
Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Analytical Method
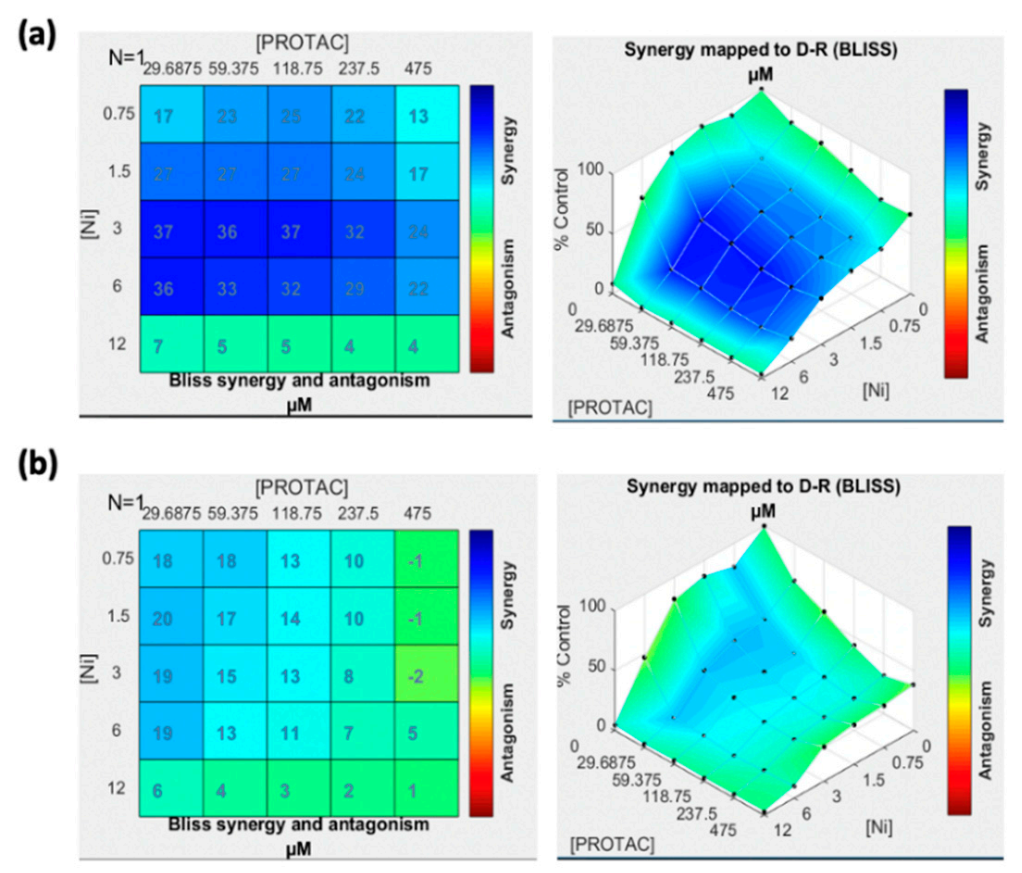
2.3. Cell Viability Assay and Effect of Drug Combination
2.4. Preparation and Characterization of ARNIPL
2.5. In Vitro Release Study
2.6. Enzyme Linked Immunosorbent Assay
