Completing the Enalaprilat Excretion Pathway—Renal Handling by the Proximal Tubule

 , , ,
, , ,
Abstract
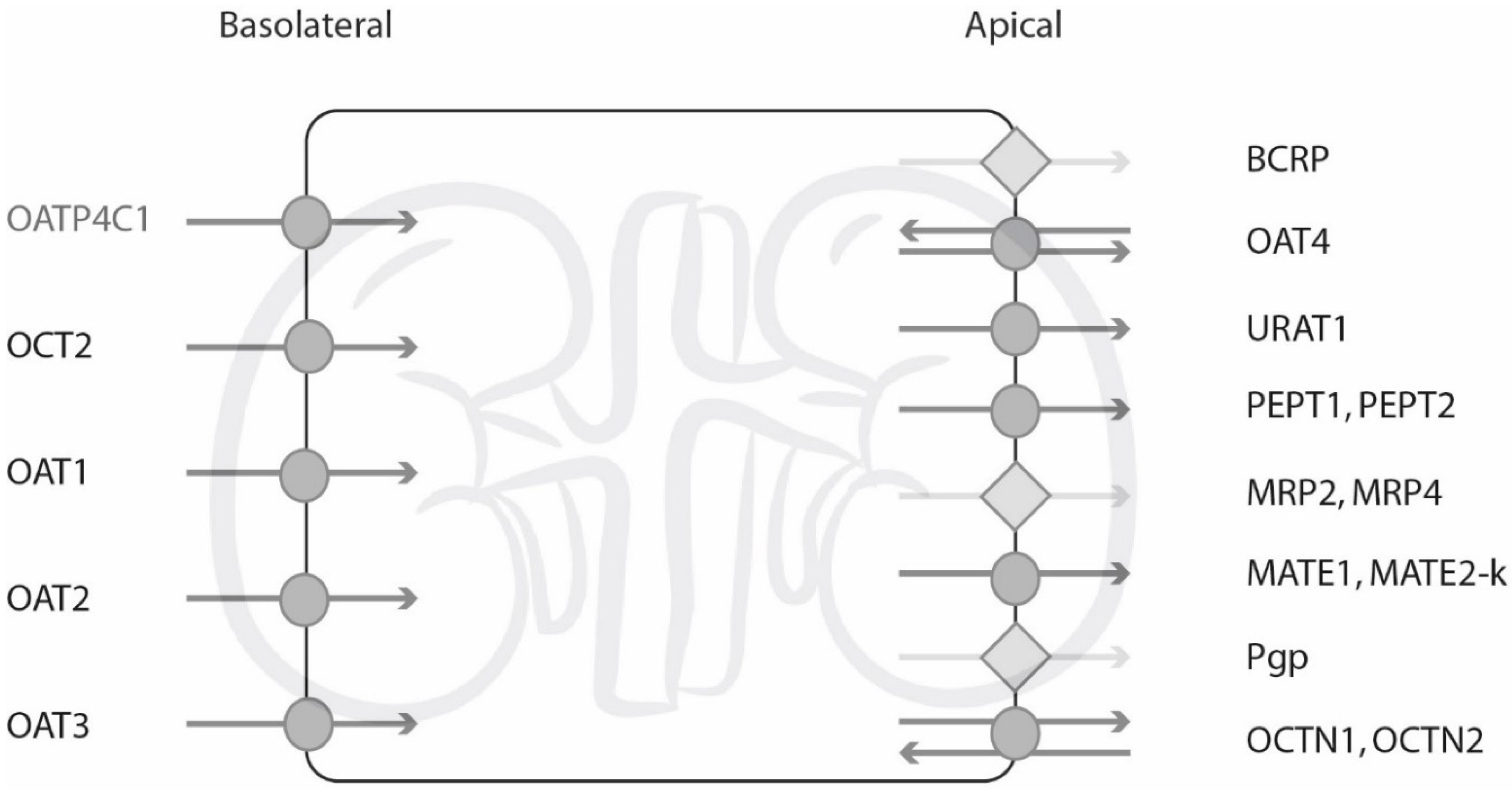
:1. Introduction
2. Material and Methods
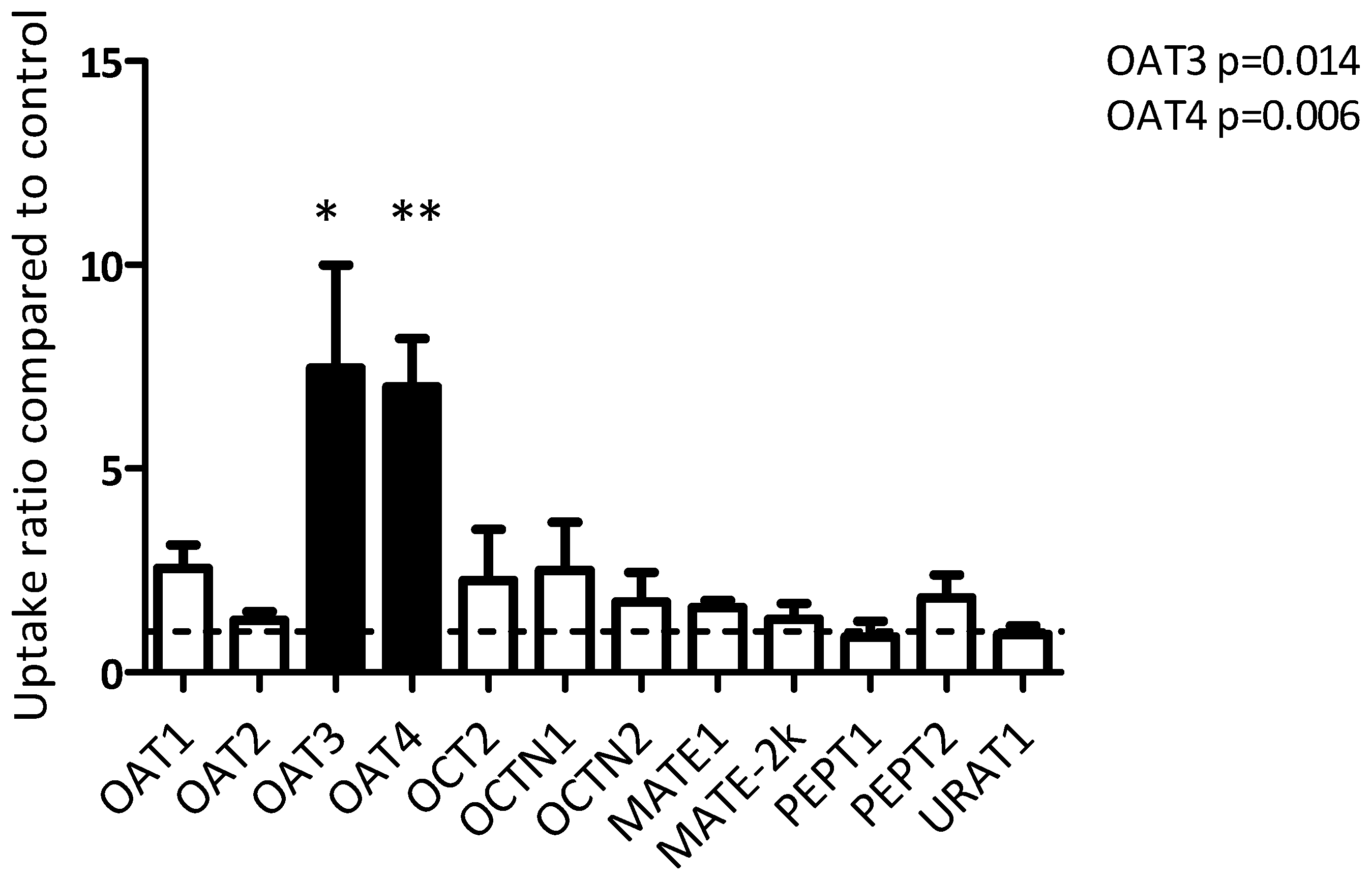
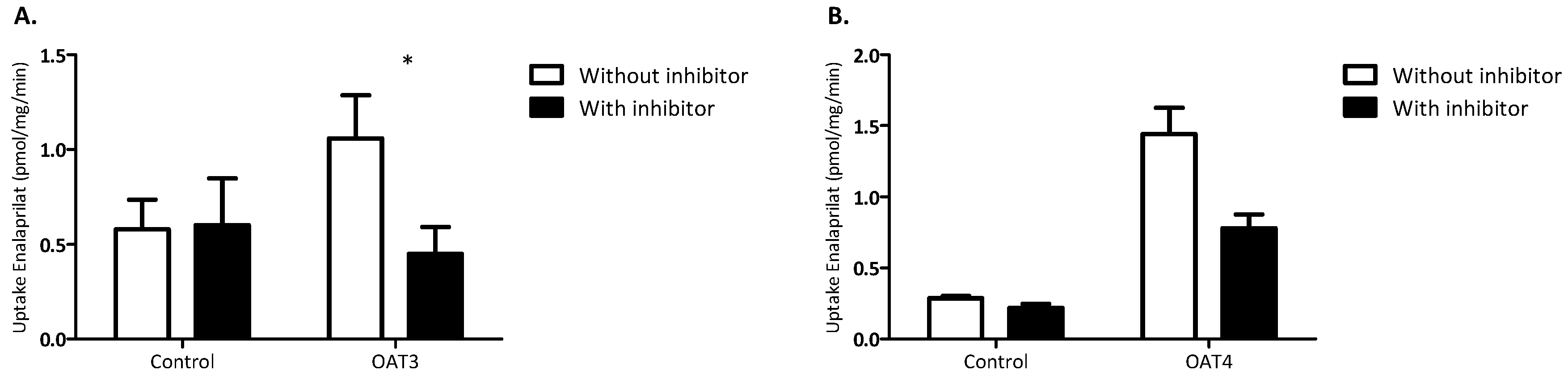
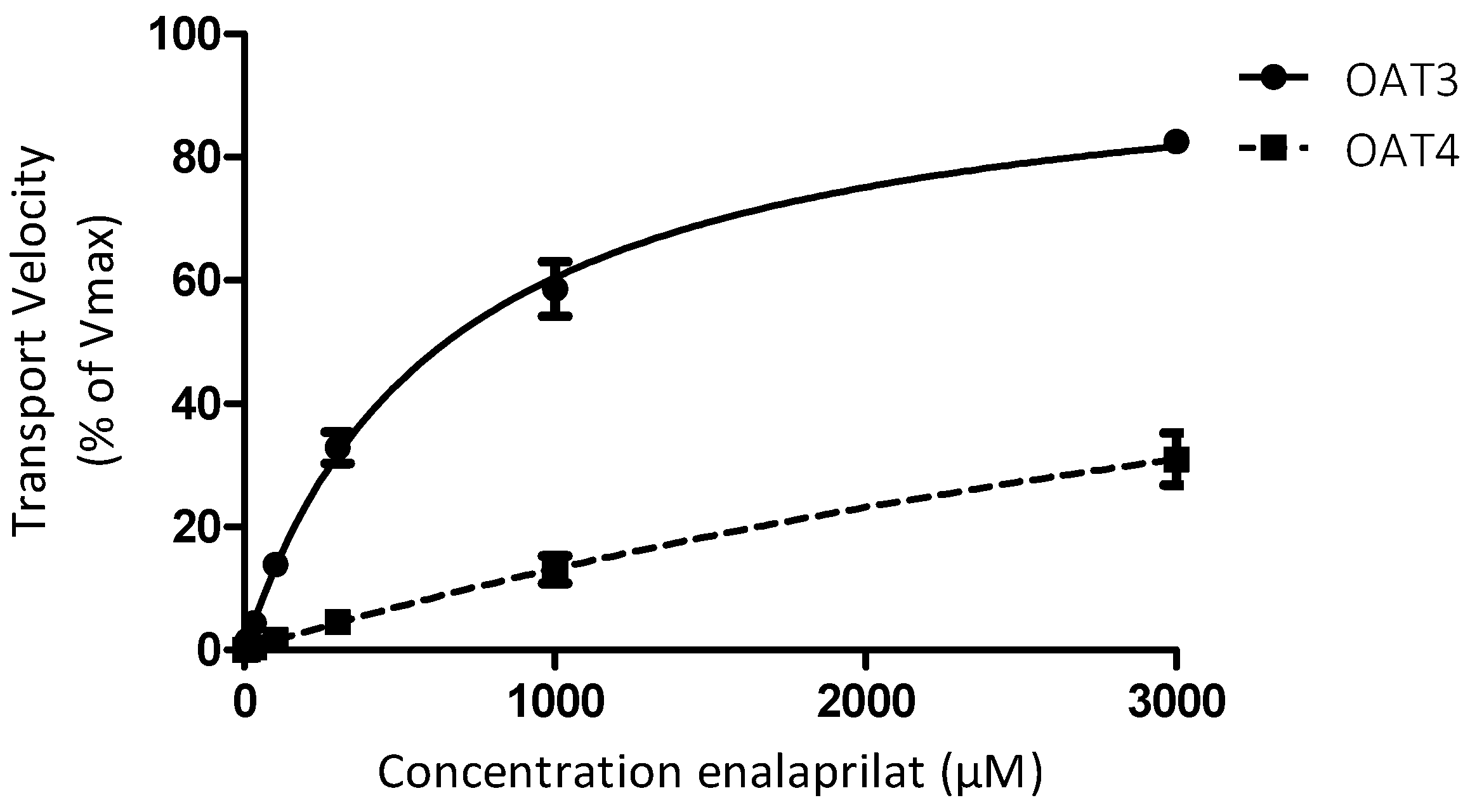
3. Results
4. Discussion
5. Limitations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| [3H]-E1S | [3H]-estrone-sulfate ammonium salt |
| [3H]-MTX | [3H]-methotrexate |
| [3H]-NMQ | [3H]-N-methyl quinidine |
| ACE | angiotensin-converting-enzyme |
| AUC | area under the curve |
| ANOVA | analysis of variance |
| BSA | bovine serum albumin |
| BCRP | breast cancer resistance protein |
| CDCF | carboxy-2′,7′-dichlorofluorescein |
| CES1 | carboxylesterase 1 |
| CI | confidence interval |
| CKD | chronic kidney diseases |
| Cmax | maximum serum concentration |
| CMV | cytomegalovirus |
| DMEM | Dulbecco’s modified Eagle medium |
| E217βG | estradiol 17β-d-glucuronide |
| EYFP | enhanced yellow fluorescent protein |
| FBS | fetal bovine serum |
| HBSS | Hanks’ balanced salt solution |
| HEK293 | human embryonic kidney 293 |
| HPLC | high-performance liquid chromatography |
| IC50 | half maximal inhibitory concentration |
| Km | substrate concentration at which half of this speed is obtained |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| MATE1 | multidrug and toxin extrusion 1 |
| MATE2k | multidrug and toxin extrusion 2k |
| MRP2 | multidrug resistance-associated protein 2 |
| MRP4 | multidrug resistance-associated protein 4 |
| OAT | organic anion transporter |
| OAT1 | organic anion transporter 1 |
| OAT3 | organic anion transporter 3 |
| OATP | organic anion transporting polypeptides |
| OCT2 | organic cation transporter 2 |
| OCTN1 | organic cation/carnitine transporter 1 |
