Most common grade ≥3 AEs were anemia, thrombocytopenia and pneumonia; 18% of patients had serious AEs related to treatment and 59% discontinued treatment due to AEs | [103] |
| I/II | AML | FLT3 | Gilteritinib in dose escalation cohorts of 20 to 450 mg daily | NR | ORR was 40% and most CRc were achieved in patients in the 120 mg/day and 200 mg/day cohorts; Median OS for all patients was 25 weeks; Better ORR was observed for patients’ FLT3 mutation-positive | Grade ≥3 AE included febrile neutropenia, anemia, thrombocytopenia, sepsis and pneumonia; Drug-related AEs leading to treatment discontinuation happened in 10% of patients; MTD was defined as 300 mg/day | [104] |
| I | AML | AURK A/B | AMG 900 was administered, after protocol amendment, at escalating doses of 30 to 75 mg daily for 7 days every 2 weeks | NR | 9% of patients, following the protocol before amendment, achieved CRi and no other responses were observed; For responders, maximum duration of response was 3 months | The most common AEs were nausea, diarrhea, febrile neutropenia and fatigue and the most common grade ≥3 AE was neutropenia; 31% of patients had serious AEs and 14% discontinued treatment due to AEs; Two deaths, respiratory failure and septic shock, were considered related to treatment | [105] |
| III | CLL | PI3K-δ | Idelalisib 150 mg twice daily | Ofatumumab administered IV in doses of 300 mg on day 1 followed by a dose of 1000 mg weekly for 7 weeks and then every 4 weeks for 16 weeks | ORR was 75.3% and only one patient had a CR; Median OS was 20.9 months and PFS was 16.3 months | Diarrhea, pyrexia, neutropenia and fatigue were the most common reported AEs; Grade ≥3 AEs happened in 91% of patients and included neutropenia, diarrhea and pneumonia; 39% of patients discontinued treatment due to AEs | [106] |
| I | AML | VEGFR; FGFR; PDGFR | Nintedanib twice daily in dosages of 100 mg, 200 mg or 300 mg in a 28-day cycle | Low-dose cytarabine from days 1 to 10 at 20 mg twice daily | 2 out of 12 patients had an objective response of CR and CRi; Median OS was 234 days | No DLTs were reported during dose escalation; Most common AEs associated with treatment were gastrointestinal and treatment-related grade ≥3 AEs were reported in 4 patients; A total of 12 serious AEs were reported, the most common being neutropenic fever | [107] |
| I | AML; CMML | AURK A; Multi-kinase activity | ENMD-2076 administered in daily escalating doses of 225, 275, 325 or 375 mg | NR | ORR was 25% with best response being CRi; Median number of cycles to best response was 2 and median DOR was 4.8 months; 48% of patients discontinued treatment due to disease progression | 96% of patients reported treatment related AEs; Some of the most common nonhematologic toxicities were fatigue, diarrhea and hypertension; DLTs were observed in all dose levels except 225 mg/day | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, C.B.; de Pinho Pessoa, F.M.C.; da Silva, E.L.; da Costa Pantoja, L.; Ribeiro, R.M.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Montenegro, R.C.; Burbano, R.M.R.; Khayat, A.S.; et al. Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations. Pharmaceutics 2021, 13, 1604. https://doi.org/10.3390/pharmaceutics13101604
Machado CB, de Pinho Pessoa FMC, da Silva EL, da Costa Pantoja L, Ribeiro RM, de Moraes Filho MO, de Moraes MEA, Montenegro RC, Burbano RMR, Khayat AS, et al. Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations. Pharmaceutics. 2021; 13(10):1604. https://doi.org/10.3390/pharmaceutics13101604
Chicago/Turabian StyleMachado, Caio Bezerra, Flávia Melo Cunha de Pinho Pessoa, Emerson Lucena da Silva, Laudreísa da Costa Pantoja, Rodrigo Monteiro Ribeiro, Manoel Odorico de Moraes Filho, Maria Elisabete Amaral de Moraes, Raquel Carvalho Montenegro, Rommel Mário Rodriguez Burbano, André Salim Khayat, and et al. 2021. "Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations" Pharmaceutics 13, no. 10: 1604. https://doi.org/10.3390/pharmaceutics13101604