2.7. Western Blot Assay
2.8. Clonogenic Assay
2.9. Vasculogenic Mimicry
2.10. Flow Cytometry for Apoptosis Analysis
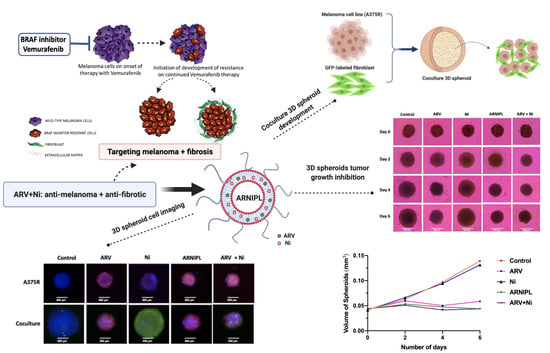
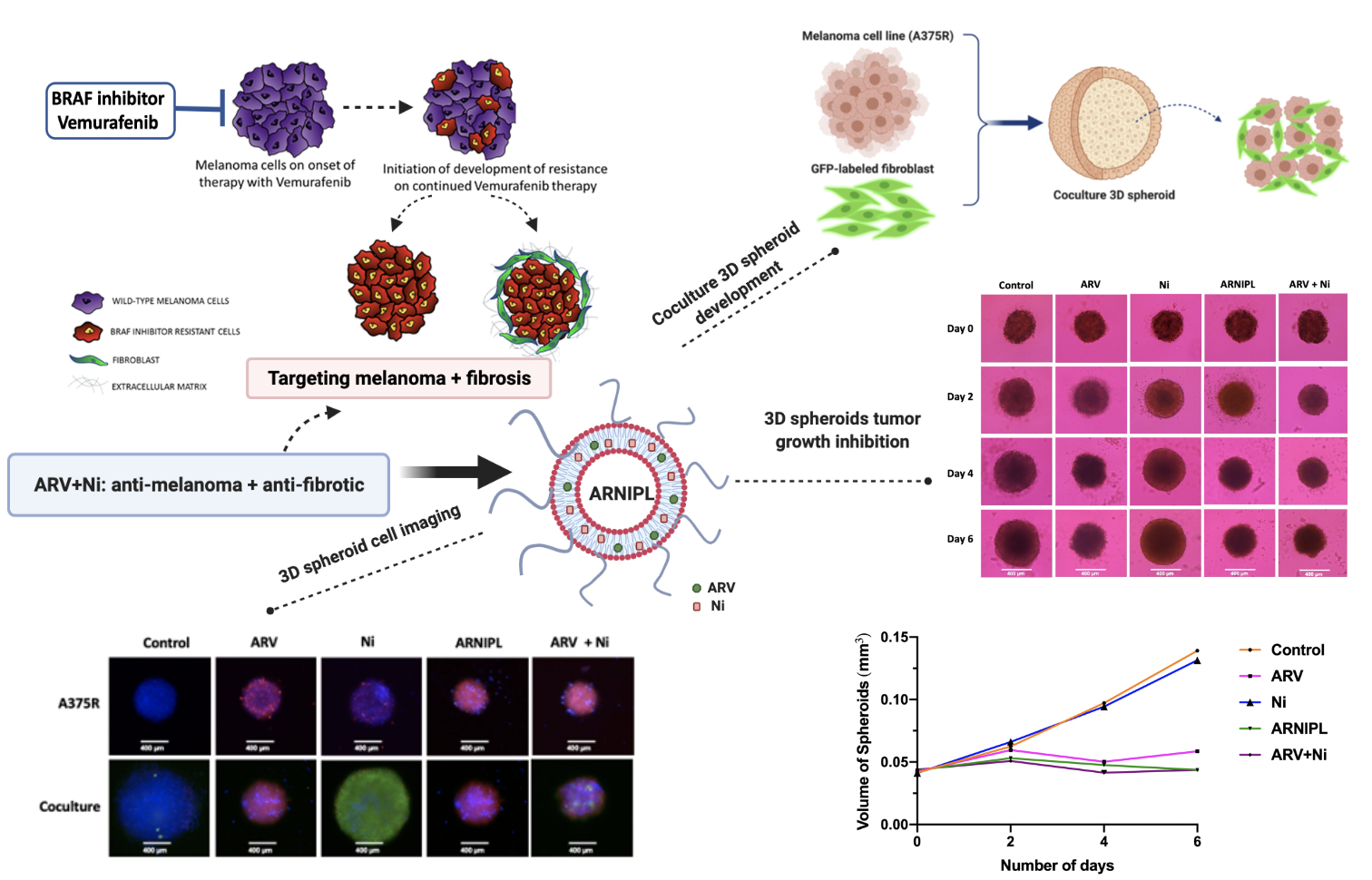
2.11. Spheroids Development and Characterization of 3D Multicellular Tumor Spheroids