| OCTN2 | organic cation/carnitine transporter 2 |
| PEPT1 | peptide transporter 1 |
| PEPT2 | peptide transporter 2 |
| P-gp | P-glycoprotein |
| SEM | standard error of the mean |
| URAT1 | urate transporter 1 |
| Vmax | maximum transport velocity |
References
- Wilkins, E.; Wilson, L.; Wickramasinghe, K.; Bhatnagar, P.; Leal, J.; Luengo-Fernandez, R.; Burns, R.; Rayner, M.; Townsend, N. European Cardiovascular Disease Statistics 2017; European Heart Network: Brussels, Belgium, 2017. [Google Scholar]
- Lopez-Sendon, J.; Swedberg, K.; McMurray, J.; Tamargo, J.; Maggioni, A.P.; Dargie, H.; Tendera, M.; Waagstein, F.; Kjekshus, J.; Lechat, P.; et al. Expert consensus document on angiotensin converting enzyme inhibitors in cardiovascular disease: The Task Force on ACE-inhibitors of the European Society of Cardiology. Eur. Heart J. 2004, 25, 1454–1470. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.J.; Fischer, U.; Mehta, Z.; Rothwell, P.M. Effects of antihypertensive-drug class on interindividual variation in blood pressure and risk of stroke: A systematic review and meta-analysis. Lancet 2010, 375, 906–915. [Google Scholar] [CrossRef]
- Knutter, I.; Wollesky, C.; Kottra, G.; Hahn, M.G.; Fischer, W.; Zebisch, K.; Neubert, R.H.; Daniel, H.; Brandsch, M. Transport of angiotensin-converting enzyme inhibitors by H+/peptide transporters revisited. J. Pharm. Exp. 2008, 327, 432–441. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cui, Y.; Chung, A.Y.; Shitara, Y.; Sugiyama, Y.; Keppler, D.; Pang, K.S. Vectorial transport of enalapril by Oatp1a1/Mrp2 and OATP1B1 and OATP1B3/MRP2 in rat and human livers. J. Pharm. Exp. 2006, 318, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Rasmussen, H.B.; Linnet, K. In vitro drug metabolism by human carboxylesterase 1: Focus on angiotensin-converting enzyme inhibitors. Drug Metab. Dispos. 