2.12. Statistics
3. Results
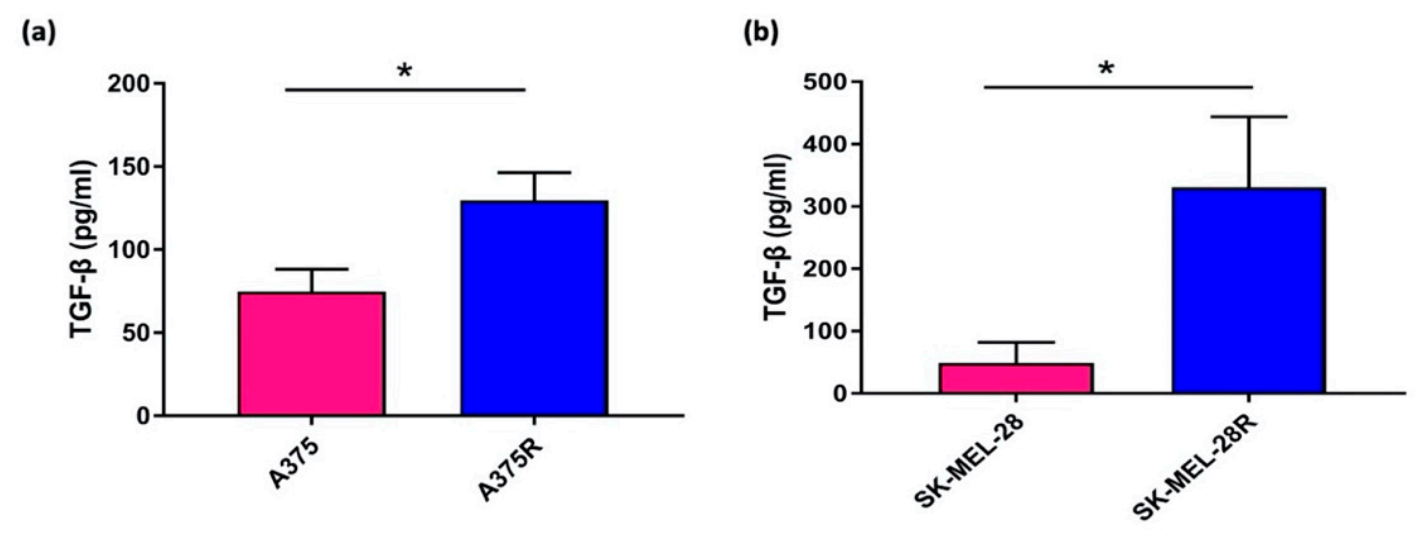
3.1. Enzyme Linked Immunosorbent Assay
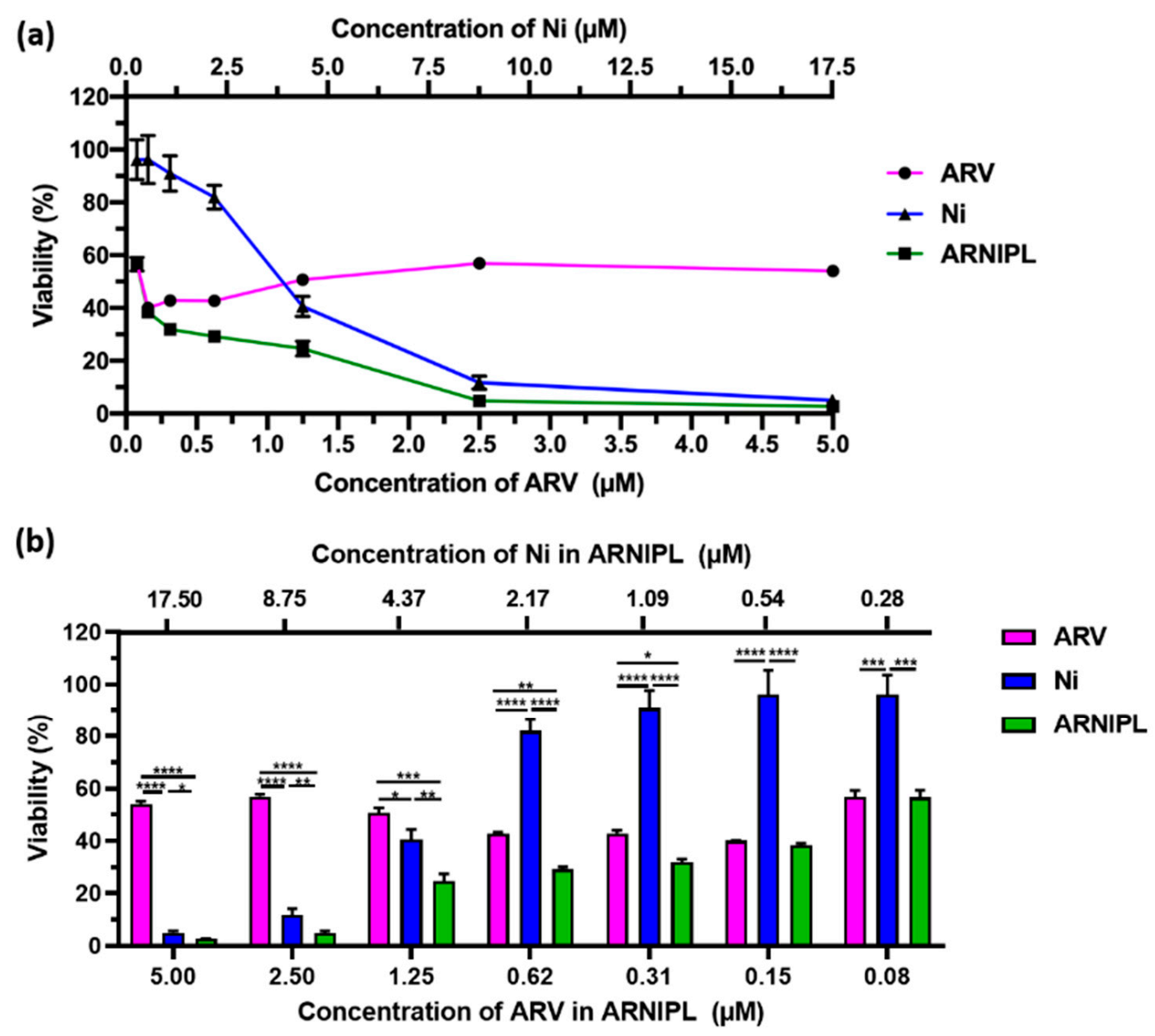
3.2. Cell Viability Assay and Effect of Drug Combination
3.3. Characterization and Stability of ARNIPL
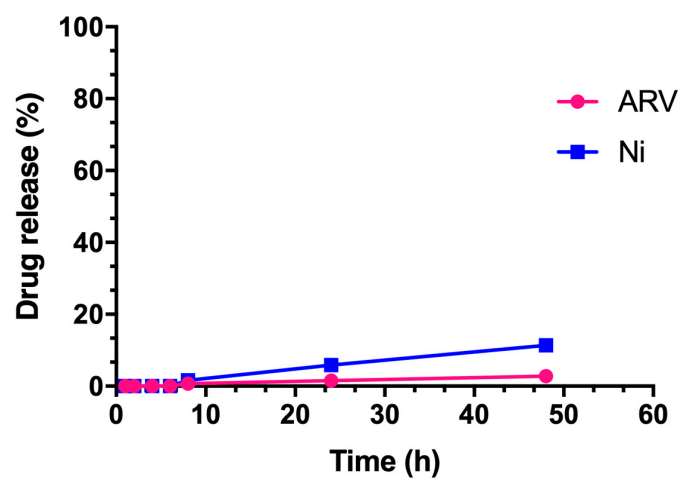
3.4. In Vitro Release Study
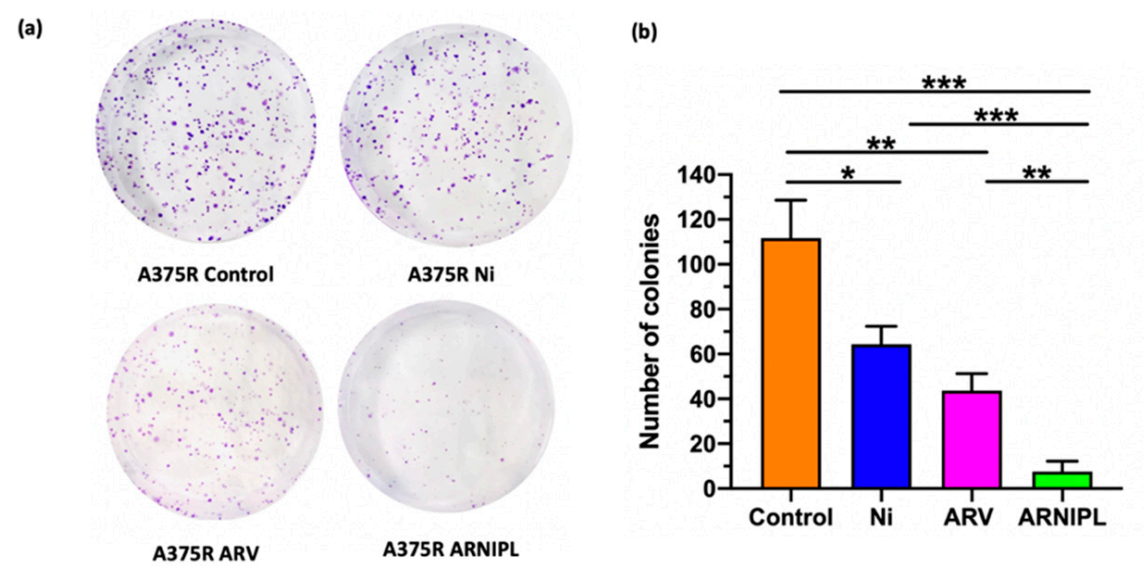
3.5. Clonogenic Assay
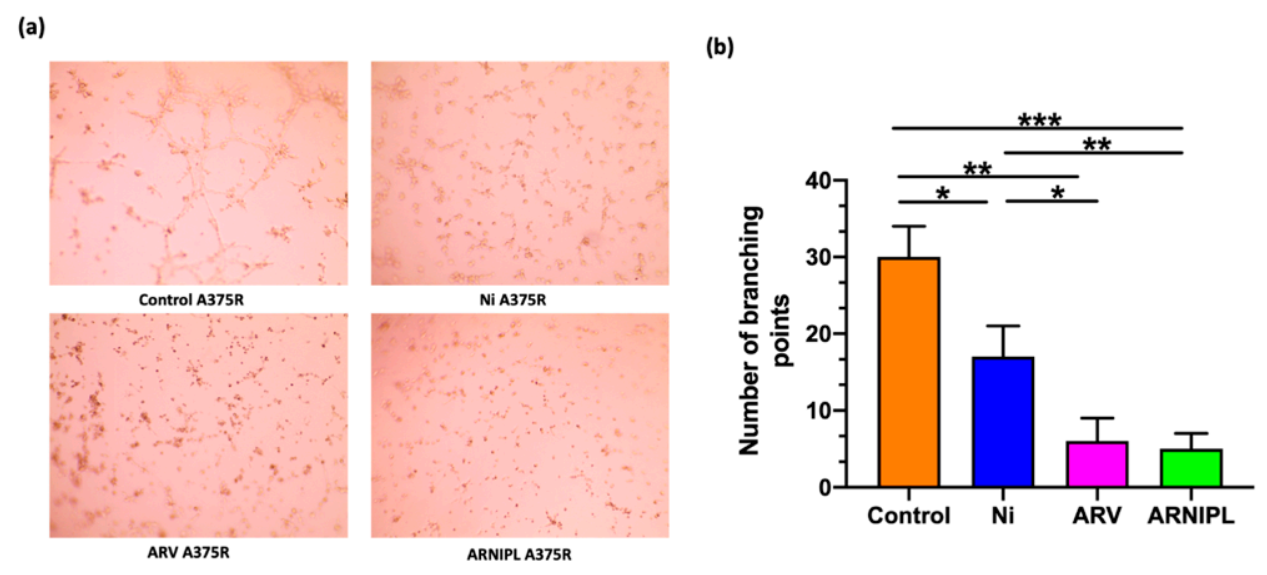
3.6. Vasculogenic Mimicry
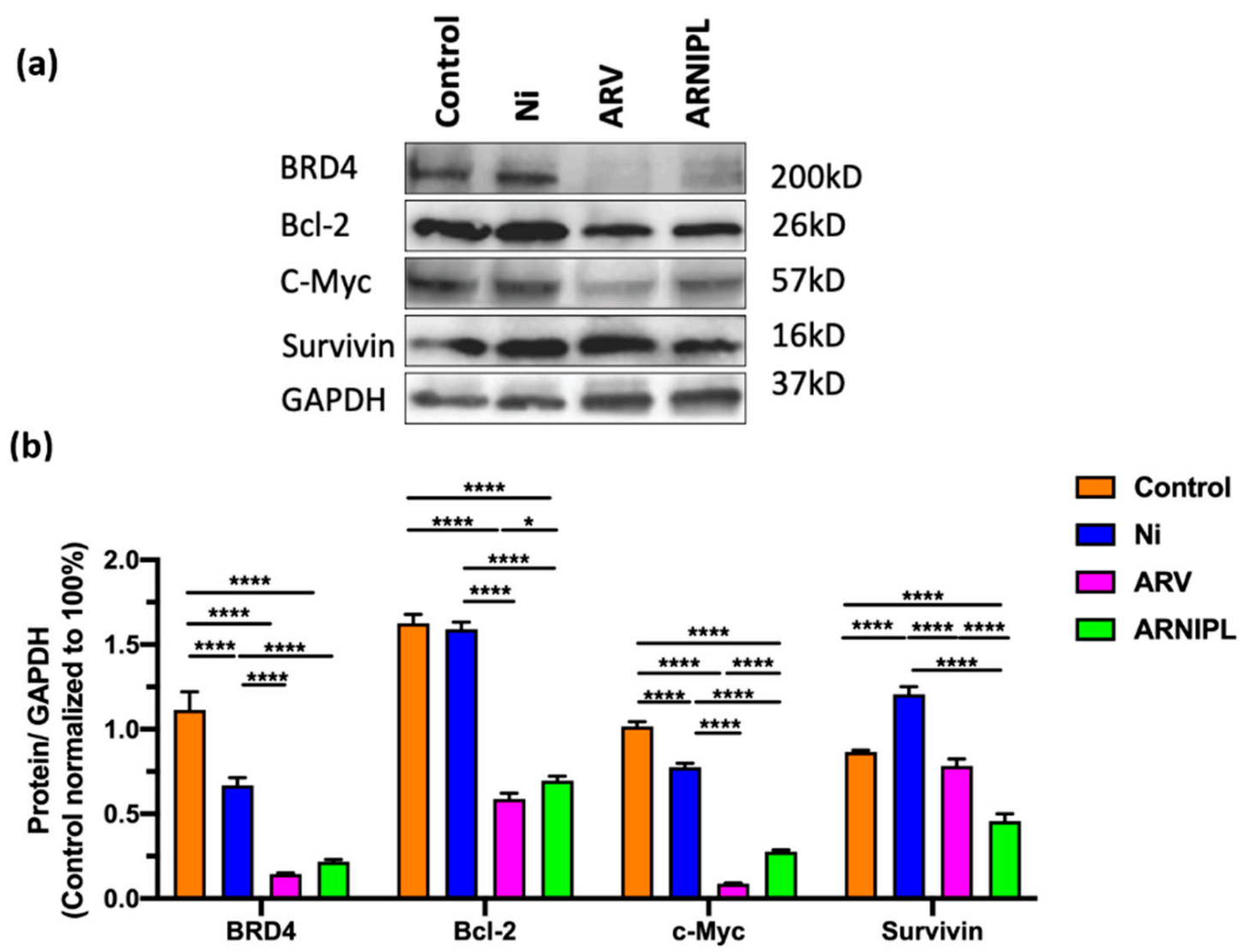
3.7. Western Blot Assay
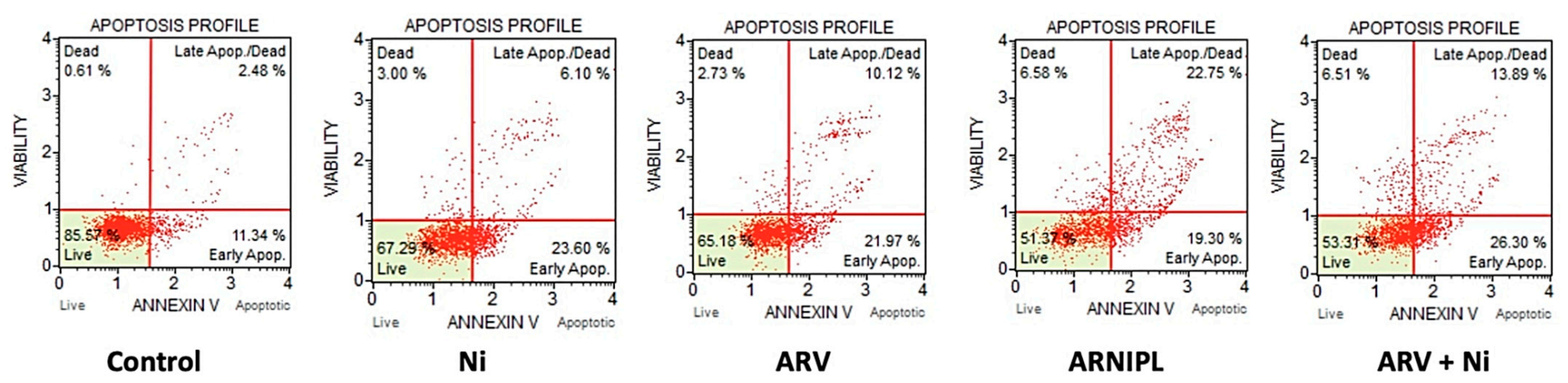