2014, 42, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Holenarsipur, V.K.; Gaud, N.; Sinha, J.; Sivaprasad, S.; Bhutani, P.; Subramanian, M.; Singh, S.P.; Arla, R.; Paruchury, S.; Sharma, T.; et al. Absorption and cleavage of enalapril, a carboxyl ester prodrug, in the rat intestine: In vitro, in situ intestinal perfusion and portal vein cannulation models. Biopharm. Drug Dispos. 2015, 36, 385–397. [Google Scholar] [CrossRef]
- Ferslew, B.C.; Kock, K.; Bridges, A.S.; Brouwer, K.L. Role of multidrug resistance-associated protein 4 in the basolateral efflux of hepatically derived enalaprilat. Drug Metab. Dispos. 2014, 42, 1567–1574. [Google Scholar] [CrossRef] [Green Version]
- Mujais, S.K.; Quintanilla, A.; Zahid, M.; Koch, K.; Shaw, W.; Gibson, T. Renal handling of enalaprilat. Am. J. Kidney Dis. 1992, 19, 121–125. [Google Scholar] [CrossRef]
- Noormohamed, F.H.; McNabb, W.R.; Lant, A.F. Pharmacokinetic and pharmacodynamic actions of enalapril in humans: Effect of probenecid pretreatment. J. Pharm. Exp. 1990, 253, 362–368. [Google Scholar]
- Zhang, D.; Hop, C.; Patilea-Vrana, G.; Gampa, G.; Seneviratne, H.K.; Unadkat, J.D.; Kenny, J.R.; Nagapudi, K.; Di, L.; Zhou, L.; et al. Drug Concentration Asymmetry in Tissues and Plasma for Small Molecule-Related Therapeutic Modalities. Drug Metab. Dispos. 2019, 47, 1122–1135. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The organic anion transporter (OAT) family: A systems biology perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Ni, Y.; Duan, Z.; Zhou, D.; Liu, S.; Wan, H.; Gui, C.; Zhang, H. Identification of Structural Features for the Inhibition of OAT3-Mediated Uptake of Enalaprilat by Selected Drugs and Flavonoids. Front. Pharm. 2020, 11, 802. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Te Brake, L.H.; van den Heuvel, J.J.; Buaben, A.O.; van Crevel, R.; Bilos, A.; Russel, F.G.; Aarnoutse, R.E.; Koenderink, J.B. Moxifloxacin Is a Potent In Vitro Inhibitor of OCT- and MATE-Mediated Transport of Metformin and Ethambutol. Antimicrob. Agents Chemother. 2016, 60, 7105–7114. [Google Scholar] [CrossRef] [Green Version]
- El-Sheikh, A.A.; van den Heuvel, J.J.; Koenderink, J.B.; Russel, F.G. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J. Pharm. Exp. 2007, 320, 229–235. [Google Scholar] [CrossRef] [Green Version]
- El-Sheikh, A.A.; Greupink, R.; Wortelboer, H.M.; van den Heuvel, J.J.; Schreurs, M.; Koenderink, J.B.; Masereeuw, R.; Russel, F.G. Interaction of immunosuppressive drugs with human organic anion transporter (OAT) 1 and OAT3, and multidrug resistance-associated protein (MRP) 2 and MRP4. Transl. Res. 2013, 162, 398–409. [Google Scholar] [CrossRef]
- Lempers, V.J.; van den Heuvel, J.J.; Russel, F.G.; Aarnoutse, R.E.; Burger, D.M.; Brüggemann, R.J.; Koenderink, J.B. Inhibitory Potential of Antifungal Drugs on ATP-Binding Cassette Transporters P-Glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob. Agents Chemother. 2016, 60, 3372–3379. [Google Scholar] [CrossRef] [Green Version]
- van der Velden, M.; Bilos, A.; van den Heuvel, J.; Rijpma, S.R.; Hurkmans, E.G.E.; Sauerwein, R.W.; Russel, F.G.M.; Koenderink, J.B. Proguanil and cycloguanil are organic cation transporter and multidrug and toxin extrusion substrates. Malar. J. 2017, 16, 422. [Google Scholar] [CrossRef]
- Henjakovic, M.; Hagos, Y.; Krick, W.; Burckhardt, G.; Burckhardt, B.C. Human organic anion transporter 2 is distinct from organic anion transporters 1 and 3 with respect to transport function. Am. J. Physiol. Ren. Physiol. 2015, 309, F843–F851. [Google Scholar] [CrossRef] [Green Version]