3.8. Apoptosis Assay
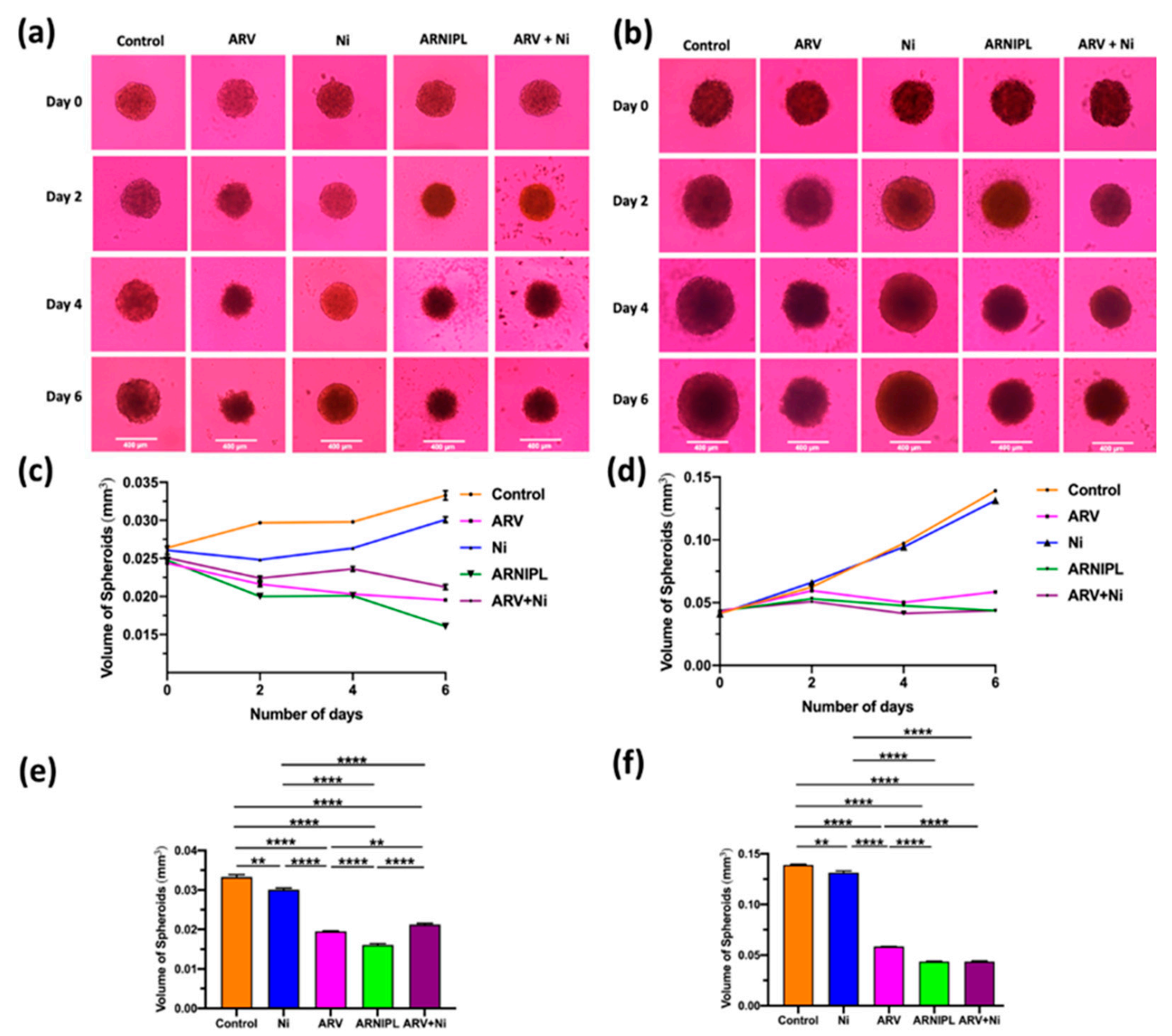
3.9. Determination of ARNIPL Efficacy in 3D Tumor Spheroids
3.9.1. Development and Characterization of 3D Tumor Spheroids
3.9.2. Enzyme Linked Immunosorbent Assay in 3D Spheroids
3.9.3. 3D Cell Viability Study
3.9.4. 3D Tumor Spheroid Live and Dead Cell Imaging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Key Statistics for Melanoma Skin Cancer. Available online: https://www.cancer.org/cancer/melanoma-skin-cancer/about/key-statistics.html (accessed on 27 May 2021).
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, J.L.; Layos, L.; Buges, C.; de Los Llanos Gil, M.; Vila, L.; Martinez-Balibrea, E.; Martinez-Cardus, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma-A mini review. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2021, 1865. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollins, R.; Kim, K.; Tsao, C. The Emerging Epigenetic Landscape in Melanoma. In Human Skin Cancer, Potential Biomarkers and Therapeutic Targets; IntechOpen: London, UK, 2016. [Google Scholar]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Sun, R.; Zhao, X.; Zhu, D.; Zhao, X.; Gu, Q.; Dong, X.; Zhang, D.; Zhang, Y.; Li, Y.; et al. C-myc overexpression drives melanoma metastasis by promoting vasculogenic mimicry via c-myc/snail/Bax signaling. J. Mol. Med. 2017, 95, 53–67. [Google Scholar] [CrossRef]
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; Gonzalez-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W.; et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013, 73, 6264–6276. [Google Scholar] [CrossRef] [Green Version]
- Prochownik, E.V.; Vogt, P.K. Therapeutic Targeting of Myc. Genes Cancer 2010, 1, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Aird, F.; Kandela, I.; Mantis, C.; Reproducibility Project: Cancer, B. Replication Study: BET bromodomain inhibition as a therapeutic strategy to target c-Myc. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Cermakova, K.; Hodges, H.C. Next-generation drugs and probes for chromatin biology: From targeted protein degradation to phase separation. Molecules 2018, 23, 1958. [Google Scholar] [CrossRef] [Green Version]
- Rathod, D.; Fu, Y.; Patel, K. BRD4 PROTAC as a novel therapeutic approach for the treatment of vemurafenib resistant melanoma: Preformulation studies, formulation development and in vitro evaluation. Eur. J. Pharm. Sci. 2019, 138. [Google Scholar] [CrossRef]
- Ruiter, D.; Bogenrieder, T.; Elder, D.; Herlyn, M. Melanoma-stroma interactions: Structural and functional aspects. Lancet Oncol. 2002, 3, 35–43. [Google Scholar] [CrossRef]
- Lee, J.T.; Herlyn, M. Microenvironmental influences in melanoma progression. J. Cell. Biochem. 2007, 101, 862–872. [Google Scholar] [CrossRef]
- Flach, E.H.; Rebecca, V.W.; Herlyn, M.; Smalley, K.S.; Anderson, A.R. Fibroblasts contribute to melanoma tumor growth and drug resistance. Mol. Pharm. 2011, 8, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Krstic, J.; Trivanovic, D.; Mojsilovic, S.; Santibanez, J.F. Transforming growth factor-beta and oxidative stress interplay: Implications in tumorigenesis and cancer progression. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, I.V.; Wargo, J.A.; Flaherty, K.T.; Messina, J.L.; Smalley, K.S.M. Braf inhibition generates a host-tumor niche that mediates therapeutic escape. J. Investig. Dermatol. 2015, 135, 3115–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, M.H.; Croteau, W.; Mullins, D.W.; Brinckerhoff, C.E. The BRAF(V600E) inhibitor, PLX4032, increases type I collagen synthesis in melanoma cells. Matrix Biol. 2015, 48, 66–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Hu, Y.; Pang, Z. Modulating the tumor microenvironment to enhance tumor nanomedicine delivery. Front. Pharmacol. 2017, 8, 952. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel mechanisms for the antifibrotic action of nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Huwyler, J.; Drewe, J.; Krahenbuhl, S. Tumor targeting using liposomal antineoplastic drugs. Int. J. Nanomed. 2008, 3, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, D.; Bansal, A.; Sharma, R.; Dhawan, S. Advanced nanomedicine: Present contributions and future expectations. Am. J. Phytomed. Clin. Ther. 2013, 1, 124–139. [Google Scholar]
- Tran, S.; DeGiovanni, P.J.; Piel, B.; Rai, P. Cancer nanomedicine: A review of recent success in drug delivery. Clin. Transl. Med. 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Rathod, D.; Abo-Ali, E.M.; Dukhande, V.V.; Patel, K. EphA2-Receptor Targeted PEGylated nanoliposomes for the treatment of BRAF(V600E) mutated parent- and Vemurafenib-Resistant Melanoma. Pharmaceutics 2019, 11, 504. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Narayanan, S.; Balasubramanian, G.; Sethuraman, S.; Krishnan, U.M. Dual drug loaded nanoliposomal chemotherapy: A promising strategy for treatment of head and neck squamous cell carcinoma. Eur. J. Pharm. Biopharm. 2016, 99, 73–83. [Google Scholar] [CrossRef]
- Sen, K.; Banerjee, S.; Mandal, M. Dual drug loaded liposome bearing apigenin and 5-Fluorouracil for synergistic therapeutic efficacy in colorectal cancer. Colloids Surf. B Biointerfaces 2019, 180, 9–22. [Google Scholar] [CrossRef]
- Ahmed, K.S.; Changling, S.; Shan, X.; Mao, J.; Qiu, L.; Chen, J. Liposome-based codelivery of celecoxib and doxorubicin hydrochloride as a synergistic dual-drug delivery system for enhancing the anticancer effect. J. Liposome Res. 2020, 30, 285–296. [Google Scholar] [CrossRef]