- van Aubel, R.A.; Smeets, P.H.; Peters, J.G.; Bindels, R.J.; Russel, F.G. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: Putative efflux pump for urinary cAMP and cGMP. J. Am. Soc. Nephrol. 2002, 13, 595–603. [Google Scholar]
- Ivanyuk, A.; Livio, F.; Biollaz, J.; Buclin, T. Renal Drug Transporters and Drug Interactions. Clin. Pharm. 2017, 56, 825–892. [Google Scholar] [CrossRef]
- Yin, J.; Wang, J. Renal drug transporters and their significance in drug-drug interactions. Acta Pharm. Sin. B 2016, 6, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Ebner, T.; Ishiguro, N.; Taub, M.E. The Use of Transporter Probe Drug Cocktails for the Assessment of Transporter-Based Drug-Drug Interactions in a Clinical Setting-Proposal of a Four Component Transporter Cocktail. J. Pharm. Sci. 2015, 104, 3220–3228. [Google Scholar] [CrossRef]
- Windass, A.S.; Lowes, S.; Wang, Y.; Brown, C.D. The contribution of organic anion transporters OAT1 and OAT3 to the renal uptake of rosuvastatin. J. Pharm. Exp. 2007, 322, 1221–1227. [Google Scholar] [CrossRef]
- Hasannejad, H.; Takeda, M.; Taki, K.; Shin, H.J.; Babu, E.; Jutabha, P.; Khamdang, S.; Aleboyeh, M.; Onozato, M.L.; Tojo, A.; et al. Interactions of human organic anion transporters with diuretics. J. Pharm. Exp. 2004, 308, 1021–1029. [Google Scholar] [CrossRef]
- Bindschedler, M.; Degen, P.; Flesch, G.; de Gasparo, M.; Preiswerk, G. Pharmacokinetic and pharmacodynamic interaction of single oral doses of valsartan and furosemide. Eur. J. Clin. Pharm. 1997, 52, 371–378. [Google Scholar] [CrossRef]
- Van Hecken, A.M.; Verbesselt, R.; Buntinx, A.; Cirillo, V.J.; De Schepper, P.J. Absence of a pharmacokinetic interaction between enalapril and frusemide. Br. J. Clin. Pharm. 1987, 23, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Posada, M.M.; Cannady, E.A.; Payne, C.D.; Zhang, X.; Bacon, J.A.; Pak, Y.A.; Higgins, J.W.; Shahri, N.; Hall, S.D.; Hillgren, K.M. Prediction of Transporter-Mediated Drug-Drug Interactions for Baricitinib. Clin. Transl. Sci. 2017, 10, 509–519. [Google Scholar] [CrossRef]