- Lee, R.; Choi, Y.J.; Jeong, M.S.; Park, Y.I.; Motoyama, K.; Kim, M.W.; Kwon, S.H.; Choi, J.H. Hyaluronic acid-decorated glycol chitosan nanoparticles for pH-sensitive controlled release of doxorubicin and celecoxib in nonsmall cell lung cancer. Bioconjugate Chem. 2020, 31, 923–932. [Google Scholar] [CrossRef]
- Mu, L.M.; Ju, R.J.; Liu, R.; Bu, Y.Z.; Zhang, J.Y.; Li, X.Q.; Zeng, F.; Lu, W.L. Dual-functional drug liposomes in treatment of resistant cancers. Adv. Drug Deliv. Rev. 2017, 115, 46–56. [Google Scholar] [CrossRef]
- Saraswat, A.L.; Maher, T.J. Development and optimization of stealth liposomal system for enhanced in vitro cytotoxic effect of quercetin. J. Drug Deliv. Sci. Technol. 2020, 55. [Google Scholar] [CrossRef]
- Hussain, Z.; Khan, S.; Imran, M.; Sohail, M.; Shah, S.W.A.; de Matas, M. PEGylation: A promising strategy to overcome challenges to cancer-targeted nanomedicines: A review of challenges to clinical transition and promising resolution. Drug Deliv. Transl. Res. 2019, 9, 721–734. [Google Scholar] [CrossRef]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Ding, X.; Huang, T.; Zhang, S.; Li, Y.; Xu, L.; Chen, G.; Ying, Y.; Wang, Y.; Feng, Z.; et al. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am. J. Transl. Res. 2019, 11, 5728–5739. [Google Scholar]
- Spender, L.C.; Ferguson, G.J.; Liu, S.; Cui, C.; Girotti, M.R.; Sibbet, G.; Higgs, E.B.; Shuttleworth, M.K.; Hamilton, T.; Lorigan, P.; et al. Mutational activation of BRAF confers sensitivity to transforming growth factor beta inhibitors in human cancer cells. Oncotarget 2016, 7, 81995–82012. [Google Scholar] [CrossRef] [Green Version]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Rathod, D.; Patel, K. Protein kinase C inhibitor anchored BRD4 PROTAC PEGylated nanoliposomes for the treatment of vemurafenib-resistant melanoma. Exp. Cell Res. 2020, 396. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Zhou, H.; Fan, G.; Li, Q. Molecular mechanisms and anticancer therapeutic strategies in vasculogenic mimicry. J. Cancer 2019, 10, 6327–6340. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Enomoto, K.; Zhao, L.; Zhu, Y.J.; Willingham, M.C.; Meltzer, P.; Qi, J.; Cheng, S.Y. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clin. Cancer Res. 2017, 23, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Sahni, J.M.; Gayle, S.S.; Bonk, K.L.; Vite, L.C.; Yori, J.L.; Webb, B.; Ramos, E.K.; Seachrist, D.D.; Landis, M.D.; Chang, J.C.; et al. Bromodomain and extraterminal protein inhibition blocks growth of triple-negative breast cancers through the suppression of aurora kinases. J. Biol. Chem. 2016, 291, 23756–23768. [Google Scholar] [CrossRef] [Green Version]
- Berenguer-Daize, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef] [PubMed]
- Saraswat, A.; Patki, M.; Fu, Y.; Barot, S.; Dukhande, V.V.; Patel, K. Nanoformulation of PROteolysis TArgeting Chimera targeting ‘undruggable’ c-Myc for the treatment of pancreatic cancer. Nanomedicine 2020, 15, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, H.; Gage, J.; Leonard, F.; Srinivasan, S.; Souza, G.R.; Dave, B.; Godin, B. Three-dimensional in vitro co-culture model of breast tumor using magnetic levitation. Sci. Rep. 2014, 4, 6468. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Ni | ARV | ARNIPL-Ni | ARNIPL-ARV |
|---|---|---|---|---|
| IC50 (μM) | 4.35 ± 0.47 | 0.13 ± 0.08 | 0.24 ± 0.05 **** | 0.07 ± 0.06 |
| Group | Size | Zeta Potential | EE of ARV (%) | EE of Ni (%) | DL of ARV (%w/w) | DL of Ni (%w/w) |
|---|---|---|---|---|---|---|
| ARNIPL (without citric acid) | 138.4 ± 6.66 | −25.2 ± 4.41 | 79.68 ± 7.60 | 21.67 ± 2.15 | 0.80 ± 0.08 | 0.43 ± 0.04 |
| ARNIPL (with citric acid) | 99.62 ± 4.78 | −5.34 ± 3.82 | 94.15 ± 3.48 | 97.16 ± 2.33 | 0.94 ± 0.03 | 1.94 ± 0.05 |
| ARNIPL (optimized) | 111.1 ± 6.55 | +13.9 ± 6.62 | 97.80 ± 3.20 | 96.86 ± 2.63 | 1.96 ± 0.05 | 3.87 ± 0.11 |
| %SF ± S.D. | Ni | ARV | ARNIPL |
|---|---|---|---|
| A375R | 47.5 ± 4.19 | 37.5 ± 3.81 | 18.0 ± 2.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Saraswat, A.; Wei, Z.; Agrawal, M.Y.; Dukhande, V.V.; Reznik, S.E.; Patel, K. Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma. Pharmaceutics 2021, 13, 1005. https://doi.org/10.3390/pharmaceutics13071005
Fu Y, Saraswat A, Wei Z, Agrawal MY, Dukhande VV, Reznik SE, Patel K. Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma. Pharmaceutics. 2021; 13(7):1005. https://doi.org/10.3390/pharmaceutics13071005
Chicago/Turabian StyleFu, Yige, Aishwarya Saraswat, Zenghui Wei, Manas Yogendra Agrawal, Vikas V. Dukhande, Sandra E. Reznik, and Ketan Patel. 2021. "Development of Dual ARV-825 and Nintedanib-Loaded PEGylated Nano-Liposomes for Synergistic Efficacy in Vemurafnib-Resistant Melanoma" Pharmaceutics 13, no. 7: 1005. https://doi.org/10.3390/pharmaceutics13071005