- Schwartz, J.B.; Taylor, A.; Abernethy, D.; O’Meara, M.; Farmer, J.; Young, J.; Nelson, E.; Pool, J.; Mitchell, J.R. Pharmacokinetics and pharmacodynamics of enalapril in patients with congestive heart failure and patients with hypertension. J. Cardiovasc. Pharm. 1985, 7, 767–776. [Google Scholar] [CrossRef]
- Hagos, Y.; Stein, D.; Ugele, B.; Burckhardt, G.; Bahn, A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J. Am. Soc. Nephrol. 2007, 18, 430–439. [Google Scholar] [CrossRef]
- Drozdzik, M.; Busch, D.; Lapczuk, J.; Müller, J.; Ostrowski, M.; Kurzawski, M.; Oswald, S. Protein Abundance of Clinically Relevant Drug Transporters in the Human Liver and Intestine: A Comparative Analysis in Paired Tissue Specimens. Clin. Pharm. 2019, 105, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, K.L.; Aleksunes, L.M.; Brandys, B.; Giacoia, G.P.; Knipp, G.; Lukacova, V.; Meibohm, B.; Nigam, S.K.; Rieder, M.; de Wildt, S.N. Human Ontogeny of Drug Transporters: Review and Recommendations of the Pediatric Transporter Working Group. Clin. Pharm. 2015, 98, 266–287. [Google Scholar] [CrossRef] [Green Version]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.M.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L.R. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper From the International Transporter Consortium. Clin. Pharm. 2018, 104, 900–915. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | Model Substrate (Assay) | Transport, Approx. Fold of Control (p-Value) | Inhibition of Transport Shown (Inhibitor) | Reference |
|---|---|---|---|---|
| OAT1 | [3H]-MTX (cells) | 3 fold (p < 0.05) | Yes (probenecid) | El Sheikh et al., 2013 [17] |
| OAT3 | [3H]-MTX (cells) | 3 fold (p < 0.05) | Yes (probenecid) | El Sheikh et al., 2013 [17] |
| OCT2 | Cycloguanil (cells) | 12 fold (p < 0.03) | Yes (multiple, inhibition of ASP most significant for Quinine) | Van der Velden et al., 2017 [19] |
| MATE1 | Cycloguanil (cells) | 4 fold (p < 0.01) | - | Van der Velden et al., 2017 [19] |
| MATE2K | Cycloguanil (cells) | 5 fold (p < 0.001) | - | Van der Velden et al., 2017 [19] |
| MRP2 | [3H]-MTX (vesicles) | 4 fold | - | Lempers et al., 2016 [18] |
| MRP4 | [3H]- E217βG (vesicles) | 5 fold | - | Lempers et al., 2016 [18] |
| P-GP | [3H]-NMQ (vesicles) | 5 fold | - | Lempers et al., 2016 [18] |
| BCRP | [3H]-E1S (vesicles) | 11 fold | - | Lempers et al., 2016 [18] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smeets, N.J.L.; Litjens, C.H.C.; van den Heuvel, J.J.M.W.; van Hove, H.; van den Broek, P.; Russel, F.G.M.; Koenderink, J.B.; de Wildt, S.N. Completing the Enalaprilat Excretion Pathway—Renal Handling by the Proximal Tubule. Pharmaceutics 2020, 12, 935. https://doi.org/10.3390/pharmaceutics12100935
Smeets NJL, Litjens CHC, van den Heuvel JJMW, van Hove H, van den Broek P, Russel FGM, Koenderink JB, de Wildt SN. Completing the Enalaprilat Excretion Pathway—Renal Handling by the Proximal Tubule. Pharmaceutics. 2020; 12(10):935. https://doi.org/10.3390/pharmaceutics12100935
Chicago/Turabian StyleSmeets, Nori J. L., Carlijn H. C. Litjens, Jeroen J. M. W. van den Heuvel, Hedwig van Hove, Petra van den Broek, Frans G. M. Russel, Jan B. Koenderink, and Saskia N. de Wildt. 2020. "Completing the Enalaprilat Excretion Pathway—Renal Handling by the Proximal Tubule" Pharmaceutics 12, no. 10: 935. https://doi.org/10.3390/pharmaceutics12100935